

Abstract
Oxidative stress is a pathway of injury that is common to almost all neurological conditions. Hence, methods to scavenge radicals have been extensively tested for neuroprotection. However, saving neurons alone may not be sufficient in treating CNS disease. Here, we tested the cytoprotective actions of the glutathione precursor GCEE in brain endothelium. First, oxidative stress was induced in a human brain microvascular endothelial cell line by exposure to H2O2. Addition of GCEE significantly reduced formation of reactive oxygen species, restored glutathione levels which were reduced in the presence of H2O2, and decreased cell death during H2O2-mediated injury. Next, we asked whether GCEE can also protect brain endothelial cells against oxygen-glucose deprivation (OGD). As expected, OGD disrupted mitochondrial membrane potentials. GCEE was able to ameliorate these mitochondrial effects. Concomitantly, GCEE significantly decreased endothelial cell death after OGD. Lastly, our in-vivo experiments using a mouse model of brain trauma show that post-trauma (10 min after CCI) administration of GCEE by intraperitoneal injection results in a decrease in acute blood-brain barrier permeability. These data suggest that the beneficial effects of GCEE on brain endothelial cells and microvessels may contribute to its potential efficacy as a neuroprotective agent in traumatic brain injury.
Keywords: GCEE, glutathione, oxidative stress, endothelial, neuroprotection, blood-brain-barrier
Introduction
Oxidative stress is a major pathogenic mechanism in almost all CNS diseases (Andersen 2004, Hall et al. 2010, Mustafa et al. 2010, Roberts et al. 2009, Ayer & Zhang 2008). The brain is especially vulnerable because it contains high amounts of polyunsaturated fatty acids, which are targets of lipid peroxidation. In addition, it has lower levels of glutathione (Commandeur et al. 1995), glutathione peroxidase, and superoxide dismutases than other organ systems (Dringen 2000), giving it a relatively low antioxidant capacity. Therapeutic strategies that target free radicals are a mainstay of investigations of potential neuroprotective agents (Hall et al. 2010, Mustafa et al. 2010, Roberts et al. 2009, Besson 2009). However, recent studies suggest that protecting neurons alone may not be enough. The entire neurovascular unit, including neuronal, glial and vascular compartments, must be protected (Lok et al. 2007, Abbott et al. 2006, Simard & Nedergaard 2004, Zlokovic 2005).
In particular, microvascular endothelial cells of the brain are important targets of oxidative stress because of their high content of polyunsaturated fatty acids. Endothelial cells participate in key functions within the neurovascular unit, including delivery of oxygen, autoregulaton of regional cerebral blood flow, and trophic support for neurons. Thus, endothelial dysfunction has deleterious consequences for other cell types in the brain, and prevention of oxidative stress in brain endothelium may be an extremely important therapeutic target for a wide range of neurological disorders.
A well-known strategy to combat the effects of oxidative damage is that of restoring glutathione (GSH) levels in the target organ. For example, increasing GSH levels with the administration of N-acetylcysteine is a clinically proven therapy used in the treatment of acetaminophen-induced hepatoxicity. The synthetic agent, gamma-glutamylcysteinyl ethyl ester (GCEE) has been shown to be an effective way for boosting endogenous levels of the key antioxidant GSH and thus blocking oxidative neuronal damage in many experimental models (Drake et al. 2002). In the present study, we asked whether GCEE can reduce oxidative injury in brain endothelium.
Materials and Methods
Cell Culture
A human brain microvascular endothelial cell line was cultured in RPMI 1640 (Invitrogen, Carlsbad, CA,) + 10% FBS + 10% NuSerum + 2 nM L-Glutamine + 1 mM Sodium Pyruvate + MEM non essential Amino Acids + MEM vitamins + Penicillin/Streptomycin (P/S) 100 units/ml (Complete Media). These cells have been previously characterized for brain endothelial phenotypes (Callahan et al. 2004). All experimental procedures were conducted in serum-free media. Although serum-containing media is necessary during the growth phase of the endothelial cells, serum-free media is used during the experiment to reduce the confounding effect of growth factors which may be present in variable amounts in different preparations of serum-containing media.
Measurement of Reactive Oxygen Species (ROS) Formation
Cells were grown to 70% confluence. For measuring ROS formation, the cells were placed in serum-free RPMI media without phenol red to prevent interference of the red dye with the assay for measuring ROS. Cells were pretreated with vehicle or GCEE (Bachem, Torrance, CA) for 1 hr at 37C. The media was then removed and the cells were incubated with 10uM dichlorofluorobromane (DCF) (Carlsbad, CA) for 30 min at 37C. After 30 min, the media containing DCF was removed and the wells were rinsed twice with serum-free RPMI. Afterwards, the following conditions were applied: vehicle alone, 100uM H2O2 +vehicle, or 100uM H2O2 + 750uM GCEE. ROS formation was examined using a plate reader (SpectraMax M5, Bio-Tek Instruments, Inc) with excitation at 488nm and emission at 525nm. Values were recorded at 2.5 min intervals for 30 min.
Measurement of Glutathione (GSH)
Cells were grown to 70% confluence and then pretreated with GCEE in serum-free RPMI media without phenol red for 1h at 37C. Cells were then treated with vehicle alone, 100uM H2O2, or 100uM H2O2 + 750uM GCEE for 1h and 2h. Media was removed and the cells were rinsed twice with serum-free RPMI without phenol red and incubated with 40uM of monochlorobimane (mBCl) (St. Louis, MO) for 30 min at 37C. GSH was measured after 30 min with a plate reader with excitation at 360nm and emission at 460nm.
Induction of Oxidative Injury with H2O2
Cells were plated in wells pre-coated with 10ug/ml of human fibronectin factor (BD Biosciences). When 70–80% confluent, cells were placed in serum-free media with vehicle or 100uM H2O2 for 18 h. Vehicle or GCEE was added 1 h prior to the addition of H2O2 and present in the media for the entire duration of H2O2 incubation. After 18 h, determination of cell survival was made using MTT assay, LDH assay, or quantification of cells that exclude Trypan blue.
MTT Assay
The MTT assay measures mitochondrial activity and is also commonly used as a measure of cell viability. This assay measures the mitochondrial reduction of 3-(4.5-dimethylthiazol-2-yl) 2,5-diphenyl-tetrazolium bromide (MTT) (Sigma, St. Louis, MO) to a blue formazan product. At the completion of each experimental condition, cells were placed in media with 0.4% MTT. After 60 min (37°C), media was removed and cells were dissolved in DMSO. Formazan formation was measured by reading absorbance at 570nm with a reference setting of 630nm on a microplate reader. The optical density (OD) of each sample was expressed as a percentage of the OD of the non-injured control.
LDH Assay
Cytotoxicity was quantified by a standard measurement of lactate dehydrogenase (LDH) release using the LDH assay kit (Roche, Indianapolis, IN).
Trypan Blue Exclusion Test
After removing the media, 50ul of 0.05% trypsin was added to each well. After 3 min, serum-containing media was added to inactivate trypsin. The media and cells were centrifuged at 1000rpm for 5 min and the pellet was resuspended in PBS with 50% trypan blue. A visible light microscope was used to count cells.
Oxygen/Glucose Deprivation
When 70–80% confluent, cells were pretreated with vehicle or GCEE prepared in serum-free RPMI media 1 h prior to OGD. The OGD protocol consisted of the following: The media was changed to DMEM without glucose and without glutamine, with the addition of vehicle or GCEE. Hypoxia was induced by placing the cells in a chamber (Billups-Rothenberg) and perfusing the chamber with 90% N2, 5%H2, and 5%CO2 for 30 minutes. The chamber was then sealed and kept at 37°C for 8h. At the end of 8h, the media was changed to serum-free RPMI (with glucose and glutamine), again with vehicle or GCEE, and the cells were re-oxygenated in the incubator at 37°C for 16h. After 16h, cell survival was determined using MTT assay.
Measurement of Mitochondrial Membrane Potentials
To analyze the breakdown of mitochondrial membrane potentials by fluorescence-activated cell sorting/scanning (FACS) analysis, cells were subjected to OGD for 8h and re-oxygenation for 16h. Vehicle or GCEE was added 1h prior to OGD and present during OGD/reoxygenation. At the end of the 24h, media was aspirated and cells were washed, trypsinized, re-suspended in PBS and stained with 500nM tetramethylrhodamine ethylester (TMRE) (Invitrogen, Carlsbad, CA) for 30 min at 25C in the dark. Cells were washed again with PBS and fluorescence was measured in the red channel.
Controlled Cortical Impact
The trauma protocol was approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and complied with the NIH Guide for the Care and Use of Laboratory Animals. Male C57Bl/6 mice (10 weeks of age), weighing 22 to 26 kg, were used. They were given food and water ad libidum and were housed in pathogen free facilities with 12h day/night cycles. The controlled cortical impact (CCI) model was used as previously described with minor modifications (You et al. 2007). Mice were anesthetized with 4% isoflurane (Anaquest, Memphis, TN) in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH) and positioned in a stereotaxic frame. Anesthesia was maintained using 2–3% isoflurane. A right craniotomy was made using a portable drill and 5 mm trephine over the right parieto-temporal cortex, and the bone flap was removed. Mice were subjected to CCI using a pneumatic cylinder with a 3-mm flat-tip impounder, velocity 6 m/s, depth of 0.6mm, and 150ms impact duration. Following CCI, the scalp was sutured closed and mice were returned to their cages to recover from anesthesia. Vehicle (sterile water) or GCEE at a dose of 150mg/kg body weight was administered by intraperitoneal injection 10 min after trauma. An injection volume of 100 microliters was used for vehicle and GCEE injections. Investigators were blinded to treatment type (vehicle vs. GCEE) during surgery and data acquisition. Vehicle- and drug-treated mice were subjected to CCI concomitantly. Equal numbers (7 each) of male C57Bl/6 mice (n=7 per group) were used. One mouse died 25 min after CCI and one mouse had inadequate injection of Evans blue. These two mice, both of which were found during the analysis to belong to the vehicle group, were excluded from the data analysis. The final analysis included data from 5 vehicle-injected and 7 GCEE-injected mice.
Determination of Blood-brain-barrier permeability after CCI
Evans Blue (2% wt/vol in PBS) in a volume of 4ml/kg was given by tail vein injection 30 min after CCI and allowed to circulate for 1h 30min. 2h after CCI, the mice were sacrificed under deep anesthesia by intracardiac perfusion with 60cc PBS, at the end of which clear PBS was seen to return to the right atrium in all the animals. The brains were removed; the hemispheres were separated, then homogenized in 50% trichloroacetic acid (wt/vol) solution and centrifuged at 10,000rpm for 20 min. The supernatant, which contains the extracted dye, was diluted with ethanol (1:3), and 100ul from each sample was read using a fluorescence plate reader (ex/em:620 nm/680 nm). Calculations were based on Evans blue external standards (25–2000 ng/ml) in the solvent 50% TCA /ethanol (1:3). The amount of Evans blue was quantified according to a linear standard curve and is expressed as grams of Evans blue per hemisphere.
GCEE Dosage for CCI
The dosage of 150mg/kg (150ug/g) body weight was based on previous reports (Drake et al. 2003, Lai et al. 2008), in which GCEE administration by intraperitonal injection was correlated with improved outcomes in animals models of oxidative stress or brain trauma. This dose is comparable to the dose of the related compound N-acetylcysteine used clinically for the treatment of acetaminophen overdose in humans.
Statistical Analysis
All of the in-vitro experiments were done in duplicate or triplicate. Quantitative data for all experiments were analyzed using ANOVA followed by Tukey HSD for multiple comparisons, and expressed as mean +/− SD. (Post-hoc tests were not performed for the analysis of Evans blue extravasation because there were fewer than 3 groups.) Statistical significance was set at p<0.05.
Results
GCEE decreases formation of reactive oxygen species (ROS) in brain microvascular endothelial cells during oxidative injury with H2O2
Oxidative stress was induced by incubation of human brain microvascular endothelial cells (HBMECs) with 100uM H2O2. GCEE (750uM) or vehicle was added to cells 1h prior to and during H2O2 incubation. Reactive oxygen species (ROS) formation increased almost immediately after addition of 100uM H2O2, and reached a plateau at twice the baseline level by 10 min. Addition of GCEE along reduced the level of ROS formation to baseline (p<0.01) (Fig.1).
Fig. 1. GCEE decreases formation of reactive oxygen species (ROS) during oxidative injury with H2O2.
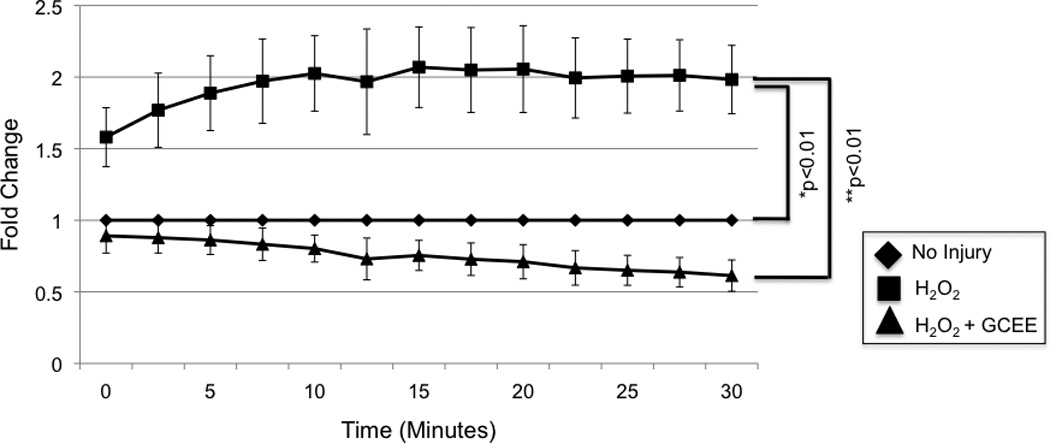
1h incubation with 100uM H2O2 resulted in an increase of ROS to twice the baseline level. Incubation with GCEE for 1 h prior to and during H2O2 incubation reduced the level of ROS formation to baseline (*p<0.01, comparing addition of GCEE vs. GCEE to cells incubated with H2O2). Values are expressed as fold change in ROS (+/− SD) compared to un-injured controls. n = 3 experiments
GCEE restores glutathione levels in brain microvascular endothelial cells during oxidative injury induced by H2O2
Incubation of HBMECs with 100uM H2O2 for 1h resulted in a decrease in intracellular GSH levels to 78% of un-injured cells (p<.01). Addition of GCEE (750uM) 1h prior to and during H2O2 incubation resulted in preservation of GSH levels that are 98% of un-injured cells (p<0.05) (Fig. 2). Similar results were obtained when HBMECs were incubated with H2O2 for 2h (data not shown).
Fig. 2. GCEE restores glutathione levels in brain microvascular endothelial cells during oxidative injury induced by H2O2.
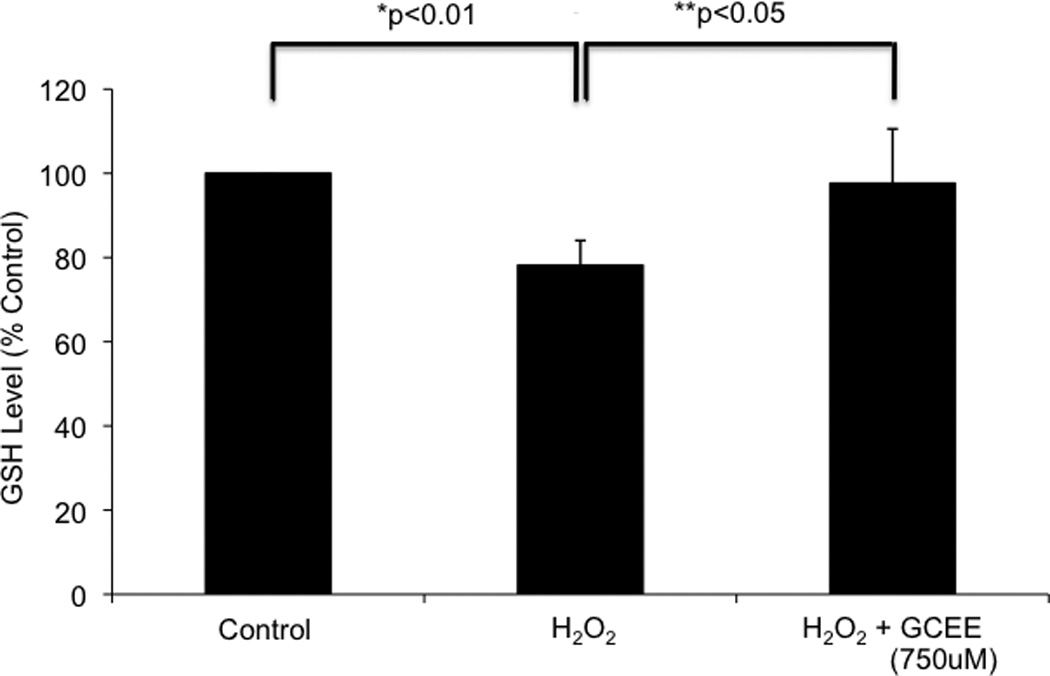
1h incubation with 100uM H2O2 resulted in a decrease in GSH levels to 78% of baseline (*p<0.01). Incubation with GCEE for 1h prior to and during H2O2 incubation prevented this decrease, resulting in GSH levels that are 98% of baseline (**p<0.05, comparing the addition of GCEE vs. vehicle). Values are expressed as fold change in GSH levels (+/− SD) compared to un-injured controls. n = 4 experiments
GCEE increases survival of brain microvascular endothelial cells during oxidative injury with H2O2
Oxidative stress was induced by incubation of HBMECs with 100uM H2O2 for 18h. H2O2 reduced cell survival (Fig. 3a), measured by MTT assay, to 32% of the normal baseline found in untreated cells (p<0.05) (Fig. 3b). Addition of GCEE (500 – 1000uM) significantly restored MTT levels to normal range (p<.05) (Fig. 3b). These results on cell survival were validated with an LDH cytotoxicity assay (Fig. 3c) and with determination of cell viability using Trypan blue exclusion (Fig 3d).
Fig. 3. GCEE increases viability of brain microvascular endothelial cells during oxidative injury with H2O2.
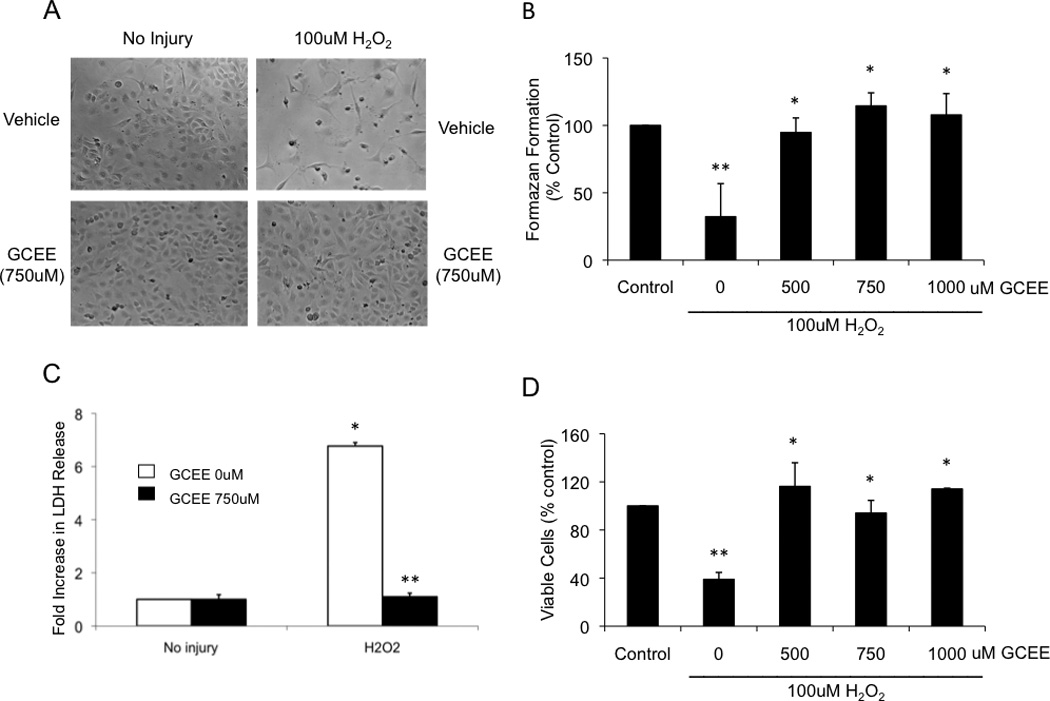
The viability of brain microvascular endothelial cells was followed by light microscopy (a), mitochondrial activity (b), lactate dehydrogenase release (c) and trypan blue exclusion test (d), as described in Materials and methods. The cells were treated for 18h either with vehicle or with 100uM H2O2 with the indicated concentration of GCEE. In all of these assays, the relevant parameter formazen formation, mitochondrial activity, % LDH release, and number of viable cells were expressed as a percentage compared to untreated cells. n=3 experiments for mitochondrial activity and for LDH release; n=2 for measurement of trypan-blue exclusion test; *p<.05; **p<.05.
GCEE improves endothelial mitochondrial membrane potential during oxygen-glucose deprivation
To rule out the possibility that GCEE protected endothelial cells through a direct chemical reaction with H2O2, we further tested the endothelial protective actions of GCEE in a model of oxygen-glucose deprivation (OGD), which mimics metabolic stress. After 8h OGD and 16h re-oxygenation, mitochondrial membrane potentials were decreased to 60% compared to that of untreated cells (p< 0.05). Addition of GCEE (750uM) 1h prior to and during OGD/re-oxygenation significantly restored mitochondrial membrane potentials to 86% of control levels (p<0.05) (Fig.4b).
Fig. 4. GCEE improves endothelial mitochondrial membrane potential and increases viability of HBMECs during oxygen-glucose deprivation.
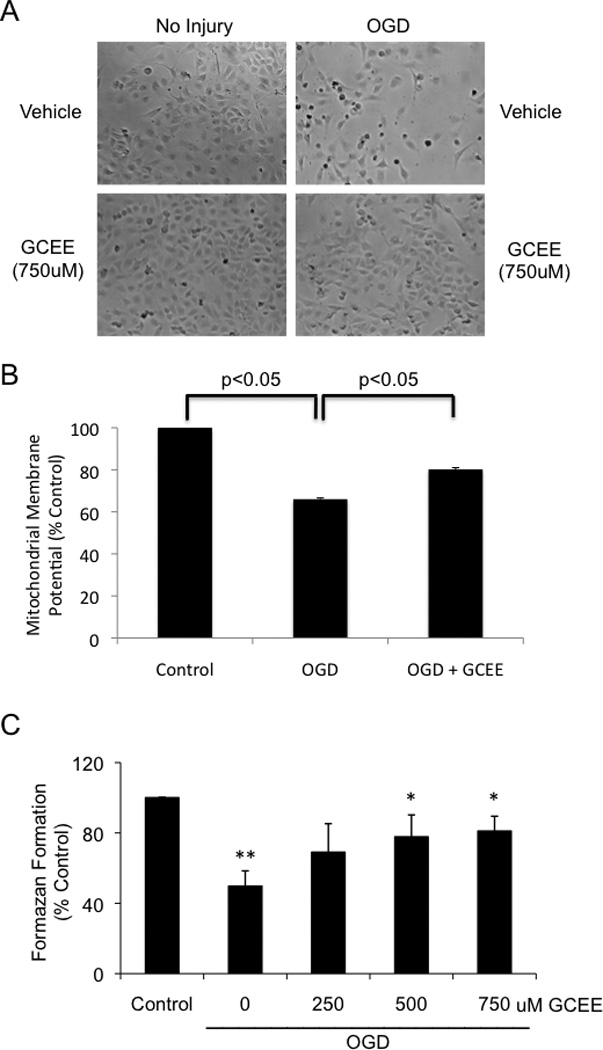
The effect of OGD on brain microvascular endothelial cells was followed by light microscopy (a), measurements of endothelial mitochondrial membrane potentials (b), and mitochondrial activity (c). The cells were treated with 8h OGD followed by 16h reoxygenation with the addition of 750uM GCEE or vehicle. n = 3 experiments for measurements of mitochondrial membrane potential, n = 4 experiments for mitochondrial activity; *p<.05.
GCEE increases survival of brain microvascular endothelial cells during oxygen-glucose deprivation
8h of OGD followed by 16h of re-oxygenation reduced cell survival to less than 50% of control levels (p<0.05). Addition of GCEE (500uM and 750uM) 1h prior to and during OGD and re-oxygenation significantly increased cell survival (p<0.05) up to 81% of normoxia controls (Fig. 4a, 4c).
GCEE decreases acute blood-brain-barrier permeability in a mouse model of traumatic brain injury
In a mouse CCI model of traumatic brain injury, acute blood-brain-barrier permeability was assessed by measuring Evans blue dye extravasation 2h after CCI. There was a 43% reduction in Evans blue dye extravasation in mice that were given GCEE by intraperitonal injection 10 min after CCI (p<.01).
Discussion
The presented data provide evidence that GCEE (250uM to 1000uM) protects brain microvascular endothelial cells in two in-vitro models of endothelial injury: incubation with H2O2 to simulate oxidative stress, and OGD to simulate energetic failure. During H2O2-induced injury, GCEE (750uM) decreased ROS formation and preserved intracellular GSH levels at an early time point (1h), before cell death normally occurs. In OGD-induced injury, GCEE (750uM) improved endothelial mitochondrial membrane potentials. This beneficial effect on mitochondria is consistent with previous reports, in which GCEE reduced the formation of 3-nitrotyrosine and protein carbonyl residues, dampened the release of cytochrome c, and decreased overall mitochondrial swelling in isolated mitochondria challenged with ONOO (Drake et al. 2003). In our study, the overall effect of GCEE, measured at 18h after H2O2 incubation and 24h after OGD, is an increase in endothelial cell survival.
Our in-vivo data complement the results of the in-vitro experiments. In a controlled cortical impact model in mice, intraperitoneal injection of GCEE (150mg/kg) 10 min after trauma reduced acute blood-brain-barrier permeability. A beneficial effect of GCEE in experimental brain trauma has been reported by other groups. Administration of GCEE after CCI in mice restored brain GSH levels, reduced markers of autophagy, and improved behavioral indices (Lai et al. 2008, Clark et al. 2008). Similarly, GCEE administration to rats after TBI resulted in significantly decreased levels of protein carbonyls and 3-nitrotyrosine, indicators of oxidation and nitration which were elevated after trauma (Reed et al. 2009)).
Protecting the brain against oxidative stress has been the subject of many investigations. Reduced GSH is an important cellular antioxidant, and ameliorates the deleterious effects of reactive oxygen and nitrogen species (ROS/RNS) formed during normal and abnormal processes. Decreased brain GSH levels are associated with mitochondrial damage, which may contribute to the progression of chronic neuropathologies, including Parkinson’s and Alzheimer’s disease (Schulz et al. 2000). Parkinson’s disease is characterized by selective decreases in GSH within the affected brain region (Chinta et al. 2006, Perry et al. 1982, Perry & Yong 1986). Unfortunately, pharmacologic strategies to increase brain GSH levels have been limited by its inefficient transport across the blood-brain barrier, and its extracellular degradation into its amino acid components (Meister & Anderson 1983). GCEE is a pharmacologically important way to circumvent this problem.
GSH synthesis is mediated by gamma-glutamylcysteine synthetase and GSH synthetase. The limiting amino acid cysteine is present in micromolar levels in the brain, while glutamate and glycine are present in millimolar levels. Hence the provision of the GSH precursor gamma-glutamylcysteine has been proposed as an alternative to GSH administration (Anderson & Luo 1998). With the availability of this rate limiting substrate, GSH formation is effected through the addition of glycine by GSH synthetase (Butterfield et al. 2002). The efficiency of gamma-glutamylcysteine transport across plasma membranes is enhanced by its linkage to an ester (Nishida et al. 1996). For this reason, the substance gamma-glutamylcysteine ethyl ester (GCEE) was created as a strategy to boost intracellular GSH levels. Additionally, the provision of GCEE allows further GSH synthesis to occur by bypassing the feedback inhibition of gamma-glutamylcysteine synthesis due to GSH itself. As predicted, GCEE up-regulates GSH levels in the rodent brain (Drake et al. 2002). Additionally GCEE may have in vivo radical scavenging abilities against peroxynitrite (Drake et al. 2002).
The ability of GCEE to protect neurons has been widely described. GCEE protects rat hippocampal neurons against injury induced by the ONOO radical, in part by decreasing apoptosis (Drake et al. 2003). GCEE has been shown to cross the plasma membrane of primary neuronal cultures and protect them against beta-amyloid toxicity (Boyd-Kimball et al. 2005). In a model of Parkinson’s disease in which dopaminergic cell counts of the substantia nigra was reduced by 40% using MPTP hydrochloride, GCEE enhanced dopamine levels in surviving neurons and decreased decrease severity (Chinta et al. 2006). GCEE treatment decreased the expressions of p53, Bax, caspase-3 activity, and DNA damage in a model of status epilepticus induced by kainic acid (Yalcin et al. 2010), and reduced ROS formation in oxidatively damaged synaptosomes by kainic acid (Yalcin et al. 2010, Turunc et al. 2010).
Although there have been many reports of GCEE’s effect on neurons, this is the first report, to our knowledge, that GCEE is cytoprotective for cerebral endothelial cells. Our data suggest that GCEE protects brain cerebral endothelial cells against oxidative stress, and at least one of its mechanisms is an increase in endothelial GSH levels. Our focus on investigating the endothelial-protective properties of GCEE is spurred by recent findings that effective neuroprotective strategies must target not only neurons, but also other cell types in the brain (Lok et al. 2007, Abbott et al. 2006, Simard & Nedergaard 2004, Zlokovic 2005). Endothelial cells perform many important functions, including blood brain barrier function, cerebral blood flow regulation, and trophic support for neurons through the production of growth factors. All of these processes may be adversely affected by oxidative injury. Even in the absence of cell death, endothelial dysfunction may enhance neuroinflammation, BBB leakage and uncoupling of hemodynamic and trophic gradients (Pun et al. 2009, Hawkins & Davis 2005) GCEE’s beneficial effect on brain endothelial cells would be expected to have a positive effect on other cells in the neurovascular unit, and adds to its therapeutic potential for many types of neurological pathologies.
There are several limitations to our study. First, our in-vitro experiments used a cell line. How these data reflect the measured parameters of endothelial viability, ROS formation, and GSH levels in the in vivo situation remains to be fully determined. Secondly, the mechanisms may be multimodal. In addition to GCEE’s beneficial effects on ROS, GSH levels and mitochondrial membrane potentials, other pathways may be involved. Secondly, we have not excluded the possibility that the beneficial effects of GCEE may be partially due to the GCEE dipeptide itself, or the possibility that mBrCl fluorescence may reflect GCEE levels along with GSH levels. However, extensive validation of mBCl fluorescence as a semi-quantitative method for measuring intracellular GSH has been performed previously by various groups. mBCl has been shown to react specifically with GSH in a reaction catalyzed by glutathione-S-transferase (Fernandez-Checa & Kaplowitz 1990, Shrieve et al. 1988), and minimally with other intracellular thiols when tested in different cell types (Fernandez-Checa & Kaplowitz 1990, Sebastia et al. 2003, Shrieve et al. 1988). GSH levels measured using mBCl have been shown to correlate well with HPLC values (Fernandez-Checa & Kaplowitz 1990, Kamencic et al. 2000), and mBCl fluorescence is currently used for GSH measurements in neuronal (Fernandez-Gomez et al. 2005, Kwon et al. 2010, Sun et al. 2006) and endothelial (Huet et al. 2008) experiments. A further limitation is that our in-vitro experiments were not designed to evaluate the effect of GCEE within a BBB model. They were directed towards the effect of GCEE on ROS, GSH levels, mitochondrial membrane potential, and survival of brain endothelial cells. We have not attempted to measure in-vitro indices of barrier function.
However, in addition to endothelial cell survival, BBB preservation is critical. To address this important parameter, we used an in-vivo model of brain trauma to damage the BBB, and our data suggests that GCEE can indeed protect the BBB in vivo. The protection is incomplete however, so additional factors not remedied by GCEE may be important for maintenance of BBB integrity. Another important limitation of the in vivo experiment is that BBB permeability was evaluated only at one early time point after CCI, when peak BBB permeability is expected to occur. Additionally, it is not possible to specify whether GCEE’s effect on the BBB is due to a direct action on brain microvascular endothelial cells, or to its effects on the astrocytes and pericytes that are also components of the BBB, and whether GCEE crosses the BBB and then acts directly on the neurons.
Our data suggest that GCEE is beneficial to brain microvascular endothelial cells during oxidative stress and decreases BBB permeability after TBI. Oxidative stress contributes to almost all neurological diseases, and the effect of oxidative stress on the brain endothelium likely extends to the rest of the neurovascular unit. Our data provide proof-of-concept that targeting oxidative stress in brain endothelium may be an important part of a general neuroprotective strategy.
Fig. 5. Post-trauma administration of GCEE decreases acute BBB permeability after CCI.
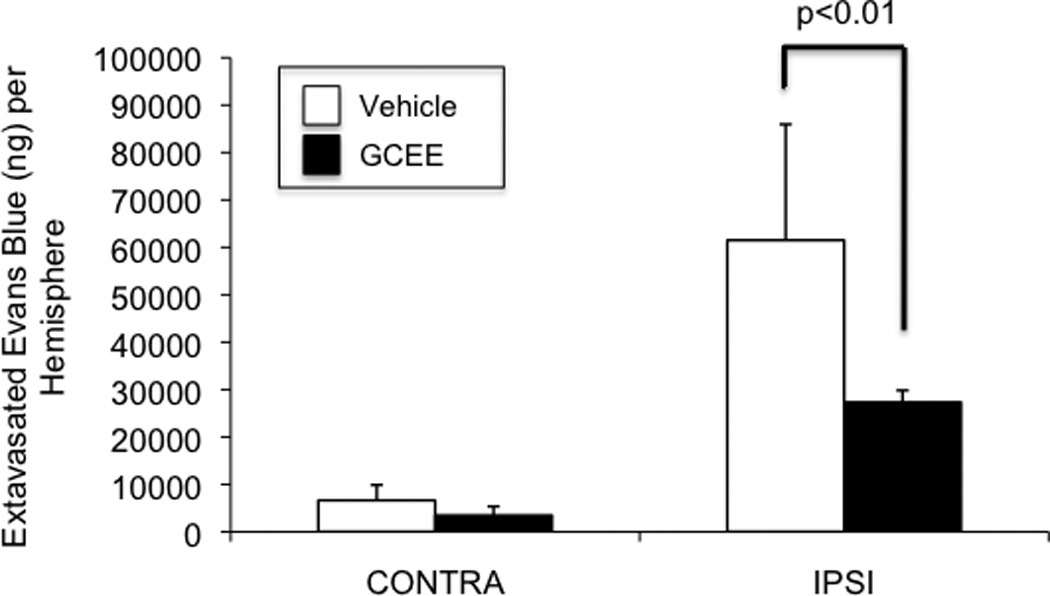
Evans blue extravasation was reduced by 43% in mice that were given GCEE by i.p. injection 10 min after CCI (p<.01). Values are expressed as grams of extravasated Evans Blue per hemisphere (+/− SD) (Fig.5). n=5 in vehicle group; n= 7 in GCEE group.
Acknowledgements
This study was supported by NIH grants (K08NS057339 to J.L., R01NS049430 to K.v.L., R01NS53560 and P01NS555104 to E.H.L.), a Scientist Development Grant from the American Heart Association (to K.v.L.), a fellowship from the Fulbright Program (to A.Y.), and by the Technological and Scientific Council of Turkey (SBAG-K69-104S280 to A.Y). The authors thank Dr. Michael J. Whalen for helpful discussions regarding the CCI model.
Abbreviations
- BBB
blood brain barrier
- CCI
controlled cortical impact
- CNS
central nervous system
- GCEE
gamma-glutamylcysteine ethyl ester
- GSH
glutathione
- HMBEC
human brain microvascular endothelial cell
- i.p.
intraperitoneal
- LDH
lactate dehydrogenase
- MTT
3-(4.5-dimethylthiazol-2-yl) 2,5-diphenyl-tetrazolium bromide
- OGD
oxygen-glucose deprivation
- TBI
traumatic brain injury
Footnotes
The authors declare no conflicts of interest.
References
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10 Suppl:S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Luo JL. Glutathione therapy: from prodrugs to genes. Semin Liver Dis. 1998;18:415–424. doi: 10.1055/s-2007-1007174. [DOI] [PubMed] [Google Scholar]
- Ayer RE, Zhang JH. Oxidative stress in subarachnoid haemorrhage: significance in acute brain injury and vasospasm. Acta Neurochir Suppl. 2008;104:33–41. doi: 10.1007/978-3-211-75718-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson VC. Drug targets for traumatic brain injury from poly(ADP-ribose) polymerase pathway modulation. Br J Pharmacol. 2009;157:695–704. doi: 10.1111/j.1476-5381.2009.00229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd-Kimball D, Sultana R, Poon HF, Mohmmad-Abdul H, Lynn BC, Klein JB, Butterfield DA. Gamma-glutamylcysteine ethyl ester protection of proteins from Abeta(1-42)-mediated oxidative stress in neuronal cell culture: a proteomics approach. J Neurosci Res. 2005;79:707–713. doi: 10.1002/jnr.20393. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Pocernich CB, Drake J. Elevated Glutathione as a Therapeutic Strategy in Alzheimer’s Disease. Drug Development Research. 2002;56:428–437. [Google Scholar]
- Callahan MK, Williams KA, Kivisakk P, Pearce D, Stins MF, Ransohoff RM. CXCR3 marks CD4+ memory T lymphocytes that are competent to migrate across a human brain microvascular endothelial cell layer. Journal of neuroimmunology. 2004;153:150–157. doi: 10.1016/j.jneuroim.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Chinta SJ, Rajagopalan S, Butterfield DA, Andersen JK. In vitro and in vivo neuroprotection by gamma-glutamylcysteine ethyl ester against MPTP: relevance to the role of glutathione in Parkinson's disease. Neurosci Lett. 2006;402:137–141. doi: 10.1016/j.neulet.2006.03.056. [DOI] [PubMed] [Google Scholar]
- Clark RS, Bayir H, Chu CT, Alber SM, Kochanek PM, Watkins SC. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy. 2008;4:88–90. doi: 10.4161/auto.5173. [DOI] [PubMed] [Google Scholar]
- Commandeur JN, Stijntjes GJ, Vermeulen NP. Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics. Pharmacol Rev. 1995;47:271–330. [PubMed] [Google Scholar]
- Drake J, Kanski J, Varadarajan S, Tsoras M, Butterfield DA. Elevation of brain glutathione by gamma-glutamylcysteine ethyl ester protects against peroxynitrite-induced oxidative stress. J Neurosci Res. 2002;68:776–784. doi: 10.1002/jnr.10266. [DOI] [PubMed] [Google Scholar]
- Drake J, Sultana R, Aksenova M, Calabrese V, Butterfield DA. Elevation of mitochondrial glutathione by gamma-glutamylcysteine ethyl ester protects mitochondria against peroxynitrite-induced oxidative stress. J Neurosci Res. 2003;74:917–927. doi: 10.1002/jnr.10810. [DOI] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Fernandez-Checa JC, Kaplowitz N. The use of monochlorobimane to determine hepatic GSH levels and synthesis. Anal Biochem. 1990;190:212–219. doi: 10.1016/0003-2697(90)90183-a. [DOI] [PubMed] [Google Scholar]
- Fernandez-Gomez FJ, Gomez-Lazaro M, Pastor D, Calvo S, Aguirre N, Galindo MF, Jordan J. Minocycline fails to protect cerebellar granular cell cultures against malonate-induced cell death. Neurobiol Dis. 2005;20:384–391. doi: 10.1016/j.nbd.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Huet O, Cherreau C, Nicco C, et al. Pivotal role of glutathione depletion in plasma-induced endothelial oxidative stress during sepsis. Crit Care Med. 2008;36:2328–2334. doi: 10.1097/CCM.0b013e3181800387. [DOI] [PubMed] [Google Scholar]
- Kamencic H, Lyon A, Paterson PG, Juurlink BH. Monochlorobimane fluorometric method to measure tissue glutathione. Anal Biochem. 2000;286:35–37. doi: 10.1006/abio.2000.4765. [DOI] [PubMed] [Google Scholar]
- Kwon KJ, Kim HJ, Shin CY, Han SH. Melatonin Potentiates the Neuroprotective Properties of Resveratrol Against Beta-Amyloid-Induced Neurodegeneration by Modulating AMP-Activated Protein Kinase Pathways. J Clin Neurol. 2010;6:127–137. doi: 10.3988/jcn.2010.6.3.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Hickey RW, Chen Y, et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab. 2008;28:540–550. doi: 10.1038/sj.jcbfm.9600551. [DOI] [PubMed] [Google Scholar]
- Lok J, Gupta P, Guo S, Kim WJ, Whalen MJ, van Leyen K, Lo EH. Cell-cell signaling in the neurovascular unit. Neurochem Res. 2007;32:2032–2045. doi: 10.1007/s11064-007-9342-9. [DOI] [PubMed] [Google Scholar]
- Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Mustafa AG, Singh IN, Wang J, Carrico KM, Hall ED. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J Neurochem. 2010;114:271–280. doi: 10.1111/j.1471-4159.2010.06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Ohta Y, Ito M, Nagamura Y, Kitahara S, Fujii K, Ishiguro I. Conversion of gamma-glutamylcysteinylethyl ester to glutathione in rat hepatocytes. Biochim Biophys Acta. 1996;1313:47–53. doi: 10.1016/0167-4889(96)00054-7. [DOI] [PubMed] [Google Scholar]
- Perry TL, Godin DV, Hansen S. Parkinson's disease: a disorder due to nigral glutathione deficiency? Neurosci Lett. 1982;33:305–310. doi: 10.1016/0304-3940(82)90390-1. [DOI] [PubMed] [Google Scholar]
- Perry TL, Yong VW. Idiopathic Parkinson's disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci Lett. 1986;67:269–274. doi: 10.1016/0304-3940(86)90320-4. [DOI] [PubMed] [Google Scholar]
- Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43:348–364. doi: 10.1080/10715760902751902. [DOI] [PubMed] [Google Scholar]
- Reed TT, Owen J, Pierce WM, Sebastian A, Sullivan PG, Butterfield DA. Proteomic identification of nitrated brain proteins in traumatic brain-injured rats treated postinjury with gamma-glutamylcysteine ethyl ester: insights into the role of elevation of glutathione as a potential therapeutic strategy for traumatic brain injury. J Neurosci Res. 2009;87:408–417. doi: 10.1002/jnr.21872. [DOI] [PubMed] [Google Scholar]
- Roberts RA, Laskin DL, Smith CV, Robertson FM, Allen EM, Doorn JA, Slikker W. Nitrative and oxidative stress in toxicology and disease. Toxicol Sci. 2009;112:4–16. doi: 10.1093/toxsci/kfp179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- Sebastia J, Cristofol R, Martin M, Rodriguez-Farre E, Sanfeliu C. Evaluation of fluorescent dyes for measuring intracellular glutathione content in primary cultures of human neurons and neuroblastoma SH-SY5Y. Cytometry A. 2003;51:16–25. doi: 10.1002/cyto.a.10003. [DOI] [PubMed] [Google Scholar]
- Shrieve DC, Bump EA, Rice GC. Heterogeneity of cellular glutathione among cells derived from a murine fibrosarcoma or a human renal cell carcinoma detected by flow cytometric analysis. J Biol Chem. 1988;263:14107–14114. [PubMed] [Google Scholar]
- Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004;129:877–896. doi: 10.1016/j.neuroscience.2004.09.053. [DOI] [PubMed] [Google Scholar]
- Sun X, Shih AY, Johannssen HC, Erb H, Li P, Murphy TH. Two-photon imaging of glutathione levels in intact brain indicates enhanced redox buffering in developing neurons and cells at the cerebrospinal fluid and blood-brain interface. J Biol Chem. 2006;281:17420–17431. doi: 10.1074/jbc.M601567200. [DOI] [PubMed] [Google Scholar]
- Turunc E, Kanit L, Yalcin A. Effect of gamma-glutamylcysteine ethylester on the levels of c-fos mRNA expression, glutathione and reactive oxygen species formation in kainic acid excitotoxicity. J Pharm Pharmacol. 2010;62:1010–1017. doi: 10.1111/j.2042-7158.2010.01122.x. [DOI] [PubMed] [Google Scholar]
- Yalcin A, Armagan G, Turunc E, Konyalioglu S, Kanit L. Potential neuroprotective effect of gamma-glutamylcysteine ethyl ester on rat brain against kainic acid-induced excitotoxicity. Free Radic Res. 2010;44:513–521. doi: 10.3109/10715761003645964. [DOI] [PubMed] [Google Scholar]
- You Z, Yang J, Takahashi K, et al. Reduced tissue damage and improved recovery of motor function after traumatic brain injury in mice deficient in complement component C4. J Cereb Blood Flow Metab. 2007;27:1954–1964. doi: 10.1038/sj.jcbfm.9600497. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]