

Abstract
Methoxyacetic acid (MAA), the active biological oxidation product of the industrial solvent ethylene glycol monomethyl ether (EGME), causes acute toxicity in several species including humans. MAA primarily affects tissues with rapidly dividing cells and high rates of energy metabolism, including testes, thymus and the fetus. Testicular toxicity, one of the most prominent consequences of EGME, and MAA, exposure, results from apoptosis of primary spermatocytes and is associated with changes in the expression of various genes and signaling pathways. This review of EGME metabolism and its organ-specific toxicities emphasizes genes and signaling pathways that are modulated by EGME exposure and their relevance to the molecular mechanisms underlying EGME and MAA toxicity. Of particular importance are the genes that code for oxidative stress response factors, protein kinases, and nuclear hormone receptors. Nuclear receptors and protein kinases regulate multiple cellular processes and are critical for signaling events required for spermatogenesis. De-regulation of their activity by EGME or MAA leads to inappropriate signaling in testicular cells. Oxidative stress in spermatocytes exposed to MAA triggers mitochondrial release of cytochrome C, activation of caspases and ultimately apoptosis. Detailed investigation of the molecular responses to MAA exposure may help elucidate the overall impact and extent of toxicity seen following EGME exposure. Finally, given the effects of EGME on multiple genes and signaling pathways in the testis, mixture studies combining EGME, or MAA, with other testicular toxicants may help identify toxicities that are aggravated by EGME exposure.
Keywords: Ethylene glycol monomethyl ether, Methoxyacetic acid, toxicity, gene expression
Introduction
Glycol ethers are widely used in the production of printing inks, varnishes, textile dyes and anti-icing additives in jet fuels. Several glycol ethers are endocrine disruptive compounds that interfere with hormonal signaling and induce toxicities in various species, including humans (Chapin and Lamb, 1984; Hardin, 1983; Li et al., 1996; Nagano, 1979). Of the various glycol ethers tested, ethylene glycol monomethyl ether (EGME), ethylene glycol monoethyl ether (EGEE) and their derivatives are particularly harmful to organs with high rates of respiration and energy metabolism, including testis, fetus, thymus and bone marrow (Beattie and Brabec, 1986). Notable toxicities include testicular degradation, developmental toxicity, leucopenia and thymocyte degradation (Boatman, 2005). EGME is produced at >106 pounds/yr in the United States (www.scorecard.org) and elicits adverse effects at much lower exposure levels than EGEE (Wier et al., 1987). Previous reviews have focused on the metabolism, dosage, routes and pharmacokinetic models of exposure and toxicity of EGME (Hardin, 1983; Johanson, 2000; Maldonado et al., 2003; Welsch, 2005). Presently, we review EGME metabolism and its organ-specific toxicities, emphasizing the genes and signaling pathways modulated upon EGME exposure and their relevance to EGME toxicity.
EGME metabolism
Oxidative metabolism of EGME to methoxyacetic acid (MAA) is a pre-requisite for EGME embryotoxicity and teratogenicity (Brown et al., 1984; Miller et al., 1982; Sleet et al., 1988). EGME is first oxidized by alcohol dehydrogenase to 2-methoxyacetaldehyde (Fig. 1). This short-lived intermediate is oxidized by aldehyde dehydrogenase to MAA, a more stable metabolite (Welsch, 2005). MAA is readily detectable in blood and other body fluids of exposed individuals and is eliminated through urine (t½elimination= ~77 hr) (Groeseneken et al., 1989). MAA is bio-activated intracellularly to a thioester by coupling with coenzyme A to form 2-methoxyacetyl~CoA, which can then enter the tricarboxylic acid cycle (Welsch, 2005). MAA toxicity can be attenuated by co-treatment with acetate, formate, D-glucose, glycine, serine and certain tricarboxylic acid pathway metabolites (Mebus et al., 1989; Welch et al., 1988; Welsch, 2005).
Fig.1. Chemical structure and metabolism of EGME in the cell.
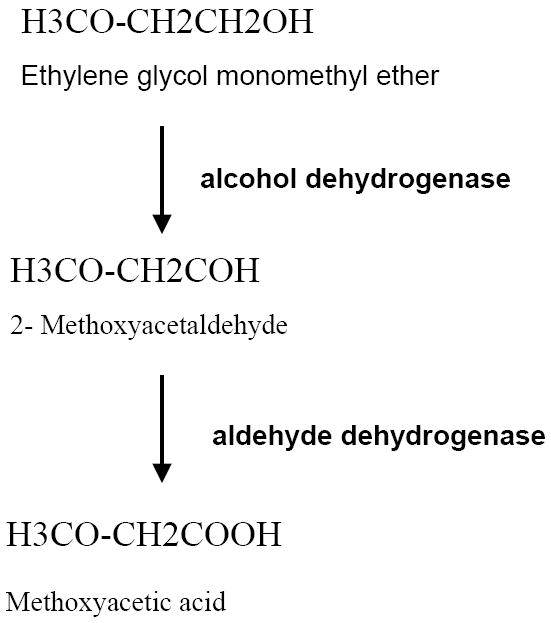
Oxidation of EGME to methoxyacetalehyde is catalysed by alcohol dehydrogenase. Methoxyacetaldehyde is further oxidized to methoxyacetic acid by aldehyde dehydrogenase.
EGME toxicity
EGME causes reproductive, developmental and hematopoietic toxicities in several species including humans (Chapin and Lamb, 1984; Hardin, 1983; Holladay et al., 1994; Miller et al., 1982). These include testicular toxicity, leucopenia, embryo lethality and malformations (Nagano, 1979). Testicular damage is a hallmark of acute EGME exposure (Chapin and Lamb, 1984; Foster et al., 1984; Li et al., 1996). In male rats, significant decreases in relative testicular weight occur at an EGME dose of 500 mg/kg/day for 2 or more days, and a single dose of 100 mg/kg/day results in degeneration of pachytene spermatocytes within 24 hr (Foster et al., 1984). Continued dosing leads to progressive depletion of spermatocytes and early spermatid populations (Foster et al., 1984) through an apoptotic mechanism (Ku et al., 1995). Calcium channel blockers such as verapamil and diltiazem prevent EGME-induced toxicity in rats in vivo, suggesting a role for calcium deregulation in EGME toxicity (Ghanayem and Chapin, 1990). In humans, the available MAA toxicity data indicate reproductive toxicity. For example, an increased prevalence of oligospermia and azoospermia and lower sperm counts per ejaculate were observed in 73 shipyard painters exposed to 2.6 mg/m3 EGME (Welch et al., 1988). EGME dose-response studies indicate a lowest observed adverse effect level (LOAEL) for testicular toxicity of 167 mg/m3 in rats (IRS, 1995) as compared to 2.6 mg/m3 in humans, indicating much greater sensitivity in humans. However, both rat and human seminiferous tubules respond to MAA at the same dosage, and in a similar manner, when treated in vitro with MAA, although the morphology of the dying spermatocytes in the two species is different (Li et al., 1996).
At high doses (>250 mg/kg), EGME has profound toxic effects on prenatal development (Nagano et al., 1981). Two developmental phases are disrupted in pregnant mice treated with EGME at 100-500 mg/kg body weight for 1-3 days: neurogenesis, specifically, closure of the anterior neural tube; and digit differentiation in fore and hind limbs (Welsch, 2005). Further, EGME targets multiple hematopoietic compartments. In adult rats and mice (Dieter, 1993) and in mice exposed in utero (Holladay et al., 1994), EGME induces thymic hypocellularity and atrophy and inhibits thymocyte maturation.
Modulation of gene expression by EGME and MAA
Investigation of gene expression changes following exposure to EGME or its active metabolite, MAA, has revealed changes in multiple genes and proteins in both spermatocytes and Sertoli cells in the testes (Fukushima et al., 2005; Jansen et al., 2004; Jindo et al., 2001; Syed and Hecht, 1998; Tirado et al., 2003; Wang and Chapin, 2000). Highlights of these findings are discussed below.
Exposure of rat pachytene spermatocytes to 5 mM EGME or 5 mM MAA up-regulates a thiol specific anti-oxidant (TSA) protein within 30 min and down regulates polo-like kinase-1 (Plk1) after 6 hr. In Sertoli cells, an oxidative stress protein homologous to A-170 is induced within 30 min, while the 3’,5’-cyclic AMP phosphodiesterase splice variant PDE 3.1 is repressed within 60 min (Syed and Hecht, 1998). These results suggest that MAA disturbs the oxidative balance in these cells. Since oxidative changes in cellular metabolism play a critical role in apoptosis (Erkkila et al., 2003), the activation of an anti-oxidant response in spermatocytes may serve as a defense mechanism to protect against MAA toxicity. Change in redox status could also cause the observed up-regulation of A-170 in Sertoli cells. A-170 is 90% identical to p62, a human oxidative stress-responsive protein involved in signal transduction in macrophages. Plk1 plays an important role in meiosis by regulating microtubule organization and spindle formation. The decrease in Plk1 expression within 6 hr of EGME or MAA treatment may be a reflection of the DNA damage and mitotic spindle disassembly that spermatocytes undergo upon MAA exposure.
Protein kinases play a role in EGME/MAA-induced toxicity. EGME-induced apoptosis in rat seminiferous tubules is significantly attenuated by the protein kinase inhibitors H-7, H-8, W-7, genistein and K-252a (Jindo et al., 2001). Further, immunohistochemical analysis demonstrated an increase in expression of several kinases in Sertoli cells surrounding the apoptotic spermatocytes 16 hr after treatment with 200 mg/kg EGME. Kinases whose activities increase significantly in Sertoli cells surrounding the dying spermatocytes include three isoforms of protein kinase C (PKC) (PKCμ, ζ and γ), Src, A-kinase anchor protein 220 kDa (AKAP 220), calmodulin kinase II (CaMK II) and myosin light chain kinase (MLCK). Local disruption of the plasma membrane of germ cells by EGME may be responsible for the alteration in expression of various kinases, as these are usually associated with the plasma membrane (Jindo et al., 2001). PKC enzymes are involved in diverse cellular functions including cell proliferation, tumor promotion, and apoptotic cell death. Activation of PKC by MAA could result from genotoxic stress and DNA damage.
Six differentially expressed RNAs were identified by suppression subtractive hybridization of cDNA libraries generated from testicular RNA of EGME-treated vs. untreated mice (Wang and Chapin, 2000). Motif analysis revealed putative PKC phosphorylation sites in the protein sequences of all of the differentially expressed cDNAs. MLCK, one of the genes activated by MAA, is a calcium/calmodulin-dependent protein kinase implicated in apoptosis. Tumor necrosis factor-induced apoptosis is blocked by an inhibitor of MLCK and apoptotic membrane blebbing is related to MLCK activity (Jindo et al., 2001). 2D-gel analysis revealed that EGME increases phosphorylation of glucose-regulated protein 94 (Grp94) inside dying spermatocytes (Jindo et al., 2001). Grp94, located in the endoplasmic reticulum (and perhaps the plasma membrane), undergoes autophosphorylation and can be induced by toxic insults such as calcium ionophores and oxidative stress. Oxidative stress induced by MAA probably causes increased synthesis and phosphorylation of Grp94.
MAA enhances the transcriptional activity of ligand-activated nuclear receptors, such as progesterone receptor, estrogen receptor and androgen receptor (AR), both in vitro and in vivo (Jansen et al., 2004; Tirado et al., 2003). These and other nuclear receptors play critical roles in physiology and development, and their modulation by MAA could lead to gonadal toxicities. The modulation of nuclear receptor transcriptional activity by MAA is proposed to proceed through the activation of protein kinases and/or by inhibition of histone deacetylases. Tirado et al (2003) investigated the effect of MAA on AR and on androgen binding protein (ABP) in Sertoli cells, since these proteins may play critical role in spermatocyte survival and maturation (De Gendt et al., 2004; Selva et al., 2000). Immunohistochemical studies revealed that MAA altered the cyclic expression of AR protein and increased ABP expression in Sertoli cells. Laser capture microdissection of seminiferous tubules exposed to MAA demonstrated that MAA rapidly decreases AR mRNA in stages VII-VIII, but significantly increases expression in stages III-IV and X-XIII. ABP expression is also stage-specifically modulated by MAA administration (Tirado, 2003). The observed variation in cyclic expression of AR and ABP in Sertoli cells could contribute to MAA-induced apoptosis of germ cells.
Microarray analysis of testicular RNA from rats treated with 2000 mg/kg EGME for 6 hr identified 71 genes that were up-regulated and 54 that were down regulated (Fukushima et al., 2005). These genes belong to ten functional categories based on Gene Ontology analysis, including spermatogenesis, apoptosis, signal transduction, transcription and enzymes. The spermatogenesis-related genes that were induced include IGFBP-3, glutathione S-transferases Yb and Pi and glutathione synthase, HSP70-2. Glutathione S-transferases Yb, Pi and glutathione synthase protect germ cells from electrophilic and oxidative stress. HSP70-2 is a molecular chaperon that plays a critical role in spermatogenesis and mice lacking this gene are not fertile (Eddy, 1999). Changes in expression of HSP70-2 may result in anomalous spermatogenesis. The expression of interleukin-1 receptor also increased upon EGME treatment. Interleukin-1, a paracrine factor, can influence spermatogenesis indirectly, by increasing expression of IGFBP-3, which inhibits IGF-1, a spermatogenesis-promoting factor. Indeed, IGFBP-3 expression was up regulated following EGME treatment as demonstrated by microarray analysis and verified by quantitative PCR (Fukushima et al., 2005).
Mechanism of MAA-induced apoptosis in germ cells
The primary targets of EGME and MAA are tissues with rapidly dividing cells, which have high rates of respiration and energy metabolism. MAA concentrations ≥3.85 mM inhibit state III respiration and the respiratory control ratio in hepatic and testicular mitochondria (Beattie and Brabec, 1986). In testes, MAA selectively depletes spermatocytes by activating apoptosis within 24 hr of exposure (Suter et al., 1998). Induction of spermatocyte apoptosis by MAA correlates with increased expression of the pro-apoptotic factors Bax and Bak as compared to the pro-survival factor Bcl-W (Yan et al., 2000). MAA triggers apoptosis through the mitochondrial pathway (Rao and Shaha, 2002). Acute MAA exposure induces release of cytochrome C, activation of caspases 9 and 3, and degradation of spermatocyte DNA (Fig. 2). Further, cytochrome C release occurs as a result of a fall in the ratio of reduced to oxidized glutatione (GSH/GSSG ratio); replenishment of GSH using precursors such as N-acetyl cysteine prevents MAA-induced pro-apoptotic events and apoptosis (Rao and Shaha, 2002) (Fig. 2).
Fig.2. Schematic representation of MAA-induced apoptosis in spermatocytes.
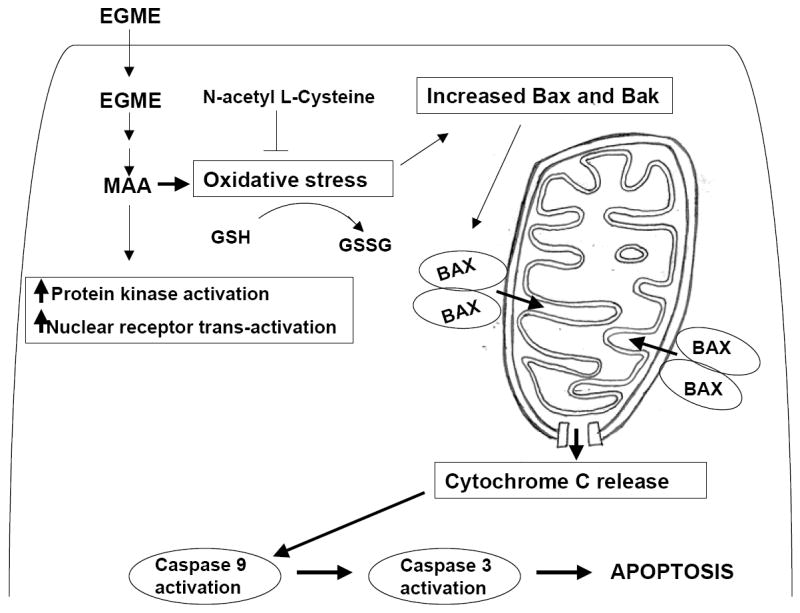
EGME is oxidized to MAA inside the spermatocyte. MAA induced oxidative stress causes a reduction in the cellular GSH to GSSG ratio, leading to increased expression and activation of proapoptotic factors BAK and BAX. The dimerization of BAK and BAX facilitates the release of cytochrome c from mitochondria which in turn causes activation of procaspase-9 to caspase-9 followed by activation of procaspase 3 to caspase 3 thereby commiting the cells to the apoptosis.
Conclusion
EGME-induced testicular toxicity is observed in multiple species and is mediated by the primary active metabolite, MAA. At the molecular level, MAA affects the expression of multiple genes and signaling pathways in both spermatocytes and Sertoli cells of the testes, and potentially other cell types as well. MAA-responsive genes include oxidative stress-response genes, protein kinases and nuclear receptors. MAA also decreases the expression of genes that promote spermatogenesis and cell survival. The induction of anti-oxidant and oxidative stress response genes suggests that MAA induces oxidative stress in target organs. Oxidative stress influences several biological and pathological processes. Many signal transduction pathways critical for development, including proliferation, differentiation and apoptosis, are redox sensitive. Thus, chemicals that induce oxidative stress may induce toxicities by de-regulation of these critical pathways leading to teratogenicity (Hansen JM, 2006). Several MAP kinase pathways are influenced by oxidative stress, including the ERK, JNK, p38 and BMK1 pathways (McCubrey et al., 2006). Protein kinases, in turn, are key mediators of steroid hormone receptor action (Lange CA, 2004). Steroid receptors, such as AR, estrogen receptor, progesterone receptor and their co-regulators, are targets for multiple kinases. Phosphorylation of these receptors occurs on multiple sites and can modulate overall transcriptional activity through effects on nuclear localization, protein stability and DNA binding (Weigel and Moore, 2007). Thus, by inducing oxidative stress in target cells, MAA can impact critical biological responses by modulating kinase and nuclear receptor activity. In germ cells, MAA-induced oxidative stress decreases the GSH/GSSG ratio, triggering mitochondrial cytochrome C release and spermatocyte apoptosis. Further investigation of the molecular responses to MAA exposure, in other testicular cells, such as Leydig cells, and also in non-testicular cells, may help elucidate the true impact and overall extent of MAA toxicity. Finally, given the ability of EGME and MAA to alter multiple signaling pathways and to severely disrupt spermatogenesis in the adult, mixture studies involving these chemicals in combination with other potential testicular toxicants seem warranted.
Acknowledgments
Supported in part by NIH grant 5 P42 ES07381, Superfund Basic Research Program at Boston University (to D.J.W.).
References
- Beattie PJ, Brabec MJ. Methoxyacetic acid and ethoxyacetic acid inhibit mitochondrial function in vitro. J Biochem Toxicol. 1986;1(3):61–70. doi: 10.1002/jbt.2570010307. [DOI] [PubMed] [Google Scholar]
- Boatman RJ. International industry initiatives to improve the glycol ether health effects knowledge base. Toxicol Lett. 2005;156(1):39–50. doi: 10.1016/j.toxlet.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Brown NA, Holt D, Webb M. The teratogenicity of methoxyacetic acid in the rat. Toxicol Lett. 1984;22(1):93–100. doi: 10.1016/0378-4274(84)90051-1. [DOI] [PubMed] [Google Scholar]
- Chapin RE, Lamb JCt. Effects of ethylene glycol monomethyl ether on various parameters of testicular function in the F344 rat. Environ Health Perspect. 1984;57:219–224. doi: 10.1289/ehp.8457219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gendt K, Swinnen JV, Saunders PT, Schoonjans L, Dewerchin M, Devos A, Tan K, Atanassova N, Claessens F, Lecureuil C, Heyns W, Carmeliet P, Guillou F, Sharpe RM, Verhoeven G. A Sertoli cell-selective knockout of the androgen receptor causes spermatogenic arrest in meiosis. Proc Natl Acad Sci U S A. 2004;101(5):1327–1332. doi: 10.1073/pnas.0308114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieter M. NTP technical report on the toxicity studies of Ethylene Glycol Ethers: 2-Methoxyethanol, 2-Ethoxyethanol, 2-Butoxyethanol (CAS Nos. 109-86-4, 110-80-5, 111-76-2) Administered in Drinking Water to F344/N Rats and B6C3F1 Mice. Toxic Rep Ser. 1993;26:1–G15. [PubMed] [Google Scholar]
- Eddy EM. Role of heat shock protein HSP70-2 in spermatogenesis. Reviews of reproduction. 1999;4(1):23–30. doi: 10.1530/ror.0.0040023. [DOI] [PubMed] [Google Scholar]
- Erkkila K, Suomalainen L, Wikstrom M, Parvinen M, Dunkel L. Chemical anoxia delays germ cell apoptosis in the human testis. Biol Reprod. 2003;69(2):617–626. doi: 10.1095/biolreprod.102.013920. [DOI] [PubMed] [Google Scholar]
- Foster PM, Creasy DM, Foster JR, Gray TJ. Testicular toxicity produced by ethylene glycol monomethyl and monoethyl ethers in the rat. Environ Health Perspect. 1984;57:207–217. doi: 10.1289/ehp.8457207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima T, Yamamoto T, Kikkawa R, Hamada Y, Komiyama M, Mori C, Horii I. Effects of male reproductive toxicants on gene expression in rat testes. J Toxicol Sci. 2005;30(3):195–206. doi: 10.2131/jts.30.195. [DOI] [PubMed] [Google Scholar]
- Ghanayem BI, Chapin RE. Calcium channel blockers protect against ethylene glycol monomethyl ether (2-methoxyethanol)-induced testicular toxicity. Exp Mol Pathol. 1990;52(3):279–290. doi: 10.1016/0014-4800(90)90069-p. [DOI] [PubMed] [Google Scholar]
- Groeseneken D, Veulemans H, Masschelein R, Van Vlem E. Experimental human exposure to ethylene glycol monomethyl ether. Int Arch Occup Environ Health. 1989;61(4):243–247. doi: 10.1007/BF00381421. [DOI] [PubMed] [Google Scholar]
- Hardin BD. Reproductive toxicity of the glycol ethers. Toxicology. 1983;27(2):91–102. doi: 10.1016/0300-483x(83)90014-8. [DOI] [PubMed] [Google Scholar]
- Holladay SD, Comment CE, Kwon J, Luster MI. Fetal hematopoietic alterations after maternal exposure to ethylene glycol monomethyl ether: prolymphoid cell targeting. Toxicol Appl Pharmacol. 1994;129(1):53–60. doi: 10.1006/taap.1994.1228. [DOI] [PubMed] [Google Scholar]
- Jansen MS, Nagel SC, Miranda PJ, Lobenhofer EK, Afshari CA, McDonnell DP. Short-chain fatty acids enhance nuclear receptor activity through mitogen-activated protein kinase activation and histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2004;101(18):7199–7204. doi: 10.1073/pnas.0402014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindo T, Wine RN, Li LH, Chapin RE. Protein kinase activity is central to rat germ cell apoptosis induced by methoxyacetic acid. Toxicol Pathol. 2001;29(6):607–616. doi: 10.1080/019262301753385933. [DOI] [PubMed] [Google Scholar]
- Johanson G. Toxicity review of ethylene glycol monomethyl ether and its acetate ester. Crit Rev Toxicol. 2000;30(3):307–345. doi: 10.1080/10408440091159220. [DOI] [PubMed] [Google Scholar]
- Ku WW, Wine RN, Chae BY, Ghanayem BI, Chapin RE. Spermatocyte toxicity of 2-methoxyethanol (ME) in rats and guinea pigs: evidence for the induction of apoptosis. Toxicol Appl Pharmacol. 1995;134(1):100–110. doi: 10.1006/taap.1995.1173. [DOI] [PubMed] [Google Scholar]
- Li LH, Wine RN, Chapin RE. 2-Methoxyacetic acid (MAA)-induced spermatocyte apoptosis in human and rat testes: an in vitro comparison. J Androl. 1996;17(5):538–549. [PubMed] [Google Scholar]
- Maldonado G, Delzell E, Tyl RW, Sever LE. Occupational exposure to glycol ethers and human congenital malformations. Int Arch Occup Environ Health. 2003;76(6):405–423. doi: 10.1007/s00420-003-0441-x. [DOI] [PubMed] [Google Scholar]
- Mebus CA, Welsch F, Working PK. Attenuation of 2-methoxyethanol-induced testicular toxicity in the rat by simple physiological compounds. Toxicol Appl Pharmacol. 1989;99(1):110–121. doi: 10.1016/0041-008x(89)90116-6. [DOI] [PubMed] [Google Scholar]
- Miller RR, Carreon RE, Young JT, McKenna MJ. Toxicity of methoxyacetic acid in rats. Fundam Appl Toxicol. 1982;2(4):158–160. doi: 10.1016/s0272-0590(82)80039-0. [DOI] [PubMed] [Google Scholar]
- Nagano K. Testicular atrophy of mice induced by ethylene glycol mono alkyl ethers (author’s transl) Sangyo Igaku. 1979;21(1):29–35. doi: 10.1539/joh1959.21.29. [DOI] [PubMed] [Google Scholar]
- Nagano K, Nakayama E, Oobayashi H, Yamada T, Adachi H, Nishizawa T, Ozawa H, Nakaichi M, Okuda H, Minami K, Yamazaki K. Embryotoxic effects of ethylene glycol monomethyl ether in mice. Toxicology. 1981;20(4):335–343. doi: 10.1016/0300-483x(81)90040-8. [DOI] [PubMed] [Google Scholar]
- Rao AV, Shaha C. N-acetylcysteine prevents MAA induced male germ cell apoptosis: role of glutathione and cytochrome c. FEBS Lett. 2002;527(1-3):133–137. doi: 10.1016/s0014-5793(02)03196-4. [DOI] [PubMed] [Google Scholar]
- Selva DM, Tirado OM, Toran N, Suarez-Quian CA, Reventos J, Munell F. Meiotic arrest and germ cell apoptosis in androgen-binding protein transgenic mice. Endocrinology. 2000;141(3):1168–1177. doi: 10.1210/endo.141.3.7383. [DOI] [PubMed] [Google Scholar]
- Sleet RB, Greene JA, Welsch F. The relationship of embryotoxicity to disposition of 2-methoxyethanol in mice. Toxicol Appl Pharmacol. 1988;93(2):195–207. doi: 10.1016/0041-008x(88)90120-2. [DOI] [PubMed] [Google Scholar]
- Suter L, Meier G, Bechter R, Bobadilla M. Flow cytometry as a sensitive tool to assess testicular damage in rat. Arch Toxicol. 1998;72(12):791–797. doi: 10.1007/s002040050575. [DOI] [PubMed] [Google Scholar]
- Syed V, Hecht NB. Rat pachytene spermatocytes down-regulate a polo-like kinase and up-regulate a thiol-specific antioxidant protein, whereas sertoli cells down-regulate a phosphodiesterase and up-regulate an oxidative stress protein after exposure to methoxyethanol and methoxyacetic acid. Endocrinology. 1998;139(8):3503–3511. doi: 10.1210/endo.139.8.6123. [DOI] [PubMed] [Google Scholar]
- Tirado OM, Martinez ED, Rodriguez OC, Danielsen M, Selva DM, Reventos J, Munell F, Suarez-Quian CA. Methoxyacetic acid disregulation of androgen receptor and androgen-binding protein expression in adult rat testis. Biol Reprod. 2003;68(4):1437–1446. doi: 10.1095/biolreprod.102.004937. [DOI] [PubMed] [Google Scholar]
- Wang W, Chapin RE. Differential gene expression detected by suppression subtractive hybridization in the ethylene glycol monomethyl ether-induced testicular lesion. Toxicol Sci. 2000;56(1):165–174. doi: 10.1093/toxsci/56.1.165. [DOI] [PubMed] [Google Scholar]
- Welch LS, Schrader SM, Turner TW, Cullen MR. Effects of exposure to ethylene glycol ethers on shipyard painters: II. Male reproduction. Am J Ind Med. 1988;14(5):509–526. doi: 10.1002/ajim.4700140503. [DOI] [PubMed] [Google Scholar]
- Welsch F. The mechanism of ethylene glycol ether reproductive and developmental toxicity and evidence for adverse effects in humans. Toxicol Lett. 2005;156(1):13–28. doi: 10.1016/j.toxlet.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Wier PJ, Lewis SC, Traul KA. A comparison of developmental toxicity evident at term to postnatal growth and survival using ethylene glycol monoethyl ether, ethylene glycol monobutyl ether and ethanol. Teratog Carcinog Mutagen. 1987;7(1):55–64. doi: 10.1002/tcm.1770070108. [DOI] [PubMed] [Google Scholar]
- Yan W, Suominen J, Samson M, Jegou B, Toppari J. Involvement of Bcl-2 family proteins in germ cell apoptosis during testicular development in the rat and pro-survival effect of stem cell factor on germ cells in vitro. Mol Cell Endocrinol. 2000;165(1-2):115–129. doi: 10.1016/s0303-7207(00)00257-4. [DOI] [PubMed] [Google Scholar]