

Abstract
Bortezomib, a therapeutic agent for multiple myeloma (MM) and mantle cell lymphoma, suppresses proteosomal degradation leading to substantial changes in cellular transcriptional programs and ultimately resulting in apoptosis. Transcriptional regulators required for bortezomib-induced apoptosis in MM cells are largely unknown. Using gene expression profiling, we identified 36 transcription factors that displayed altered expression in MM cells treated with bortezomib. Analysis of a publically available database identified Kruppel-like family factor 9 (KLF9) as the only transcription factor with significantly higher basal expression in MM cells from patients who responded to bortezomib compared with nonresponders. We demonstrated that KLF9 in cultured MM cells was up-regulated by bortezomib; however, it was not through the induction of endoplasmic reticulum stress. Instead, KLF9 levels correlated with bortezomib-dependent inhibition of histone deacetylases (HDAC) and were increased by the HDAC inhibitor LBH589 (panobinostat). Furthermore, bortezomib induced binding of endogenous KLF9 to the promoter of the proapoptotic gene NOXA. Importantly, KLF9 knockdown impaired NOXA up-regulation and apoptosis caused by bortezomib, LBH589, or a combination of theses drugs, whereas KLF9 overexpression induced apoptosis that was partially NOXA-dependent. Our data identify KLF9 as a novel and potentially clinically relevant transcriptional regulator of drug-induced apoptosis in MM cells.
Introduction
Multiple myeloma (MM) is a plasma cell disorder that accounts for approximately 10% of all hematologic malignancies.1,2 Although the introduction of novel agents in the past decade has increased median overall survival of myeloma patients from 30 months to 45-72 months, the disease still remains incurable.3–5 One of these agents, bortezomib (Velcade, PS-341), significantly increased overall survival in patients with relapsed or refractory multiple myeloma when used as a single agent in comparison to high-dose dexamethasone, one of the standard therapies for this disease.1–5 Bortezomib acts via inhibition of proteasome-mediated protein degradation, ultimately causing death in cells from many types of malignancies, including MM cells.3–5 Bortezomib apoptosis-inducing activity has been attributed in part to the alterations in the expression of several BCL2 family proteins,6 among which the BH3-only protein NOXA appears to play an important role.7–9 NOXA triggers apoptosis by binding to the prosurvival molecule MCL1, thus preventing it from sequestering proteins BAX, BAK and BIM, which are all critical inducers of apoptosis.9–12 It has been reported that bortezomib increases NOXA protein levels by suppressing its proteosomal degradation8 and by transcriptional activation of its gene.8,13
Recently, several transcription factors including C-MYC,13,14 ATF3,15 ATF4,15,16 and p5314 have been shown to functionally participate in bortezomib-induced death in cells from several solid tumor lines. However, the roles of at least some of these factors in bortezomib cytotoxicity appear to vary among cells from different tumor types or even among cell lines from the same type of tumors. For instance, C-MYC was implicated in bortezomib toxicity in A375 melanoma cells,14 HCT116 colon carcinoma cells,14 and HeLa cells14 but not in SK-Mel-28 melanoma cells15 or SH-SY5Y neuroblastoma cells.16 Inhibition of p53 tumor suppressor gene was shown to be dispensable for bortezomib-dependent apoptosis in cells from several melanoma lines7 but was required for it in the abovementioned A375, HCT116, and HeLa cells.14 Depletion of ATF4, a mediator of the endoplasmic reticulum (ER) stress response15,17 rendered HeLa cells15 and SH-SY5Y cells16 resistant to bortezomib-induced cell death, however, ATF4 was required for resistance to bortezomib in MCF7 cells.18
In MM cells, only one transcription factor, NF-κB, has long been considered as a major target of bortezomib.19,20 On the other hand, recent studies suggested that inhibition of NF-κB cannot fully account for the bortezomib cytotoxicity in MM cells,21,22 nor even be required for it.23 Three other transcription factors have been shown to participate in bortezomib-induced cytotoxicity in MM cells. In one study, knocking down C-MYC led to partial suppression of apoptosis induced by bortezomib or combination of bortezomib and histone deacetylase inhibitor SAHA.24 It was suggested that C-MYC contributed to the bortezomib-dependent formation of aggresomes24 and transcriptional activation of NOXA.24,13 Another paper demonstrated that transcriptional factor JUN induced apoptosis in MM cells by direct up-regulation of the expression of a transcription regulator early growth response protein 1 (EGR1).25,26 The authors reported that both transcription factors control bortezomib-induced apoptosis presumably because of their ability to suppress expression of antiapoptotic protein survivin.26
Given the complexity of bortezomib-dependent pathways in the cell, the current knowledge approximately transcription factors critical for bortezomib-induced toxicity in MM cells is incomplete. In answer to this problem, here we present data on identification and characterization of KLF9, a novel and potentially clinically relevant transcriptional regulator of bortezomib-induced apoptosis in MM cells.
Methods
Cell lines and reagents
Multiple myeloma cells MM1.S and RPMI-8226 were cultured in RPMI-1640 medium supplemented with 10% FBS, 100 units/mL penicillin G + 100 μg/mL streptomycin, 0.1mM nonessential amino acids, and 1mM sodium pyruvate. All cell culture agents were purchased from Invitrogen. Bortezomib was purchased from LC Laboratories, and LBH589 was purchased from Selleck Chemicals.
siRNAs, shRNAs, lentiviral constructs, and infection
For overexpression experiments, human KLF9 cDNAs was cloned under the control of the CMV promoter into the lentiviral vectors pLV-SV40-puro (a gift from Dr Peter Chumakov, Cleveland Clinic). The pLKO-1 lentiviral vectors containing control shRNA was purchased from Sigma-Aldrich. The following sequences corresponding to human KLF9 gene were used for generating pLKO-1-based shRNAs: KLF9-1a 5′-ACAGTCTGGAAAGTCCAGAT-3′; KLF9-1b 5′-GCTTGTTGGACCTGAACAAGT-3′; KLF9-2a 5′-CAGTCGAATAAACTTGCGACCGC-CACGTG; KLF9-2b 5′-TGAGGAGTGACCACCTCACAAAGCACGCC-3′. Human JUN gene-specific shRNAs in pLKO-1 vector were purchased from Sigma-Aldrich. shRNA sequences are as follows: JUN-1 5′-CGGACCTTATGGCTACAGTAA-3′; JUN-2 5′-CGCAAACCTCAGCAACT-TCAA-3′. NOXA-specific shRNA and the lentiviral infection protocol have been previously described.8 For the optimal suppression of KLF9 levels, cells were simultaneously infected with viruses containing 2 shRNAs: KLF9-1a and KLF9-1b, or KLF9-2a and KLF9-2b. The equivalent amounts of control virus were used in all experiments.
Q-RT-PCR
Total RNA was isolated from cells using the RNeasy Mini Kit (QIAGEN). cDNA was prepared using the cDNA reverse transcription kit (Invitrogen). Quantitative RT-PCR (Q-RT-PCR) was performed on 7900HT Fast Real-Time PCR System (Applied Biosystems) using SYBR Green PCR Master Mix (Applied Biosystems) and the following PCR primers: KLF9 (5′-TGGCTGTGGGAAAGTCTATGG-3′ and 5′-CTCGTCTGAGCGGGAG-AAACT-3′); NOXA (PMA1P1) (5′-AGCTGGAAGTCGAGTGTGCT-3′ and 5′-TCCTGAGCAGAAGAGTTTGGA-3′); ACTB (5-TCATGAAGTGTGACGTGGAC-ATC-3′ and 5′-CAGGAGGAGCAATGATCTTGATCT-3′). PCR data were analyzed using sequence detection software (SDS) 2.4 (Applied Biosystems).
Immunoblotting
The following antibodies were used: KLF9 (sc-12 996 and sc-12 994), NOXA (sc-56 169), tubulin (sc-8035), HDAC1 (sc-8410), all from Santa Cruz Biotechnology; HDAC2 (ab7029) from Abcam, acetylated histone H3 (06-599) from Millipore; cleaved caspase 3 (#9661), JUN (#9165), and histone H3 (#9717) from Cell Signaling.
Membranes were developed using alkaline phosphatase-conjugated secondary antibodies and Pierce Fast Western blot Kit and the Alpha-Innotech FluorChem HD2 imaging system. For the detection of endogenous NOXA levels, Pierce SuperSignal West Femto Substrate was used. The ImageQuant 2.0 program package was used for signal quantification. An equivalent area in each lane was used to determine the background signal and this was subtracted from the value for the protein signal in that lane.
Immunofluorescence
MM1.S and RPMI-8826 cells were incubated for 36 hours with 5 or 10nM of bortezomib, respectively, followed by methanol fixation, permeabilization, and centrifugation onto glass slides. Cells were incubated with ubiquitin antibodies (sc-8017, Santa Cruz Biotechnology) followed by incubation with Alexa Flour 590 antibodies (Molecular Probe). Coverslips were mounted using Prolong antifade reagent containing DAPI (Molecular Probes). Images were captured using a Leica DMI 4000 B microscope.
ELISA
One million MM cells were plated into fresh media and media supernatant was probed periodically with the Human Lambda ELISA Quantitation Kit (Bethyl Laboratories) to detect secreted λ light chain according to the manufacturer's instructions.
Luciferase reporter assay
The pGL3-basic plasmid containing [-198; 157] region of the promoter of human NOXA gene upstream of firefly luciferase gene was previously described.13 The following PCR primers were used to amplify DNA corresponding to the [-92;157] region of the NOXA promoter (5′-AGGTACCCAGGGGCGGGCCGGGCGTCT-3′ and 5′-CAGATCTCCCACTCA-GCGACAGAGC-3′). The PCR product was cloned into the pGL3-basic vector (Promega) and the insert was verified by sequencing. pGL-3 vectors containing different regions of the NOXA promoter were mixed at 10:1 ratio with the pRL-SV40 plasmid (expressing the renilla luciferase gene). The plasmid mixture was transfected separately with or without KLF9-expressing vector (pLV-SV40-puro-KLF9) into HEK 293 cells using Superfect reagent (QIAGEN). Forty-eight hours after transfection, firefly and renilla-dependent luciferase activities were determined using the dual-luciferase assay kit (Promega). Firefly-dependent luciferase emission was normalized with respect to renilla signals.
Apoptosis detection assay
Apoptosis was detected using APO LOGIX Carboxyfluorescein FAM-DEVD-FMK for caspase 3 kit according to the manufacturer's recommendations. After staining, cells were observed under the fluorescent microscope and the apoptotic rates were determined by counting positive and negative cells in multiple view fields (more than 50 cells per sample).
Chromatin immunoprecipitation
Interactions between KLF9 and the NOXA promoter were assessed using the EZ-Chip kit from Millipore according to the manufacturer's recommendations with the following antibodies from Santa Cruz Biotechnology: normal goat IgG (sc-2755), KLF9 (sc-12 996 or sc-12 994). Interactions between acetylated histone H3 and KLF9 promoter were assessed using the same kit, normal rabbit IgG (sc-2027) and antibodies against acetylated histone H3 (Millipore). The following primers were used for the analysis of KLF9 binding to the DNA: GAPDH promoter (5′-TACTAGCGGTTTTACGGGCG-3′) and (5′-TCGAACAGGAGCAGAGAGCGA-3′); NOXA distal promoter region (5′-TTCAAGCGATTCTC-GTGCCTCA-3′ and 5′-ACGCCTGTAATCCCAGCTCTTT-3′); NOXA proximal promoter region (5′- GGCGTCTAGTTTCCCTACGTCA-3′ and 5′-AGATGCCAACTACACACGGTGA -3′); KLF9 distal promoter region (5′-CCCTGTCCTCTCATCCATAACT-3′ and 5′-GCAACCAGCCTTCCAATCAA-3′); KLF9 proximal promoter region (5′-TGATATGTTCTGGCTACAGGCCCA-3′ and 5′-CAAACAAACCAACCGGAAGGAGCA-3′).
Microarray analysis
Total cellular RNA was isolated using RNeasy Mini Kit (QIAGEN). Five micrograms from each sample was used for production of biotinylated cRNA as described in the Affymetrix GeneChip analysis instruction manual (Affymetrix). RNA conversion into cDNA, preparation of double-stranded DNA, preparation of biotinylated cRNA and hybridization to Affymetrix GeneChip Array was done at the University of Michigan Microarray Core Facility. The raw experimental microarray data were normalized with the Affymetrix Microarray Suite (MAS 5.0) based on the housekeeping gene method. Expression values obtained were adjusted to the intensity of the corresponding expression value of 100 housekeeping genes. A linear model specifically designed for microarray analysis was applied to the data and then differentially expressed probe sets were detected using a nested F test approach. An adjusted P value of .005 was used to assess significance, adjusting for multiple comparisons using false discovery rate.
Databases and statistical analysis
Gene expression data were obtained from gene expression profiles (generated at Millenium Pharmaceuticals) of myeloma cells collected from 239 patients before their enrollment in phase 2 and phase 3 trials of Bortezomib or Dexamethasone. The dataset along with its annotations were downloaded from NCBI's gene expression omnibus (GEO) database (GSE9782). Data were processed using MAS5.0 (Affymetrix). The dataset includes 169 patients treated with bortezomib (85 responders, 84 nonresponders) and the 70 patients treated with dexamethasone (28 responders, 42 nonresponders). The responder and nonresponder populations within each therapy group were compared using the 2-tailed Wilcoxon/Mann-Whitney test to determine statistical significance. All statistical analysis was performed using the statistical computational environment R Version 2.13.2 (http://www.r-project.org/). Gene expression profile data for patients who were treated total therapy 2 (TT2) or total therapy 3 (which includes bortezomib) was also downloaded from GEO database (GSE2658). Based on annotations, gene expression data were obtained from CD138 positive plasma cells isolated from patients before treatment with TT2 or TT3 and patient survival recorded 69.24 months after treatment. Gene expression data for the relevant genes was extracted as previously detailed, and classified on the basis of survival.
Results
KLF9 is induced by bortezomib in MM cells and its higher expression correlates with response to bortezomib in MM patients
To identify transcription factors required for bortezomib-induced death in MM cells, we performed microarray-based analyses of global gene expression in MM1.S multiple myeloma cells in the presence or absence of a clinically relevant dose of bortezomib (5nM, 24 hours). The microarray data revealed 36 sequence-specific DNA binding factors, the expression of which changed by more than 2-fold in response to bortezomib in 2 independent experiments. The identified genes are listed in supplemental Table 1 (available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Recently, Chen et al correlated high basal mRNA levels of 2 transcription factors (JUN and EGR1) in MM cells from patients before the therapy with higher overall survival of patients receiving TT3 versus TT2 regimens (the sets of drugs used in these regimens differ only by bortezomib present in TT3 regimen).26 In a separate report, bortezomib has been shown to up-regulate JUN mRNA levels in cultured cells.27 Collectively, these papers suggest that basal levels of a drug-inducible transcription factor in patients' MM cells may predict the patients' response to a drug.
Based on these premises, we evaluated the prognostic value of the 36 bortezomib-induced genes identified in our initial screen using recently described microarray-based global expression dataset that was generated for myeloma cells collected from patients with relapsed myeloma before treatment with bortezomib or high dose dexamethasone.28 This database, in combination with patient clinical response and survival data, served as a valuable tool for establishing correlations between gene expression profiles and clinical outcome.28
The patient database contained information for 35 of 36 genes presented in supplemental Table 1 (HES6 was not present in the patient database). Our analysis revealed that microarray values of one of the genes, Kruppel-like factor 9 (KLF9), correlated with patient response to bortezomib with the highest statistical significance among the 35 genes according to Mann-Whitney testing (Figure 1, supplemental Table 2). KLF9 microarray values were higher in patients who responded to bortezomib therapy, whereas no correlation was observed between responders and nonresponders to dexamethasone (Figure 1, supplemental Table 2). In general, KLF9 functions are relatively understudied. Previously, it was implicated in the control of different aspects of animal development and regulation of proliferation and differentiation of B cells.29–34 A role for KLF9 in response to chemotherapeutic agents has never been proposed.
Figure 1.

Microarray values for KLF9 expression in MM patients correlate with patient response to bortezomib, but not dexamethasone. Expression of KLF9 in patient myeloma cells was determined from gene expression profile arrays that were generated at Millenium Pharmaceuticals from patients before Bortezomib (Bz; n = 163) or dexamethasone therapy (Dex; n = 67), and deposited in the Gene expression omnibus database (GSE9782). Using the dataset annotations, KLF9 expression among responders (R) or nonresponders (NR) within Bz (n = 85 for R, n = 78 for NR) or Dex (n = 28 for R, n = 39 for NR) groups were plotted on the horizontal axis against the log transformed normalized affymetrix expression units on the vertical axis. Probe set numbers correspond to those in supplemental Table 2. P values between R and NR are shown above the corresponding therapy group.
To validate the microarray data with respect to KLF9 expression, we performed Q-RT-PCR and Western blotting in untreated MM1.S cells or cells treated with 5 or 10nM of bortezomib for 24 hours. Our data demonstrated that bortezomib increased both KLF9 mRNA and protein levels in a dose-dependent manner (Figure 2A-B). In addition, as previously shown,7,8 we detected a bortezomib-dependent increase in the expression of the proapoptotic protein NOXA (Figure 2B). Recently, Kikuchi et al reported that bortezomib inhibits expression of class I histone deacetylases (HDAC) including HDAC1 and HDAC2, that ultimately results in elevated amounts of acetylated histones.35 Inhibition of HDACs has been shown earlier to induce transcription of multiple genes.36 Thus, we hypothesized that transcriptional up-regulation of KLF9 by bortezomib occurs at least in part via an HDAC inhibition-dependent mechanism. To test this hypothesis, we first confirmed that treatment of MM1.S cells with bortezomib resulted in suppression of HDAC1 and HDAC2 and elevation of acetylated lysine 9 and/or 14 of histone H3 (H3Ac; Figure 2B). Next, we performed ChIP with H3Ac-specific antibodies from untreated MM1.S cells or cells treated with 5nM of bortezomib for 24 hours. After precipitation, the chromatin was de-crosslinked and the purified DNA was probed in Q-PCR with primers spanning sequences proximal to the human KLF9 mRNA start site (86; 269 bps), distal to this site (-2739; -2592 bps) or with primers corresponding to the bortezomib-insensitive promoter (GAPDH). We demonstrated substantial enrichment of the KLF9 proximal promoter region in chromatin precipitated with H3Ac-specific antibodies from bortezomib-treated cells (Figure 2C), suggesting that acetylation of histone H3 may be involved in bortezomib-induced up-regulation of KLF9 mRNA. To establish that direct inhibition of histone deacetylation leads to the transcriptional activation of KLF9, MM1.S cells were treated with the deacetylase inhibitor LBH589 (panobinostat),37 a novel potent anti-myeloma agent, followed by RNA and total protein extraction, and identification of KLF9 mRNA and protein levels, respectively. In agreement with previous reports,37,38 treatment of MM1.S cells with LBH589 led to a significant increase in the amounts of H3Ac and up-regulation of NOXA (Figure 2E). We also detected an increase in the KLF9 mRNA and protein levels (Figure 2D-E). On the other hand, treatment of LBH589 resulted in more potent up-regulation of histone H3 acetylation than bortezomib treatment (Figure 2B-E, supplemental Figure 1), and yet the amounts of KLF9 mRNA were induced less efficiently by LBH589 than by bortezomib (Figure 2A-D). Therefore, enhanced histone acetylation may only partially account for KLF9 up-regulation by bortezomib, thus suggesting that additional bortezomib-dependent pathways are involved in the up-regulation of KLF9.
Figure 2.

Bortezomib and LBH589 up-regulate KLF9 in MM1.S cells. (A) KLF9/ACTB Q-RT-PCR signal ratios were obtained from total cellular RNAs corresponding to each group of MM1.S cells treated with indicated amounts of bortezomib (BTZ) for 24 hours. Signal ratios were normalized by the corresponding ratio in untreated cells (0nM). (B) MM1.S cells were treated with indicated amounts of bortezomib for 24 hours followed by Western blotting of total protein extracts with antibodies indicated on the left. Numbers below the panels show the fold increase of KLF9/tubulin and acetylated histone H3 (Ac-H3)/tubulin ratios normalized to that in the first lane (untreated cells). (C) MM1.S cells treated or not with 5nM of bortezomib for 24 hours were cross-linked, sonicated and subjected to chromatin immunoprecipitation with acetylated histone H3 (Ac-H3)–specific antibodies or nonspecific (IgG) antibodies followed by the reversal of the cross-linking and DNA isolation. Isolated DNA was used in quantitative PCR with GAPDH or KLF9 promoter-specific primers, positions of which are designated by the numbers. All PCR signals were normalized by GAPDH-specific PCR signals and by the corresponding PCR signals obtained in reactions on DNA precipitated with IgG antibodies. (D) KLF9/ACTB Q-RT-PCR signal ratios were obtained from total cellular RNAs corresponding to each group of MM1.S cells treated with indicated amounts of LBH589 (LBH) for 24 hours. Signal ratios were normalized by the corresponding ratio in untreated cells (0nM). (E) MM1.S cells were treated with the indicated amounts of LBH589 (LBH) for 24 hours followed by Western blotting of total protein extracts with antibodies indicated on the left. Numbers below the panels show the fold increase of KLF9/tubulin and acetylated histone H3 (Ac-H3)/tubulin ratios normalized to that in the first lane (untreated cells). (F) MM1.S cells were treated with the indicated amounts of tunicamycin (TM) or bortezomib (BTZ) for 24 hours followed by cell collection and Western blotting of cell total protein extracts with antibodies indicated on the left.
Induction of ER stress is considered one of the major downstream pathways of bortezomib cytotoxicity. To determine whether KLF9 can be induced by ER stress, we treated MM1.S cells with 50nM or 100nM tunicamycin, a classic ER stress inducer39 for 24 hours. To control for ER stress, we monitored expression of ATF4 protein, a marker of ER stress that is up-regulated by bortezomib and is required for bortezomib cytotoxicity in cells from several solid tumor lines.15,16 In agreement with the published data, Figure 2F demonstrates that tunicamycin significantly induced ATF4 but not NOXA.15 It also demonstrates that KLF9 is not induced by tunicamycin, suggesting that the bortezomib-dependent increase in KLF9 amounts occurs via ER stress-independent mechanisms.
KLF9 knockdown via RNA interference suppresses bortezomib and LBH589-induced death in multiple myeloma cells
To identify the functional role of KLF9 in bortezomib-induced cell death, we knocked down KLF9 in MM1.S and RPMI-8226 cells using lentivirus-based shRNAs (Figure 3A-C). At 48 hours after infection with control or KLF9 shRNAs, MM1.S and RPMI 8826 cells were treated with different doses of bortezomib or LBH589 for 48 hours. Cell death was assessed by trypan blue-exclusion assay. As shown in Figure 3B through D, KLF9 depletion resulted in a 2- to ∼ 3.5-fold inhibition of cell death caused by bortezomib and substantial reduction in bortezomib-induced levels of NOXA and cleaved caspase 3 (Figure 3A-C) thereby demonstrating a direct functional role for KLF9 in bortezomib cytotoxicity in MM cells. Depletion of KLF9 also partially rescued MM1.S and RPMI-8826 cells from death caused by LBH589, albeit not as efficiently as from death induced by bortezomib (approximately 35%-45% inhibition of cell death, Figure 3B-D). Importantly, simultaneous treatment of MM1.S or RPMI8826 cells with bortezomib and LBH589 resulted in cumulative increase in the amounts of KLF9 (Figure 3E), whereas knocking down KLF9 rendered these myeloma cells more resistant to the combinatory treatment of bortezomib and LBH589 (Figure 3F-G).
Figure 3.

KLF9 knockdown suppresses apoptosis and efficient up-regulation of NOXA by bortezomib or LBH589. (A) MM1.S cells were infected with control shRNAs (CL), KLF9 shRNAs 1 (K9-1), or KLF9 shRNAs2 (K9-2). Forty-eight hours after infection, cells were incubated with the vehicle (DMSO) or with the indicated amounts of bortezomib (BTZ) or LBH589 (LBH) for 48 hours. Total protein extracts from the cells were probed by Western blotting with antibodies designated on the left. *Endogenous NOXA and cleaved caspase 3 levels in untreated cells were not detected under the exposure conditions used to detect these proteins in cells treated with the drugs. (B) Cells described in panel A were stained with the trypan blue. The percentage of dead cells was determined by counting positive and negative cells under light microscope in multiple view fields. The 2-tailed P value is shown for the groups with close values of the means. (C) RPMI-8226 cells were infected with control shRNAs (CL) or KLF9 shRNAs 1 (K9-1) or KLF9 shRNAs2 (K9-2). Forty-eight hours after infection, cells were incubated with the vehicle (DMSO) or with the indicated amounts of bortezomib (BTZ) or LBH589 (LBH) for 48 hours. Total protein extracts from the cells were probed in Western blotting with antibodies designated on the left. *Endogenous NOXA and cleaved caspase 3 levels in untreated cells were not detected under the exposure conditions used to detect these proteins in cells treated with the drugs. (D) Cells described in panel C were stained with trypan blue. The percentage of dead cells was determined by counting positive and negative cells under light microscope in multiple view fields. The two-tailed P value is shown for the groups with close values of the means. (E) Cells were incubated with no drug (UT) or with the indicated amounts of bortezomib (BTZ), LBH589 (LBH), or combination of the same concentration of both drugs for 24 hours. After incubation, cells were collected and total protein extracts from the cells were probed in Western blotting with antibodies designated on the left. (F) Cells were infected with control shRNAs (CL) KLF9 shRNAs 1 (K9-1), or KLF9 shRNAs2 (K9-2). Forty-eight hours after infection, cells were incubated with the indicated amounts of bortezomib (BTZ), LBH589 (LBH), or combination of the same concentration of both drugs. After incubation, cells were collected and total protein extracts from the cells were probed in Western blotting with antibodies designated on the left. (G) Cells described in panel F were stained with trypan blue. The percentage of dead cells was determined by counting positive and negative cells under light microscope in multiple view fields.
Resistance to bortezomib has been recently linked to decreased protein biosynthesis24 that could be manifested in low proliferation rates and to formation of aggresomes, aggregates of ubiquitin-conjugated proteins that are accumulated in the perinuclear area because of the inhibition of the proteasome.24 To test the possibility that KLF9 depletion affects any or all of the above phenotypes, we assessed proliferation rates and the amounts of secreted λ light chain (as a measure of protein biosynthesis rates in MM cells)40,41 in untreated control and KLF9-depleted MM1.S and RPMI-8826 cells. As shown in supplemental Figure 2A and B, no significant difference was identified in either assay between control and KLF9-depleted cells. In addition, no significant difference was detected in the number of cells containing small or large bortezomib-induced aggresomes between wild-type and KLF9-depleted populations (supplemental Figure 2C-D). Therefore, inhibition of KLF9 does not affect proliferation or bortezomib-induced protein accumulation in MM cells. It is also noteworthy that KLF9 levels did not correlate with that of proliferation markers TOP2A and MKI67 in the patient database (data not shown).
KLF9 overexpression induces NOXA-dependent apoptosis in multiple myeloma cells
To determine whether KLF9 up-regulation was sufficient to cause apoptosis in MM cells, we cloned KLF9 cDNA into a lentiviral vector followed by transduction into MM1.S or RPMI-8226 cells in parallel with an empty vector control. MM cells overexpressing KLF9 demonstrated an ∼ 2.5-fold increase in NOXA mRNA and protein levels already at the second day after infection (Figure 4A-B), although no significant increase in cell death was detected at that time. However, starting at day 3 after infection, MM1.S and RPMI-8226 cells that were overexpressing KLF9 underwent apoptosis (Figure 5C) with the concomitant up-regulation of NOXA (Figure 4D). These data demonstrate that ectopic expression of KLF9 was sufficient to induce apoptosis in MM cells.
Figure 4.

KLF9 induces NOXA-dependent apoptosis. (A) MM1.S and RPMI-8226 cells were infected with control vector (V) or vector expressing KLF9 cDNA (KLF9). Cells were collected 48 hours after infection and the total protein extracts were probed in Western blotting with antibodies shown on the left. *Note that an enhanced assay was used for detecting basal levels of NOXA (see “Immunoblotting”). The percentage of dead cells was determined by counting trypan blue positive and negative cells in multiple view fields. (B) NOXA/ACTB Q-RT-PCR signal ratios were obtained in indicated cells expressing KLF9 cDNA or empty vector. Signal ratios were normalized by the corresponding ratio in cells infected with the empty vector. (C) Cells were infected as described in (A). At the indicated days after infection, cells were stained for the activated caspase 3 using APO LOGIX “Carboxyfluorescein FAM-DEVD-FMK for caspase 3” kit. The percentage of apoptotic cells was determined by counting positive and negative cells with fluorescence microscopy in multiple view fields. (D) MM1.S and RPMI-8826 cells were infected with NOXA shRNA followed by super-infection with control vector or vector expressing KLF9 cDNA. Five days after the second infection cells were collected and total cellular protein extracts were probed in Western blotting with the antibodies designated on the left. (E) Cells described in panel D were stained for the activated caspase 3 using APO LOGIX “Carboxyfluorescein FAM-DEVD-FMK for caspase 3” kit. The percentage of apoptotic cells was determined by counting positive and negative cells with fluorescence microscopy in multiple view fields. The 2-tailed P value is shown.
Figure 5.

KLF9 directly interacts with NOXA promoter in bortezomib-dependent manner. (A) Schematic representation of human NOXA promoter. Vertical lines above the central bar indicate potential KLF9 binding sites. Arrowheads correspond to the position of primers used for Q-PCR. The transcription start site (+1) is shown by the arrow. (B) MM1.S cells untreated or treated with 5nM bortezomib for 24 hours were cross-linked, lysed, and chromatin was immunoprecipitated with 2 different KLF9-specific antibodies (KLF91 or KLF92) or nonspecific (IgG) antibodies followed by the reversal of the cross-linking and DNA isolation. DNA was used in quantitative PCR with GAPDH or NOXA promoter-specific primers positions of which are designated by the numbers. All PCR signals were normalized by GAPDH-specific PCR signals and by the corresponding PCR signals obtained in reactions on DNA precipitated with IgG antibodies. (C) Control pGL3-basic vector “[0]” or vectors containing indicated regions of NOXA promoter were mixed with pRL-SV40 plasmid, expressing renilla luciferase gene in 1÷10 proportion, followed by transfection into HEK293 cells along with vector expressing KLF9 cDNA or empty vector. Forty-eight hours after transfection, cells were collected, and firefly and renilla-dependent luciferase activities were determined using the dual-luciferase assay kit (Promega). Measurements of firefly-dependent luciferase activity were normalized with respect to renilla signals. (D) Cells were infected with control (Cl) or KLF9-specific shRNAs (K9-1 or K9-2). Cells were collected 48 hours after infection and the total protein extracts were probed in Western blotting with antibodies shown on the left. *Note that an enhanced assay was used for detecting basal levels of NOXA (see“Immunoblotting”).
To identify the functional role of NOXA in KLF9-induced apoptosis, we depleted NOXA in MM1.S and RPMI-8226 cells via shRNA8 followed by infection with KLF9 cDNA-expressing lentivirus. Introduction of NOXA shRNA resulted in suppression of bortezomib or LBH589-induced apoptosis as was reported earlier7,8,37 (supplemental Figure 3), and in the partial reversal of KLF9-induced NOXA levels (Figure 4D). This was accompanied by a decrease in apoptosis rates (52%-27% in MM1.S cells; 59%-35% in RPMI-8226 cells, Figure 4E) and the amounts of cleaved caspase 3 (Figure 4D). These data indicate that cell death induced by KLF9 is at least partially NOXA-dependent.
To differentiate between direct and indirect up-regulation of NOXA by KLF9, we performed ChIP with control (normal goat IgG), or with 2 KLF9-specific goat antibodies from untreated MM1.S cells, or cells treated with 5nM bortezomib for 24 hours. After precipitation, the chromatin was de-crosslinked and the purified DNA was probed in Q-PCR with primers spanning the human NOXA mRNA start site. As shown in Figure 5A, the DNA region adjacent to the mRNA start site contains several potential KLF9 binding sites (5′-CA/GCCC-3′).42,43 Moreover, this promoter region has been shown previously to regulate NOXA transcriptional activation.13 For a negative control, we used PCR primers corresponding to the DNA region ∼ 1.1 Kbs upstream of the mRNA start site (Figure 5A) and PCR primers corresponding to a KLF9-irrelevant promoter (GAPDH). As shown in Figure 5B, the significant enrichments of NOXA proximal, but not distal, promoter regions were detected in chromatin precipitated with KLF9-specific antibodies only from bortezomib-treated cells. Furthermore, a luciferase-based reporter assay demonstrated a low, but reproducible, KLF9-dependent activation of a luciferase gene driven by the NOXA proximal promoter region (Figure 5C).
The observation that KLF9 does not interact with NOXA promoter in untreated cells, prompted us to investigate whether the basal, uninduced NOXA levels depend on basal KLF9 levels. As shown in Figure 5D, depletion of KLF9 did not affect basal expression of NOXA in agreement with the ChIP data. Collectively, our results argue that KLF9 directly interacts with the NOXA promoter in a bortezomib-dependent manner, thus providing a mechanistic explanation for the KLF9-dependent toxicity and KLF9-dependent pattern of NOXA transcriptional activation in response to bortezomib in MM cells.
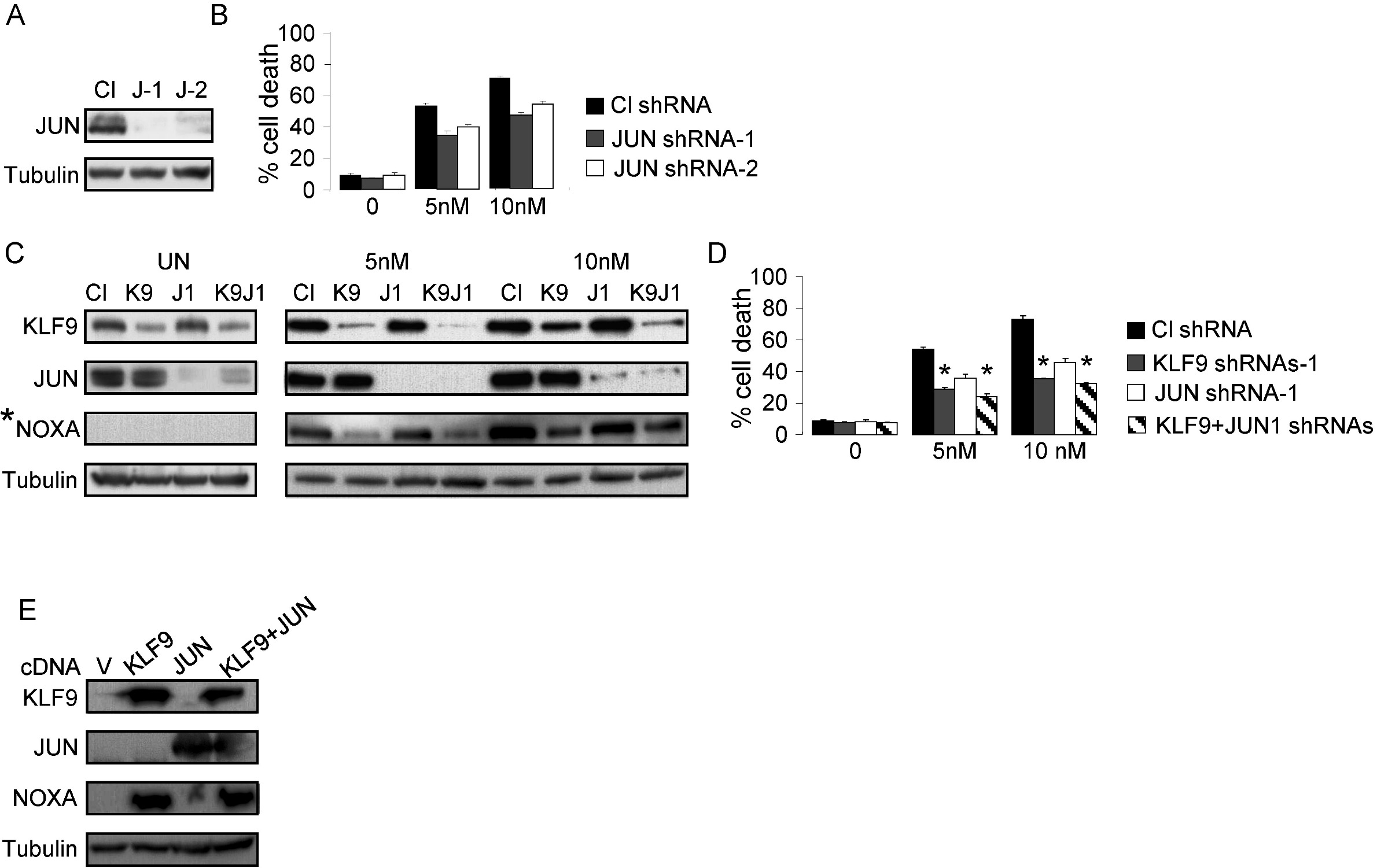
Recently, knockdown of JUN in MM cells was reported to increase their resistance to bortezomib treatment.26 To investigate effects of simultaneous inhibition of KLF9 and JUN on bortezomib-induced cell death and NOXA levels in MM cells, we depleted JUN alone or in combination with KLF9 in MM1.S cells via corresponding shRNAs and identified changes in NOXA induction and cell sensitivity to bortezomib. Inhibition of JUN with 2 independent shRNAs decreased bortezomib citotoxicity, albeit less efficiently than inhibition of KLF9 (supplemental Figure 4A-D). However, simultaneous depletion of KLF9 and JUN in MM1.S cells did not result in statistically significant decrease in cell death or in further down-regulation of bortezomib-induced NOXA levels compared with that caused by inhibition of KLF9 alone (supplemental Figure 4C-D). Moreover, ectopic expression of JUN did not up-regulate basal or KLF9-induced levels of NOXA in MM1.S cells (supplemental Figure 4E). Thus, unlike KLF9, JUN appears to be involved in NOXA-independent regulation of bortezomib toxicity in MM cells.
Discussion
Bortezomib induces substantial changes in global gene expression, and yet transcription factors responsible for the bortezomib-dependent programs in MM cells remain largely unknown. Our analyses revealed that among all bortezomib-responsive transcriptional regulators, KLF9 was the only gene with the values for all Affymetrix probesets that demonstrated a statistically significant difference between bortezomib responders and nonresponders in the patient database (supplemental Table 2). In addition, values for one of the KLF9 probesets displayed the highest statistical significance among all bortezomib-inducible transcription factors identified by us. Recently, Chen et al demonstrated that basal levels of transcriptional regulators JUN and EGR1 have prognostic values in patients receiving TT3 versus TT2 regimens (the sets of drugs used in these regimens differ only by bortezomib present in TT3 regimen).26 The study did not mention whether bortezomib would elevate JUN or EGR1 levels in cultured MM cells over the levels achieved by treating these cells with the TT2 regimen drugs; however, up-regulation of JUN by bortezomib has been reported earlier,27 whereas our current data (supplemental Table 1) demonstrated bortezomib-dependent expression of both JUN and EGR1. A possible explanation for these findings could be that bortezomib-induced killing involves elevated expression of certain transcription factors, and a critical threshold level of such factors has to be achieved to make the apoptotic decision irreversible. The cells that have higher levels of such proteins before treatment may be especially vulnerable because they may require a lower potency of the toxic impact to reach that critical threshold level. On the other hand, the cells that have lower steady-state levels of such proteins are less probably to achieve this critical threshold on exposure to the same levels of bortezomib. Interestingly, unlike JUN and EGR1, elevated basal levels of KLF9 did not correlate with OS in patients who received TT3 versus TT2 regimens (data not shown). This could be because of different roles that KLF9 may play in cell death induced by bortezomib monotherapy or combinational TT3 therapy.
We demonstrated that, unlike other transcription factors implemented in bortezomib cytotoxicity (ATF3,15 ATF4,15 and p5314), induction of ER stress (one the major downstream pathways of bortezomib) did not up-regulate KLF9 (Figure 2F), whereas an experimental antimyeloma agent LBH589 (HDAC inhibitor) induced both KLF9 mRNA and protein levels (Figure 2D-E). In view of recent findings by Kikuchi et al that bortezomib suppressed intracellular amounts of HDAC I family members35 and our own observations that bortezomib increased histone H3 acetylation at the KLF9 promoter (Figure 2C), it is plausible that up-regulation of KLF9 by bortezomib occurs via HDAC inhibition-dependent mechanisms. LBH589 is a more potent inducer of histone H3 acetylation (supplemental Figure 1). However LBH589 is less efficient at inducing KLF9 mRNA (Figure 2A-D), suggesting that additional transcriptional pathways are involved in up-regulation of KLF9 by bortezomib.
In earlier studies, KLF9 was identified as a gene that controlled animal development and cell proliferation. Thus, Klf9 homozygous knockout mice demonstrated defects of uterine growth,29 decreased litter size,30 reduced crypt stem/transit cell proliferation,31 and mild neurologic defects.32 Low levels of KLF9 were associated with enhanced proliferation of unstimulated B cells33 and differentiation of B cells.34 Recently, KLF9 has been implicated in suppression of growth of glioblastoma-initiating cells,44 and its levels were lower in human colorectal cancer tissues compared with individually matched normal mucosa tissues.45 However, no association between KLF9 and sensitivity or resistance to chemotherapeutic agents has ever been reported. Our data assigned a novel function to KLF9 as a regulator of NOXA-dependent apoptosis. We demonstrate that rapid increase in the amounts of KLF9 via bortezomib or LBH589 treatment up-regulates NOXA in MM cells. However, KLF9 knockdown impaired induction of NOXA by bortezomib more efficiently than by LBH589 (Figure 3A-C), suggesting that KLF9 cooperates with additional unidentified transcriptional regulators to activate NOXA in response to LBH589 in MM cells.
It is noteworthy that depletion of KLF9 in untreated cells did not affect the basal levels of NOXA (Figure 5D). We also failed to identify a correlation between KLF9 and NOXA basal levels in the patient dataset (data not shown). Furthermore, the ChIP data revealed that detectable KLF9 binding to the NOXA promoter occurs only in bortezomib-treated MM1.S cells (Figure 5B). Although these data argue that KLF9 is a direct bortezomib-dependent transcriptional activator of NOXA, ectopic expression of KLF9 was capable of activating NOXA in the absence of bortezomib (Figure 4A-D). Together these results suggest that increasing intracellular KLF9 levels beyond a certain threshold amounts may be sufficient to activate NOXA expression.
Depletion of endogenous NOXA via specific shRNA partially inhibited KLF9-induced apoptosis further attesting to a functional connection between KLF9 and NOXA. However, shRNA-mediated down-regulation of NOXA was not sufficient to completely reverse its induction by bortezomib. Therefore, a formal possibility exists that KLF9 induces apoptosis by both NOXA-dependent and NOXA-independent pathways. Further experiments will be needed for the identification of other KLF9 targets that are essential for its apoptosis-inducing activity.
In summary, we have identified KLF9 as a novel and potentially clinically relevant transcriptional regulator of drug-induced apoptosis in MM cells.
Supplementary Material
Acknowledgments
The authors are grateful to Dr Catherine Burkhart (Cleveland BioLabs Inc), Dr Eugene S. Kandel (Roswell Park Cancer Institute), and Dr Angela Omilian (Roswell Park Cancer Institute) for critical reading of the paper; Dr Constantine S. Mitsiades (Dana-Farber Cancer Institute); and Dr Aliaksandr Chekhau (Roswell Park Cancer Institute).
This work was supported by the following National Institutes of Health grants: R01 CA120244 to M.A.N., R01 CA121044 to K.L., and R01 CA127910 to L.H.B.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.M., D.Z., R.B., J.A.W., S.N.Z., E.E.F., and K.C.M. performed experiments; J.R.N., L.H.B., and K.P.L analyzed the results; J.R.N, Q.H., and S.L. performed statistical analysis; and M.A.N. designed the research, analyzed the results, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mikhail A. Nikiforov, Roswell Park Cancer Institute, Dept of Cell Stress Biology, BLSC L3-317, Elm & Carlton St, Buffalo, NY 14263; e-mail: mikhail.nikiforov@roswellpark.org.
References
- 1.Anderson KC. Bortezomib therapy for myeloma. Curr Hematol Rep. 2004;3(1):65. [PubMed] [Google Scholar]
- 2.Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. CA Cancer J Clin. 2000;50(1):7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- 3.Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raab MS, Podar K, Breitkreutz I, et al. Multiple myeloma. Lancet. 2009;374(9686):324–339. doi: 10.1016/S0140-6736(09)60221-X. [DOI] [PubMed] [Google Scholar]
- 5.Venner CP, Connors JM, Sutherland HJ, et al. Novel agents improve survival of transplant patients with multiple myeloma including those with high-risk disease defined by early relapse (< 12 months). Leuk Lymphoma. 2011;52(1):34–41. doi: 10.3109/10428194.2010.531409. [DOI] [PubMed] [Google Scholar]
- 6.Fennell DA, Chacko A, Mutti L. BCL-2 family regulation by the 20S proteasome inhibitor bortezomib. Oncogene. 2008;27(9):1189–1197. doi: 10.1038/sj.onc.1210744. [DOI] [PubMed] [Google Scholar]
- 7.Qin JZ, Ziffra J, Stennett L, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65(14):6282–6293. doi: 10.1158/0008-5472.CAN-05-0676. [DOI] [PubMed] [Google Scholar]
- 8.Fernández Y, Verhaegen M, Miller TP, et al. Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res. 2005;65(14):6294–6304. doi: 10.1158/0008-5472.CAN-05-0686. [DOI] [PubMed] [Google Scholar]
- 9.Gomez-Bougie P, Wuillème-Toumi S, Ménoret E, et al. Noxa up-regulation and Mcl-1 cleavage are associated to apoptosis induction by bortezomib in multiple myeloma. Cancer Res. 2007;67(11):5418–5424. doi: 10.1158/0008-5472.CAN-06-4322. [DOI] [PubMed] [Google Scholar]
- 10.Adams KW, Cooper GM. Rapid turnover of Mcl-1 couples translation to cell survival and apoptosis. J Biol Chem. 2007;282:6192–6200. doi: 10.1074/jbc.M610643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuroda J, Taniwaki M. Involvement of BH3-only proteins in hematologic malignancies. Crit Rev Oncol Hematol. 2009;71:89–101. doi: 10.1016/j.critrevonc.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Gillings AS, Balmanno K, Wiggins CM, Johnson M, Cook SJ. Apoptosis and autophagy: BIM as a mediator of tumour cell death in response to oncogene-targeted therapeutics. FEBS J. 2009;276(21):6050–6062. doi: 10.1111/j.1742-4658.2009.07329.x. [DOI] [PubMed] [Google Scholar]
- 13.Nikiforov MA, Riblett M, Tang WH, et al. Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc Natl Acad Sci U S A. 2007;104(49):19488–19493. doi: 10.1073/pnas.0708380104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen S, Blank JL, Peters T, et al. Genome-wide siRNA screen for modulators of cell death induced by proteasome inhibitor bortezomib. Cancer Res. 2010;70(11):4318–4326. doi: 10.1158/0008-5472.CAN-09-4428. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Mora-Jensen H, Weniger MA, et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci U S A. 2009;106(7):2200–2205. doi: 10.1073/pnas.0807611106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armstrong JL, Flockhart R, Veal GJ, Lovat PE, Redfern CP. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J Biol Chem. 2010;285(9):6091–6100. doi: 10.1074/jbc.M109.014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim R, Emi M, Tanabe K, Murakami S. Role of the unfolded protein response in cell death. Apoptosis. 2006;11(1):5–13. doi: 10.1007/s10495-005-3088-0. [DOI] [PubMed] [Google Scholar]
- 18.Milani M, Rzymski T, Mellor HR, et al. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with bortezomib. Cancer Res. 2009;69(10):4415–4423. doi: 10.1158/0008-5472.CAN-08-2839. [DOI] [PubMed] [Google Scholar]
- 19.Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61(7):3071–3076. [PubMed] [Google Scholar]
- 20.Ma MH, Yang HH, Parker K, et al. The proteasome inhibitor PS-341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents. Clin Cancer Res. 2003;9(3):1136–1144. [PubMed] [Google Scholar]
- 21.Hideshima T, Chauhan D, Richardson P, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277(19):16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 22.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rizzatti EG, Mora-Jensen H, Weniger MA, et al. Noxa mediates bortezomib induced apoptosis in both sensitive and intrinsically resistant mantle cell lymphoma cells and this effect is independent of constitutive activity of the AKT and NF-kappaB pathways. Leuk Lymphoma. 2008;49(4):798–808. doi: 10.1080/10428190801910912. [DOI] [PubMed] [Google Scholar]
- 24.Nawrocki ST, Carew JS, Maclean KH, et al. Myc regulates aggresome formation, the induction of Noxa, and apoptosis in response to the combination of bortezomib and SAHA. Blood. 2008;112(7):2917–2926. doi: 10.1182/blood-2007-12-130823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khachigian LM, Collins T. Early growth response factor 1: a pleiotropic mediator of inducible gene expression. J Mol Med. 1998;76(9):613–616. doi: 10.1007/s001090050258. [DOI] [PubMed] [Google Scholar]
- 26.Chen L, Wang S, Zhou Y, et al. Identification of early growth response protein 1 (EGR-1) as a novel target for JUN-induced apoptosis in multiple myeloma. Blood. 2010;115(1):61–70. doi: 10.1182/blood-2009-03-210526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shringarpure R, Catley L, Bhole D, Burger R, et al. Gene expression analysis of B-lymphoma cells resistant and sensitive to bortezomib. Br J Haematol. 2006;134(2):145–156. doi: 10.1111/j.1365-2141.2006.06132.x. [DOI] [PubMed] [Google Scholar]
- 28.Mulligan G, Mitsiades C, Bryant B, et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood. 2007;109(8):3177–3188. doi: 10.1182/blood-2006-09-044974. [DOI] [PubMed] [Google Scholar]
- 29.Velarde MC, Geng Y, Eason RR, Simmen FA, Simmen RC. Null mutation of Kruppel-like factor9/basic transcription element binding protein-1 alters peri-implantation uterine development in mice. Biol Reprod. 2005;73(3):472–481. doi: 10.1095/biolreprod.105.041855. [DOI] [PubMed] [Google Scholar]
- 30.Zeng Z, Velarde MC, Simmen FA, Simmen RC. Delayed parturition and altered myometrial progesterone receptor isoform A expression in mice null for Krüppel-like factor 9. Biol Reprod. 2008;78(6):1029–1037. doi: 10.1095/biolreprod.107.065821. [DOI] [PubMed] [Google Scholar]
- 31.Simmen FA, Xiao R, Velarde MC, et al. Dysregulation of intestinal crypt cell proliferation and villus cell migration in mice lacking Kruppel-like factor 9. Am J Physiol Gastrointest Liver Physiol. 2007;292(6):G1757–G1769. doi: 10.1152/ajpgi.00013.2007. [DOI] [PubMed] [Google Scholar]
- 32.Bonett RM, Hu F, Bagamasbad P, Denver RJ. Stressor and glucocorticoid-dependent induction of the immediate early gene kruppel-like factor 9: implications for neural development and plasticity. Endocrinology. 2009;150(4):1757–1765. doi: 10.1210/en.2008-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Good KL, Tangye SG. Decreased expression of Kruppel-like factors in memory B cells induces the rapid response typical of secondary antibody responses. Proc Natl Acad Sci U S A. 2007;104(33):13420–13425. doi: 10.1073/pnas.0703872104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Savignac M, Mellström B, Bébin AG, et al. Increased B cell proliferation and reduced Ig production in DREAM transgenic mice. J Immunol. 2010;185(12):7527–7536. doi: 10.4049/jimmunol.1000152. [DOI] [PubMed] [Google Scholar]
- 35.Kikuchi J, Wada T, Shimizu R, et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood. 2010;116(3):406–417. doi: 10.1182/blood-2009-07-235663. [DOI] [PubMed] [Google Scholar]
- 36.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20(8):615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 37.Maiso P, Carvajal-Vergara X, Ocio EM, et al. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006;66(11):5781–5789. doi: 10.1158/0008-5472.CAN-05-4186. [DOI] [PubMed] [Google Scholar]
- 38.Inoue S, Riley J, Gant TW, et al. Apoptosis induced by histone deacetylase inhibitors in leukemic cells is mediated by Bim and Noxa. Leukemia. 2007;8(8):1773–1782. doi: 10.1038/sj.leu.2404760. [DOI] [PubMed] [Google Scholar]
- 39.Linder S, Shoshan MC. Lysosomes and endoplasmic reticulum: targets for improved, selective anticancer therapy. Drug Resist Updat. 2005;8(4):199–204. doi: 10.1016/j.drup.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 40.Obeng EA, Carlson LM, Gutman DM, et al. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen PP, Nonaka M, O'Hair CH, et al. Human IgE synthesis in vitro by pokeweed mitogen-stimulated human lymphoid cells: verification with a reconfirmed epsilon-specific radioimmunoassay. J Immunol. 1984;133(4):1909–1913. [PubMed] [Google Scholar]
- 42.Pei H, Yao Y, Yang Y, et al. Krüppel-like factor KLF9 regulates PPARγ transactivation at the middle stage of adipogenesis. Cell Death Differ. 2011;18(2):315–327. doi: 10.1038/cdd.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sogawa K, Kikuchi Y, Imataka H, et al. Comparison of DNA-binding properties between BTEB and Sp1. J Biochem. 1993;114(4):605–609. doi: 10.1093/oxfordjournals.jbchem.a124224. [DOI] [PubMed] [Google Scholar]
- 44.Ying M, Sang Y, Li Y, et al. Krüppel-like family of transcription factor 9, a differentiation-associated transcription factor, suppresses Notch1 signaling and inhibits glioblastoma-initiating stem cells. Stem Cells. 2011;29(1):20–31. doi: 10.1002/stem.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang L, Lü B, Xu J, et al. Down-regulation of Krüppel-like factor 9 in human colorectal cancer. Pathol Int. 2008;58(6):334–338. doi: 10.1111/j.1440-1827.2008.02233.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}