

Abstract
The prohormone convertases PC1/3 and PC2 are eukaryotic serine proteases involved in the proteolytic maturation of peptide hormone precursors and are implicated in a variety of pathological conditions, including obesity, diabetes, and neurodegenerative diseases. In this work, we screened 45 compounds obtained by derivatization of a 2,5-dideoxystreptamine scaffold with guanidinyl and aryl substitutions for convertase inhibition. We identified four promising PC1/3 competitive inhibitors and three PC2 inhibitors that exhibited various inhibition mechanisms (competitive, noncompetitive, and mixed), with sub- and low micromolar inhibitory potency against a fluorogenic substrate. Low micromolar concentrations of certain compounds blocked the processing of the physiological substrate proglucagon. The best PC2 inhibitor effectively inhibited glucagon synthesis, a known PC2-mediated process, in a pancreatic cell line; no cytotoxicity was observed. We also identified compounds that were able to stimulate both 87 kDa PC1/3 and PC2 activity, behavior related to the presence of aryl groups on the dideoxystreptamine scaffold. By contrast, inhibitory activity was associated with the presence of guanidinyl groups. Molecular modeling revealed interactions of the PC1/3 inhibitors with the active site that suggest structural modifications to further enhance potency. In support of kinetic data suggesting that PC2 inhibition probably occurs via an allosteric mechanism, we identified several possible allosteric binding sites using computational searches. It is noteworthy that one compound was found to both inhibit PC2 and stimulate PC1/3. Because glucagon acts in functional opposition to insulin in blood glucose homeostasis, blocking glucagon formation and enhancing proinsulin cleavage with a single compound could represent an attractive therapeutic approach in diabetes.
Introduction
The prohormone convertases 1/3 and 2 (PC1/3 and PC2) are thought to be responsible for the processing of multiple peptide hormones and neuropeptide precursors within the constitutive and regulated secretory pathways. PC1/3 and PC2 are calcium-dependent serine proteases with acidic pH optima that belong to the bacterial subtilisin superfamily, which also includes the related yeast enzyme kexin (for review, see Cameron et al., 2001); these convertases share many functional and biochemical features. Their specificities toward various cleavage sites appear to be distinct, albeit overlapping, and variations in their expression levels are responsible for differential precursor processing, as exemplified by the processing of proopiomelanocortin (Day et al., 1992; Rhodes et al., 1993; Zhou et al., 1993). Proglucagon and proinsulin present other interesting examples of differential processing: the processing of proglucagon to glucagon is carried out mainly by PC2 (Rouillé et al., 1997), whereas insulin is processed from proinsulin mainly by PC1/3 (Smeekens et al., 1992).
During the past decade, important pathological conditions have been linked to the proprotein convertases, including obesity (Lloyd et al., 2006; Farooqi et al., 2007; Heni et al., 2010), diabetes (Furuta et al., 1997; Spruce et al., 2003), opportunistic diseases (Decroly et al., 1994), and hypercholesterolemia, a high-risk condition for cardiovascular disease (Arnaoutova et al., 2003). Owing to these linkages, there is increasing interest in prohormone convertases as novel targets for drug design, not only for disease intervention but also for use in determining the various physiological roles of these enzymes.
To date, most reported inhibitors against the proprotein convertase furin have been either proteins (Dahlen et al., 1998; Dufour et al., 2001; Komiyama et al., 2003; Richer et al., 2004) or peptides/peptide derivatives (Cameron et al., 2000a; Villemure et al., 2003; Basak and Lotfipour, 2005). Nonprotein, nonpeptide convertase inhibitors reported thus far are the natural products of the andrographalide family and their succinoyl ester derivatives (Basak et al., 1999); certain metal complexes (Podsiadlo et al., 2004); dicoumarol and its derivatives (Komiyama et al., 2009); and the bicyclic guanidine and pyrrolidine bis-piperazine derivatives we previously identified as PC2 inhibitors (Kowalska et al., 2009). Nonpeptide furin inhibitors based on 2,5-dideoxystreptamine have also been described (Jiao et al., 2006b). In the work presented here, we have screened 45 compounds related to these initial furin inhibitors that contain various aryl and guanidinyl substitutions on the 2,5-dideoxystreptamine scaffold. We identified four promising compounds that potently inhibit PC1/3 and three other inhibitory compounds directed against PC2. Finally, we present the possible binding modes of these inhibitors with both PCs through molecular modeling.
Materials and Methods
Recombinant Convertase Preparation.
Mouse 87-kDa PC1/3 and mouse pro-PC2 were purified from the conditioned medium of stably transfected, methotrexate-amplified Chinese hamster ovary cells as described previously (Hoshino et al., 2011). Pro-PC2 was activated before use by dilution in reaction buffer.
Synthesis of 2,5-Dideoxystreptamine Derivatives.
Forty-five compounds based on the 2,5-dideoxystreptamine scaffold were synthesized at PanThera Biopharma, LLC (Aiea, HI). Compounds 166829 [5-(2,4-bis(imidazolidin-2-ylideneamino)phenoxy)-2,4-bis(imidazolidin-2-ylideneamino)cyclohexanol] and 166830 [N1,N3-di(imidazolidin-2-ylidene)-4,6-bis(4 (imidazolidin-2-ylideneamino)phenoxy)cyclohexane-1,3-diamine] were synthesized via reaction of intermediates 166829a [2,4-diamino-5-(2,4-diaminophenoxy)cyclohexanol] and 166830a [4,6-bis(4-aminophenoxy)cyclohexane-1,3-diamine] with di-tert-butyl 2-thioxoimidazolidine-1,3-dicarboxylate, followed by deprotection with trifluoroacetic acid (TFA) (Scheme 1). The synthesis of compounds 166369 [5-(2,4-diguanidino-phenoxy)-2,4-diguanidino-cyclohexyl (4-octylphenyl)carbamate] and 166646 [5-(2,4-diguanidino-phenoxy)-2,4-diguanidino-cyclohexyl [1,1′-biphenyl]-4-ylcarbamate] was achieved by the TFA deprotection of the product from the reaction of intermediate 166369a [N,N′-di-tert-butoxycarbonyl-N″-{4-[2,4-di-(di-tert-butoxycarbonyl-guanidino)-phenoxy]-5-[di-(tert-butoxycarbonyl)-guanidino]-2-hydroxycyclohexyl}-guanidine] with 1-isocyanato-4-octylbenzene or 4-isocyanato-1,1′-biphenyl (Scheme 2). The remainder of the 45 compounds, including 166631 [N-[5-guanidino-2,4-bis-(4-guanidino-phenoxy)-cyclohexyl]-guanidine], 166550 [N-[2,4-bis(4-guanidino-phenoxy)-5-(4-guanidino-phenylamino)-cyclohexyl]-guanidine], 166811 [N-[5-guanidino-4-(4-guanidino-naphthalen-1-yloxy)-2-(4-guanidino-phenoxy)-cyclohexyl]-guanidine], 166812 [N-{2-[2,4-diguanidino-5-(4-guanidino-phenoxy)-cyclohexyloxy]-5-guanidino-phenyl}-guanidine], and 166691 [N-[5-(4-guanidino-naphthalen-1-ylamino)-2,4-bis-(4-guanidino-naphthalen-1-yloxy)-cyclohexyl]-guanidine], and intermediates 166829a, 166830a, and 166369a were prepared following the procedures described previously (Jiao et al., 2006a,b).
Scheme 1.

Synthesis of compounds 166829 and 166830.
Scheme 2.

Synthesis of compounds 166369 and 166646.
Library Screen and Enzyme Assays.
The PC2 assay was performed in 96-well polypropylene microtiter plates in a final volume of 50 μl, containing 100 mM sodium acetate, pH 5.0, 2 mM CaCl2, 0.2% octyl glucoside, 0.1% NaN3, and 0.1 mg/ml BSA. The substrate pGlu-Arg-Thr-Lys-Arg-methylcoumarinamide (pERTKR-AMC; Peptide Institute, Lexington, KY) was used at a final concentration of 100 μM. PC2 was used at a final concentration of 16 nM, which generated 0.4 fluorescence units per minute (where 1 fluorescence unit corresponds to 3.8 pmol of aminomethyl coumarin). The inhibitors, at final concentrations of 10, 25, and 50 μM, were preincubated with enzyme for 30 min at 37°C before the addition of substrate. All assays were performed in either duplicate or triplicate for 1 h at 37°C and were quantified using a Fluoroscan Ascent fluorometer (Thermo Fisher Scientific, Waltham, MA) using excitation/emission wavelengths of 380/460 nm. Data were analyzed using Prism 5 (GraphPad Inc., San Diego, CA). The PC1/3 assay was performed at a final concentration of 92 nM PC1/3, 100 mM sodium acetate, pH 5.5, 2 mM CaCl2, 0.2% octylglucoside, 0.1% NaN3, and 0.1 mg/ml BSA.
Determination of IC50 Values for Inhibitors.
For IC50 assays, compounds were placed into plates containing PC1/3 and PC2 and their corresponding buffers. Serial dilutions of compounds were performed to give final concentrations between 10 nM and 500 μM in 50 μl. After a 30-min preincubation at room temperature, pERTKR-AMC (final concentration, 100 μM) was added, and residual enzyme activities were monitored by measuring AMC fluorescence intensity. Data were analyzed using Prism 5. The sigmoidal curves obtained were fitted with different equations depending on the shape of the experimental points. We used a four-parameter logistic equation for a standard dose-response curve (eq. 1), and a five-parameter logistic equation for asymmetrical curves (eq. 2) and bell-shaped dose-response curves (eq. 3), which occurs when low doses stimulate a response and high doses inhibit this response:
where EC50 is the concentration of agonist that gives a response half-way between Bottom and Top; nH describes the steepness of the family of curves and was constrained to a constant value of −1.0; and Top and Bottom are plateaus.
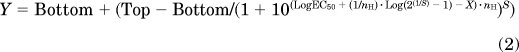 |
where Bottom and Top are the plateaus at the left and right ends of the curve, LogEC50 is the concentration that gives a half-maximal effect, and nH is the unitless slope factor, constrained to equal 1.0 (stimulation) or −1 (inhibition).
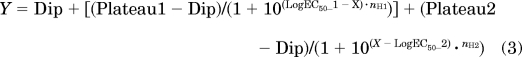 |
where Plateau1 and Plateau2 are the plateaus at the left and right ends of the curve; Dip is the plateau level in the middle of the curve; LogEC50_1 and LogEC50_2 are the concentrations that give half-maximal stimulatory and inhibitory effects, and nH1 and nH2 are the Hill slopes, which are considered to be equal to 1.0 (stimulation) and −1 (inhibition).
Enzyme Kinetics.
Studies of PC2 and PC1/3 inhibition kinetics were carried out at various concentrations of pERTKR-AMC ranging from 0 to 200 μM in the presence and absence of inhibitors. For all kinetic measurements, the compounds were preincubated with either enzyme for 30 min before the addition of substrate. All assays were performed in triplicate in 96-well microplates. Inhibition constants were determined using different equations, depending on the inhibition mechanism (Copeland, 2005). The Km values of PC1/3 and PC2 for pERTKR-AMC were 11 and 42 μM, respectively, as described previously (Cameron et al., 2000a; Kowalska et al., 2009).
Proglucagon Cleavage.
Human proglucagon, expressed from a Met-hPG/pET-9b vector (kindly supplied by R. B. Mackin, Creighton University, Omaha, NE) was purified as described previously (Bonic and Mackin, 2003). PC1/3 (3 μg) was preincubated for 1.5 h at room temperature with various concentrations of the 2,5-dideoxystreptamine derivatives 166811, 166812, 166550, and 166631 in 100 mM sodium acetate, pH 5.5, containing 2 mM CaCl2, 0.1% NaN3, 0.2% octylglucoside, and 0.1 mg/ml BSA. Human proglucagon (1.25 μg) was then added. PC2 (50 ng) was preincubated for 1 h at room temperature with different concentrations of 166830, 166829, and 166369 in 100 mM sodium acetate, pH 5, containing 2 mM CaCl2, 0.1% NaN3, 0.2% octylglucoside, and 0.1 mg/ml BSA; 3 μg of proglucagon were then added. The processing of proglucagon was carried out for 1 h for PC2 and 6 h for PC1/3 at 37°C; concentrated Laemmli sample buffer was then added, and the samples were boiled. Digestion products were separated on 18% polyacrylamide Tris-HCl gels and then Coomassie-stained.
Cell Culture.
α-TC6 cells, a mouse α cell line, were grown in Dulbecco's modified Eagle's medium containing 1 g/l d-glucose, 5% fetal calf serum, 15% horse serum (Irvine Scientific, Santa Ana, CA), and 100 U/ml penicillin/100 μg/ml streptavidin (Invitrogen, Carlsbad, CA). The cells were split into 48-well plates, incubated at 37°C/5% CO2, and used at approximately 70% confluence.
Glucagon Radioimmunoassay.
Samples from proglucagon cleavage reactions (starting amount, 2.5 μg) were digested with carboxypeptidase E (100 ng) to remove basic residues and then subjected to a commercial glucagon radioimmunoassay (GL-32K; Millipore, Billerica, MA), which uses 125I-labeled glucagon. This radioimmunoassay exhibits 0.1% cross-reactivity with oxyntomodulin, the primary gut glucagon-containing peptide. Cross-reactivity experiments using 2.5 μg of proglucagon failed to show any immunoreactivity. Samples were assayed according to the manufacturer's instructions. Radioactivity was determined using a Wallac 1470 Wizard gamma counter (PerkinElmer Life and Analytical Sciences, Wlatham, MA). The yield of immunoreactive glucagon from proglucagon in uninhibited samples was 46% of the amount expected for total hydrolysis.
For glucagon assays of α-TC6 cells, the inhibitors 166830 and (2R)-4-((R)-1-cyclohexyl-3-((S)-2-(((S)-2-(cyclohexylmethyl)piperazin-1-yl)methyl)pyrrolidin-1-yl)propan-2-yl)-2-(cyclohexylmethyl)-1-(2-(4-isobutylphenyl)propyl)piperazine (1435-6) were dissolved in 200 μl of Opti-MEM (Invitrogen) and incubated with cells at a final concentration of 75 μM for 36 h in quadruplicate wells of a 48-well plate at 37°C/5% CO2; limiting amounts of inhibitors precluded tests at higher concentrations. However, this experiment was repeated three times with similar results. Each well was then washed twice with PBS, pH 7.4. The PBS was replaced with 300 μl of 0.1 M HCl, and cells were collected by scraping, frozen on dry ice, thawed, and centrifuged. The supernatant was lyophilized and resuspended in 500 μl of RIA buffer (50 mM sodium phosphate, pH 7.6, 0.1% Triton X-100, and 0.02% sodium azide) and centrifuged again. Two microliters of α-TC6 cell extract were assayed in duplicate at two dilutions using the glucagon RIA kit described above. The experiment was repeated three times.
Viability Assay.
α-TC6 cells were split into a separate 48-well plate in quadruplicate at the same time as the experiment performed above and using the same inhibitor preparations. The medium was then changed to Opti-MEM (Invitrogen) containing inhibitors 166830 and 1435-6 (final concentration, 75 μM) and incubated for 36 h 37°C/5% CO2. Each well was washed once with PBS, pH 7.4, and 200 μl of Opti-MEM containing 20 μl of WST-1 reagent (Roche, Indianapolis, IN) were added to each well. Cells were incubated for various time periods (0.5–3 h) at 37°C. At 0, 30, 60, 90, and 180 min, the absorbance at 450 nm, which reflects the amount of formazan dye formed, was monitored with a multiwell spectrophotometer.
Statistical Analysis.
Data were analyzed using one-way analysis of variance followed by the Student-Newman-Keuls multiple comparison test, as appropriate, using the statistical software package SigmaStat (Systat Software, Inc., San Jose, CA). A probability value of p < 0.05 was considered statistically significant.
Molecular Modeling.
Homology models for prohormone convertases have been developed previously (Henrich et al., 2005) based on the X-ray crystal structure of furin (Henrich et al., 2003). In this work, we employed a refined homology model for PC1/3 and PC2; the details of our homology models will be described in a separate publication (A. B. Yongye, M. Vivoli, I. Lindberg, and K. Martinez-Mayorga, in preparation).
A search for allosteric binding sites was conducted using the SiteFinder application implemented in Molecular Operating Environment (MOE; Chemical Computing Group, Montreal, QC, Canada), and the FindSite software from the Skolnick laboratory (Brylinski and Skolnick, 2008). Agreement between the results obtained from MOE and FindSite formed the consensus set of binding sites for docking studies. LigPrep 2.2 (Schrodinger, Inc., New York, NY) was used to produce a low-energy conformation as well as different protomers for each molecule. Docking studies were conducted using Glide (Grid-based Ligand Docking with Energetics) version 5.6, from Schrödinger, Inc. (New York, NY). The dimensions of the inner and outer grids were 14 × 14 × 14 Å and 60 × 60 × 60 Å. Two levels of precision were used: standard and extra. Details of the docking protocol using the Glide program can be found in the literature (Hernández-Campos et al., 2010; F. H. Lopez-Vallejo, T. Caulfield, J. L. Medina-Franco, A. Nefzi, R. Houghten, and K. Martinez-Mayorga, in preparation). Figures 10 to 12 were generated with Maestro 9.1 and Tachyon in the VMD (Visual Molecular Dynamics) program (Humphrey et al., 1996).
Fig. 10.

Binding poses of inhibitors modeled into the PC1/3 active site. A, molecule 166811 is shown in licorice; only polar hydrogen bonds are displayed. The molecular surface of PC1/3 binding site is colored by electrostatic potential. B, overlay of docking poses obtained for four PC1/3 inhibitors.
Fig. 12.

Binding poses of inhibitors modeled into the PC2 active site and allosteric sites (best inhibitors). The arrow shows the entrance of the S1 pocket. A and B have almost the same orientation, showing the active site where compound 166830 is positioned (A) and the potential allosteric site 1 for the binding of compound 166829 (B), which is on the opposite side of the P4 (S4) subsite. C, binding poses of compound 166369 in allosteric site 3, which approaches the active site from the P4 (S4) subsite.
Results
PC1/3 and PC2 Screening.
The amino acid homologies between the catalytic domains of rat, human, and mouse PC1/3, PC2, and furin lie within a range of 51 to 68% conservation (for review, see Cameron et al., 2001). We have previously reported on a series of synthetic small molecules derived from 2,5-dideoxystreptamine as potent inhibitors of furin (Jiao et al., 2006b); because of the reasonably good conservation of the catalytic domain between furin and PC1/3 and PC2, we hypothesized that related small molecules having the proper spatial distribution of positively charged and hydrophobic groups might also represent potent inhibitors of PC1/3 and PC2. Our first goal was to identify molecules able to inhibit PC1/3 and PC2: to accomplish this, we first screened 45 derivatives of 2,5-dideoxystreptamine in a 96-well microplate format, as summarized in Fig. 1, which shows the percentage of inhibition. Four compounds, 166811, 166812, 166550, and 166631, were found to exhibit inhibitory activity against PC1/3 between 40 and 75% at a 25 μM final concentration, whereas compound 166369 stimulated PC1/3 activity by up to 50% (as shown by negative inhibition; Fig. 1A). For PC2, we identified three other compounds, 166369, 166829 and 166830, that inhibited PC2 by 50, 92, and 83%, respectively, at a final concentration of 10 μM. It is noteworthy that compounds 166691 and 166646 stimulated PC2 activity, reported as negative inhibition (Fig. 1B).
Fig. 1.

2,5-Dideoxystreptamine derivatives screening against PC1/3 (A) and PC2 (B). The compounds were tested at final concentrations of 10, 25, and 50 μM; in this figure, we show the screening experiment performed at 25 μM for PC1/3 and at 10 μM for PC2. The percentage of inhibition was calculated from the equation (1 − Vi/V0) × 100, where Vi and Vo are the enzyme rates (fluorescence units per minute) in the presence and in the absence of inhibitors, respectively. The percentage of inhibition is expressed as the mean ± S.D. and was determined in triplicate.
Kinetics of Inhibition for PC1/3 and PC2.
Affinities for all of the inhibitors shown in Fig. 2 were initially characterized by determining IC50 values (Motulsky and Christopoulos, 2004). Compounds 166811, 166812, 166550, and 166631 inhibited PC1/3 with IC50 values of 5, 20, 33, and 22 μM, respectively, whereas compounds 166829, 166830, and 166369 yielded IC50 values of 4, 2, and 8 μM, respectively, against PC2 (Fig. 2). Structures of these compounds are also shown in Fig. 2.
Fig. 2.

Structures of the most active dideoxystreptamine compounds. The structures of the most potent PC inhibitors; the relative 50% inhibitory concentration (IC50) values, determined using inhibitor concentrations ranging between 0 and 500 μM, are shown under each compound (mean ± SD). A, PC1/3; B, PC2.
To obtain information on the mode of action of the best inhibitors, Lineweaver-Burk plots were generated, as shown in Figs. 3 and 4. With regard to PC1/3 inhibition, these data suggest that all four compounds are competitive inhibitors, with Ki values of 0.5 ± 0.04 μM for 166811, 1.5 μM ± 0.1 for 166812, 2.7 ± 0.2 μM for 166550, and 2.0 ± 0.2 μM for 166631, respectively (Fig. 3). On the other hand, Lineweaver-Burk plots yielded a different type of inhibitory mechanism against PC2: 166830 exhibited competitive inhibition with a Ki of 2.0 ± 0.2 μM; 166829 behaved as a noncompetitive inhibitor with a Ki of 11 ± 0.1 μM; and 166369 showed a mixed-type inhibition with a Ki of 7.0 ± 2 μM (Fig. 4).
Fig. 3.

Inhibition kinetics for the most potent PC1/3 inhibitors. Lineweaver-Burk plot shows competitive inhibition for compounds 166811 (A), 166812 (B), 166550 (C), and 166631 (D) against PC1/3. The experiment was performed using 0 (●), 5 (■), 10 (▴), and 50 μM (▾) concentrations of inhibitors, and was carried out in duplicate.
Fig. 4.

Lineweaver-Burk plots for the most potent PC2 inhibitors: 166829 (A), 166830 (B), and 166369 (C). Kinetic assays were performed in duplicate using the following concentrations: 0 (●), 5 (■), 10 (▴), or 50 μM (▾).
Specificity of Inhibitors.
To explore the specificity of the dideoxystreptamine derivatives for PC1/3 and PC2, the most potent compounds were counter-screened against other convertases. The four best inhibitors against PC1/3 showed IC50 values ranging between 5 and 33 μM, whereas these values were greater than 500 μM when tested against PC2, indicating good discrimination (Table 1). However, these same compounds were also excellent furin inhibitors, with IC50 values against furin ranging between 0.65 and 5 μM. On the other hand, the three best PC2-inhibiting compounds, 166829, 166830, and 166369, inhibited neither furin nor PC1/3; dose-response assays yielded IC50 values above 50 μM, as shown in Table 1.
TABLE 1.
Cross-inhibition of furin, PC1/3, and PC2 by the best 2,5-dideoxystreptamine derivatives
| Compounds | IC50 |
|||||
|---|---|---|---|---|---|---|
| Furin | PC1/3 | PC2 | ||||
| μM | ||||||
| 166631 | 2 | [R2 = 0.96] | 22 | [R2 = 0.99] | >500 | |
| 166550 | 5 | [R2 = 0.99] | 33 | [R2 = 0.99] | >500 | |
| 166811 | 1.5 | [R2 = 0.98] | 5 | [R2 = 0.98] | >500 | |
| 166812 | 0.65 | [R2 = 0.98] | 20 | [R2 = 0.97] | >500 | |
| 166829 | >50 | >50 | [R2 = 0.76] | 4 | [R2 = 0.97] | |
| 166830 | >50 | >50 | [R2 = 0.84] | 2 | [R2 = 0.99] | |
| 166369 | >50 | >50 | [R2 = 0.99] | 4.5 | [R2 = 0.98] | |
87- versus 66-kDa Form of PC1/3.
At low concentrations, compound 166369 stimulated 87-kDa PC1/3, but at high concentrations, inhibition was observed (Fig. 5A). To better understand the mechanism of such stimulation, we tested the same compound against the 66-kDa form of PC1/3, which is formed in the acidic environment of secretory granules within neuroendocrine cells, and differs from the 87-kDa form by the absence of 21 kDa at the C terminus (for review, see Cameron et al., 2001). The bell-shaped curve for the 166369 reaction with 87-kDa PC1/3 showed that below 20 μM, this compound activated fluorogenic substrate cleavage; inhibition occurred between 20 μM and 1 mM. However, this same compound had a purely inhibitory effect on 66-kDa PC1/3, with an IC50 of 50 μM (Fig. 5A). Thus, the presence of the C-terminal domain profoundly affects the interaction of 166369 with PC1/3. Dose-response plots of the reaction of this same compound, 166369, with PC2 showed pure inhibition, with an IC50 of 8 μM (Fig. 5B).
Fig. 5.

Dose-response curves for 166369 against various forms of PC1/3. Assays were performed in triplicate using the compound 166369 in the concentration range 0 to 500 μM against the 87-kDa (●) and 66-kDa (○) forms of PC1/3 (A) and against PC2 (B).
PC2 Stimulators.
We found that two compounds, 166691 and 166646, were able to stimulate PC2 activity, as shown in Fig. 6. The structures of these two compounds are characterized by the presence of six aryl groups in 166691 and three in 166646 (Fig. 6A) on the dideoxystreptamine scaffold. The bell-shaped curve for the reaction of 166691 with PC2 indicates that at concentrations below 50 μM, the rate of pERTKR-AMC cleavage increased up to 200%, whereas at concentrations greater than 50 μM, this compound behaved as an inhibitor (Fig. 6B, 1). Likewise, compound 166646 also activated PC2-mediated cleavage of fluorogenic substrate at concentrations lower than 50 μM (Fig. 6B, 2).
Fig. 6.

Structures and dose-response curves of compounds 166691 and 166646 against PC2. The structures show the various aryl groups in different positions on the dideoxystreptamine scaffold (A). For compound 166691, the assay was performed in the concentration range 0 to 500 μM (B, 1), whereas for 166646, the dose-response curve was carried out using concentrations between 0 and 100 μM (B, 2).
Proglucagon Cleavage.
The strongly inhibitory compounds identified above were further evaluated for their efficacy in inhibiting PC1/3- and PC2-dependent processing of the physiologically relevant substrate proglucagon. The four best inhibitors of PC1/3 (166812, 166811, 166550, and 166631) were preincubated with this enzyme at different concentrations; proglucagon was then added, and incubation continued. Figure 7A shows that all four compounds inhibited proglucagon cleavage, as reflected by the strong maintenance of the 20-kDa proglucagon band, which was nearly absent in the control incubation performed in the absence of inhibitors. Although these compounds exhibited varying degrees of efficacy, the order of potency is in agreement with results obtained with the fluorogenic substrate; however, concentrations higher than 25 μM were required to block proglucagon processing.
Fig. 7.

Inhibition of proglucagon processing in vitro- proglucagon gel assay. A, proglucagon (PG) processing by PC1/3 in the presence and absence of the best inhibitors. B, proglucagon processing by PC2 in the presence and absence of the best inhibitors. Note that only early proglucagon cleavages are detected by this method.
In contrast, the three best PC2 inhibitors (166830, 166829, and 166369) blocked proglucagon processing with similar degrees of efficacy, as defined by maintenance of the precursor band and the disappearance of products below the 6-kDa marker. All three compounds completely inhibited PC2-mediated cleavage of proglucagon at concentrations above 10 μM (Fig. 7B).
The inhibition of PC1/3 and PC2 cleavage of proglucagon in gel assays required much higher inhibitor concentrations than inhibition of hydrolysis of the synthetic substrate pERTKR-AMC. These differences are likely to be due to the varying ratios of enzyme to substrate used in each kind of assay. Differences in substrate preference, and numbers of cleavage sites in each substrate, may also play a role.
Glucagon Radioimmunoassay of Proglucagon Cleavage In Vitro.
SDS-polyacrylamide gel electrophoresis is incapable of resolving the known PC2 product glucagon because of its small size; therefore, we analyzed inhibition of PC2-mediated proglucagon processing using a highly specific glucagon radioimmunoassay after carboxypeptidase E digestion to remove terminal basic residues (Fig. 8). Whereas the best PC2 inhibitors seemed to have similar degrees of efficacy against proglucagon when analyzed by SDS-polyacrylamide gel electrophoresis, the glucagon radioimmunoassay revealed differences. Compounds 166829 and 166369 inhibited immunoreactive glucagon production by 40 and 84%, respectively, at concentrations of 5 μM, whereas compound 166830 at the same concentration inhibited glucagon formation by 93%. These differences agree with results obtained for fluorogenic substrate cleavage, confirming that the ranking of the best PC2 inhibitors is 166830 > 166369 > 166829 for both proglucagon cleavages to immunoreactive glucagon and for pERTKR-AMC hydrolysis. We used the same assay to compare the pyrrolidine bis-piperazine 1435-6 compound, previously identified by our group as a potent PC2 inhibitor (Kowalska et al., 2009), with the present compounds. We found that the potency of compound 1435-6 in blocking PC2-mediated generation of immunoreactive glucagon from recombinant proglucagon was much weaker than that of 166830, 166369, and 166829 (Fig. 8). Thus, the dideoxystreptamine derivatives reported here represent improved inhibitors.
Fig. 8.

Inhibition of proglucagon processing in vitro glucagon radioimmunoassay. Effect of 2,5-dideoxystreptamine derivatives (166829, 166830, and 166369) and the pyrrolidine bis-piperazine 1435-6 on PC2-mediated cleavage of glucagon from proglucagon, as measured by RIA. The experiment was carried out in duplicate using a highly specific glucagon RIA; inhibitor concentrations used were 0.25, 1, 2.5, 5, 10, and 50 μM. The percentage of glucagon production was calculated from the equation (Ci/C0) × 100, where Ci and Co are the concentrations of glucagon obtained in the presence and absence of inhibitors, respectively.
Glucagon Radioimmunoassay in α-TC6 Cells.
α-TC6 pancreatic cells naturally express PC2 and synthesize glucagon. We tested the best PC2 inhibitor found here, 166830, and a previously identified PC2 inhibitor, the pyrrolidine bis-piperazine 1435-6, for their ability to block PC2-mediated synthesis of glucagon within cells. In cultures incubated for 36 h in the presence of the dideoxystreptamine derivative 166830, cellular glucagon concentrations in α-TC6 cell extracts were less than half of those in vehicle-treated wells (p < 0.0001); at the same concentration, the pyrrolidine bis-piperazine 1435-6 reduced glucagon synthesis by only 24% (Fig. 9A). Thus, both in vitro and in a cellular context, the 166830 dideoxystreptamine derivative reported here represents a superior PC2 inhibitor against glucagon synthesis.
Fig. 9.

Inhibition of glucagon synthesis by the 2,5-dideoxystreptamine derivative 166830 and the pyrrolidine bis-piperazine 1435-6 in α-TC6 cells. A, glucagon RIA. α-TC6 cultures were incubated with compounds 166830 and 1435-6 at a final concentration of 75 μM for 36 h. Cell extracts were then collected for total glucagon determination by RIA. Data represent the mean ± S.D. from quadruplicate wells from a representative experiment. *, significantly less (p < 0.0001) than the values obtained from control cultures incubated with the vehicle, Opti-MEM. B, cytotoxicity assay. In a parallel experiment, the WST-1 assay was used to determine the viability of α-TC6 cells after treatment with the inhibitors used above at the same final concentrations. The experiment was carried out using quadruplicate wells, and the data represent the mean ± S.D.
Determination of Potential Cytotoxicity of PC2 Inhibitors on α-TC6 Cells.
The potential cytotoxicity of the two compounds was monitored in parallel quadruplicate wells using the water-soluble tetrazolium salt WST-1. After 36 h of incubation with each compound, the WST-1 reagent was added and the plates incubated at 37°C. At times 0, 30, 60, 90 and 180 min, the absorbance at 450 nm was monitored, reflecting the formation of formazan product, which is directly proportional to the number of living cells present. As shown in Fig. 9B, neither compound exhibited any cytotoxic effects compared with Opti-MEM vehicle. In a separate experiment, viability tests were performed in triplicate using the LIVE/DEAD Viability/Cytotoxicity Kit (Invitrogen), which discriminates live from dead cells by simultaneous staining with green-fluorescent calcein acetoxymethyl ester (indicative of intracellular esterase activity) and red-fluorescent ethidium homodimer-1 (indicative of loss of plasma membrane integrity). This experiment confirmed the lack of cytotoxicity of the 166380 inhibitor on α-TC6 cells at 100 μM final concentration (data not shown).
Modeling of PC1/3 and PC2.
Automated docking was performed for seven inhibitors. For PC1/3, the four competitive inhibitors were modeled into the active site. For PC2, binding of the three best inhibitors was examined at the active site as well as in putative allosteric binding sites.
Docking of PC1/3 Inhibitors.
The PC1/3 inhibitors examined in this work possess similar potency and share a common structural scaffold; not surprisingly, similar docking poses and docking scores were also obtained. The maximum docking score difference was less than 3 kcal/mol. The binding site residues within 3 Å of each inhibitor tested, obtained from our models, are listed in Table 2; residues within 2 Å are in bold. All inhibitors modeled were near the catalytic Ser267, except 166550. However, compound 166550 is positioned close to Glu90; this interaction is absent with the other inhibitors.
TABLE 2.
Residues within the PC1/3 binding pocket predicted to be in contact with the most potent PC1/3 inhibitors
Residues within 2 Å are shown in bold.
| 166631 | His93, Leu126, Val130, Glu135, Ser152, Trp153, Gly154, Pro155, Asn156, Asp157, Glu163, Gly164, Ala191, Ser192, Gly193, Asn194, Asp205, Tyr207, Thr208, Ser267 |
| 166550 | Glu90, Leu126, Gly128, Val130, Glu135, Trp153, Gly154, Pro155, Asn156, Asp157, Glu163, Gly164, Ala191, Gly193, Asn194, Asp205, Tyr207 |
| 166811 | His93, Leu126, Val130, Glu135, Ser152, Trp153, Gly154, Pro155, Asn156, Asp157, Glu163, Gly164, Ala191, Gly193, Asn194, Asp205, Tyr207, Ser267 |
| 166812 | His93, Leu126, Val130, Glu135, Ser152, Trp153, Gly154, Pro155, Asn156, Asp157, Glu163, Gly164, Ala191, Ser192, Gly193, Asn194, Asp205, Tyr207, Thr208, Ser267 |
The orientation of compound 166811 in the active site of PC1/3 is shown in Fig. 10A; three guanidinyl groups from the inhibitor interact with the protein. From one side of the inhibitor, a guanidinyl group at the para-position of the naphthyl group interacts with Asp205 and Asp157. The para-guanidinyl group on the phenyl group makes contacts with Glu135 and Tyr207. In addition, one guanidinyl group attached to the cyclohexyl ring orients toward Asn156. These interactions were common to all modeled inhibitors. Figure 10 also shows the possibility of increasing PC1/3 binding affinity by the incorporation of negatively charged substituents in the naphthyl group to interact with the imidazole group of His93 and the amino group of Asn194. Overlay of the binding poses obtained for these inhibitors is shown in Fig. 10B. The cyclohexyl ring overlays the center of the active site and orients the side chains in the PC1/3 cavity. It is noticeable that one of the para-guanidinylated phenyl groups in compound 166550 orients toward Glu90, which may act as a surrogate for the Ser267 interaction found in other inhibitors. To reach Glu90, compound 166550 is slightly displaced compared with the other PC1/3 inhibitors. Finally, the second guanidinyl group attached to the cyclohexyl ring orients toward the solvent and, according to this model, does not interact with any residue in the PC1/3 binding pocket.
Identification of PC2 Allosteric Binding Sites.
A consensus set of six possible allosteric binding sites were obtained; three were located in the outer periphery of the protein and were discarded. Of the remaining three, allosteric sites 1 and 3 provided the best docking scores, as described below. Figure 11 shows the homology model of PC2. In this representation, the catalytic domain, the P domain, the active binding site, and the most relevant allosteric sites suggested by our analysis are labeled. Spheres are used to represent the central locations of the putative allosteric binding sites (brown/blue spheres) and the active site (red sphere). It is noteworthy that allosteric site 1 (shown as a blue sphere) and the active site are positioned close to one another. This close proximity would permit inhibitors to possibly occupy both sites. All modeled compounds were docked at the two most relevant allosteric binding sites (1 and 3) and the active binding site (Fig. 12). The resulting docking scores for the active site and allosteric sites are summarized in Table 3. The difference in docking scores between the allosteric sites and the active site for the competitive inhibitor 166830 measured less than 1 kcal/mol. The predicted binding affinity of compound 166829 was greater at allosteric site 3. From our docking models, inhibitor 166369 exhibited approximately equal preference for the active site and the allosteric site. Furthermore, allosteric site 3 is positioned between the two domains (P and catalytic); this allosteric site may possibly disturb domain association, which could interrupt enzymatic function. Residues within 3 Å of each inhibitor at putative binding sites are listed in Table 4; residues within 2 Å are in bold.
Fig. 11.

Molecular representation of PC2. Overall view. Spheres represent the locations of binding sites: red, active site; orange, allosteric binding sites; and blue, a key allosteric binding site.
TABLE 3.
Docking scores for PC2 inhibitors at different binding sites.
| Compound | Active site | Allosteric Site 1 | Allosteric Site 3 | Type of Inhibition | Ki |
|---|---|---|---|---|---|
| kcal/mol | μM | ||||
| 166830 | −5.9 | −6.7 | −6.6 | Competitive | 2.1 ± 0.2 |
| 166829 | −5.3 | −6.3 | −9.0 | Noncompetitive | 11.7 ± 1.1 |
| 166369 | −9.0 | −9.8 | −8.9 | Mixed | 7.3 ± 2.3 |
TABLE 4.
Residues present at different binding sites predicted to be in contact with PC2 inhibitors
Residues within 2 Å are shown in bold.
| Active Site | Allosteric Site 1 | Allosteric Site 3 | |
|---|---|---|---|
| 166830 | Phe93 His96 Leu129 Asp130 Gln131 Pro132 Phe133 Thr135 Glu139 Trp157 Gly158 Asp161 Asp167 Gly168 Arg170 Asp203 Tyr210 Glu233 |
Asp55 His96 Arg99 Leu129 Ser156 Trp157 Gly158 Pro159 Asp161 Trp194 Ala195 Ser196 Gly197 Asp198 Asp208 Ala211 Tyr231 Arg267 Ser269 Gly270 Ser272 |
|
| 166829 | Gly168 Pro169 Leu174 Ala178 Gly209 Ser212 Ser213 Met214 Trp215 Arg353 His355 Gln396 Lys439 Trp440 Pro441 His480 Met478 |
||
| 166369 | Asp55 Asp56 Phe93 Asn94 His96 Leu129 Phe133 Thr135 Glu139 Trp157 Gly158 Pro159 Thy160 Asp161 Asp167 Arg170 Trp194 Ala195 Gly197 Asp198 Asp203 Asp208 Glu233 Ser272 |
Asp55 Asp91 Trp92 Phe93 Ser95 His96 Arg99 Ser156 Asp198 Tyr231 Asp259 Leu260 Tyr261 Asn263 His268 Ser269 Ser272 |
Discussion
The multiple roles for prohormone convertases in human pathophysiology (for review, see Fugère and Day, 2005) make them prime targets for the development of therapeutic drugs. A variety of PC inhibitors have been described during the past decade; these consist of proteins, peptides, and nonpeptide small molecules (Angliker et al., 1993; Cameron et al., 2000a; Fugère et al., 2002). The use of macromolecule inhibitors such as proteins and larger peptides is hampered by their poor permeability and lesser specificity and stability. By contrast, small-molecule inhibitors exhibit long-lasting metabolic and proteolytic stability, enhanced bioavailability, and easier syntheses (Bogdanovic and Langlands, 2005), making them more attractive as potential therapeutic agents. Indeed, several types of small molecule convertase inhibitors have been reported thus far, for example diterpenes (Basak et al., 1999), heterocyclic compounds (Brinkerhoff et al., 2002; Podsiadlo et al., 2004), and dicoumarol and derivatives (Komiyama et al., 2009). However, the most potent small molecule convertase inhibitors described to date are inhibitors based on the 2,5-dideoxystreptamine scaffold, which exhibit nanomolar Ki values against furin (Jiao et al., 2006b).
It has been much more difficult to identify potent and stable small molecule inhibitors of PC1/3 and PC2 than of furin. Although inhibitors with Ki values in the nanomolar range have previously been described for PC1/3, these are either endogenous inhibitory peptides/propeptides (Boudreault et al., 1998a; Cameron et al., 2000b; Qian et al., 2000), substrate analog peptides (Becker et al., 2010), or amidated hexapeptides (Apletalina et al., 1998). To date, the only nonpeptide PC1/3 inhibitors described are certain andrographolide derivatives, which only weakly inhibit PC1/3 (Basak et al., 1999). PC2 is known to have a potent natural inhibitor in the form of the 7B2 carboxyl-terminal peptide (Apletalina et al., 2000a); it is also inhibited by the cystatin-related epididymal spermatogenic protein (Cornwall et al., 2003). The only previous report of small molecule PC2 inhibitors are certain pyrrolidine bis-piperazines and bicyclic guanidines, which inhibit this enzyme with micromolar potency (Kowalska et al., 2009).
In the work described here, we screened 45 compounds obtained by derivatization of the 2,5-dideoxystreptamine scaffold with guanidinyl and aryl substitutions in various positions. We show that 2,5-dideoxystreptamine derivatives can selectively inhibit PC1/3 and PC2 with inhibition constants in the sub- to low micromolar range, depending on the nature and position of the substituents on the 2,5-dideoxystreptamine core structure. Overall, the binding poses obtained from our docking models suggest common interactions between the various inhibitors with PC1/3. In particular, we identified the spatial locations of three guanidinyl groups that seem to define their potency. These pharmacophore features closely resemble those previously reported for furin (Jiao et al., 2006b), explaining their poor selectivity between PC1/3 and furin. Our models also lead us to suggest structural modifications to enhance potency, such as the incorporation of a hydrogen bond acceptor at the ortho or para position of the naphthyl group of compound 166811. We also found that one of the guanidinyl groups directly attached to the cyclohexyl ring does not interact with any residues in the PC1/3 binding pocket. These observations agree well with the binding poses of related compounds previously modeled into the furin active site (Jiao et al., 2006b).
Kinetic studies showed that the hydrolysis of fluorogenic substrate by PC1/3 is competitively inhibited by compounds 166811, 166812, 166550, and 166631, suggesting similar interactions between these molecules and the PC1/3 active site, a conclusion supported by the similar binding poses obtained from docking studies. It is noteworthy that only compound 166830 seems to interact directly with the PC2 active site, whereas the inhibition kinetics of 166829, a noncompetitive inhibitor, and 166369, which exhibits a mixed-type inhibition, imply the involvement of allosteric sites. Indeed, our molecular modeling and docking experiments suggest the possible presence of three such allosteric sites. Experimental confirmation of these allosteric sites will allow the development of structure-activity relationships and increase our understanding of PC regulation.
A comparison of the mechanisms and specificity of inhibition of PC1/3, PC2, and furin by the various 2,5-dideoxystreptamine derivatives revealed striking differences. Whereas PC2 was inhibited only by compounds 166829, 166830, and 166369, with different mechanisms that in some cases suggest allostery, PC1/3 and furin were both competitively inhibited—albeit to different extents—by compounds 166811, 166812, 166550, and 166631. The similar inhibitor specificity of furin and PC1/3 agrees with the observation that these two convertases are much more closely related to each other than they are to PC2 (Oliva et al., 2000). This relative selectivity showing good differentiation of PC1/3 from PC2 inhibitors is promising for the development of PC2-specific inhibitors. Although the differentiation of PC1/3 inhibitors from furin inhibitors continues to represent a difficult problem, as also reported by other groups (Jiao et al., 2006b; Komiyama et al., 2009; Becker et al., 2010), it is possible that lesser selectivity could represent an advantage under certain circumstances, if it is necessary for effective inhibition of several PCs with redundant functions.
Glucagon radioimmunoassays revealed that a 2.5 μM concentration of the best PC2 dideoxystreptamine inhibitor, compound 166830, was able to inhibit immunoreactive glucagon production from proglucagon (a late PC2-mediated processing step) by more than 50%; our previous best PC2 inhibitor, the pyrrolidine bis-piperazine 1435-6, did not achieve this level of inhibition even at 50 μM. We were excited to find that compound 166830 was also able to block PC2 activity in cell culture, as assessed by glucagon radioimmunoassay. This is the first demonstration of prohormone convertase inhibition by a small-molecule inhibitor within cells and clearly shows that glucagon synthesis can be pharmacologically inhibited in this pancreatic glucagon-synthesizing cell line. These results make this compound an attractive target for further chemical modification to increase inhibitory potency, which might be therapeutically useful in lowering blood sugar in diabetics.
It is interesting to note that certain compounds, such as 166691 and 166646, stimulated PC2 activity by up to 50%. Structural examination of these compounds provides clues as to underlying reasons for inhibition and stimulation. The presence of multiple aryl groups, such as those present on compounds 166691 and 166646, and probably their spatial distribution on the scaffold, seem to be associated with stimulation. We have previously reported that l-polyarginines (Cameron et al., 2000a) and bicyclic guanidines are also able to activate PC2 at very low concentrations, though pyrrolidine bis-piperazine-based inhibitors do not (Kowalska et al., 2009). We speculate that bicyclic guanidines and the aryl-derivatized dideoxystreptamine compounds described here may allosterically bind an exosite, effecting a conformational change that enhances enzyme activity. One such exosite near the PC2-specific P4 canopy sequence is known to contribute to binding of the 7B2 CT peptide, a tight-binding, PC2-specific inhibitor (Benjannet et al., 1998; Zhu et al., 1998; Apletalina et al., 2000b). However, experimental testing by mutagenesis will be required to confirm direct binding of these molecules to predicted allosteric sites. PC2 stimulation represents a biochemical effect that might have eventual therapeutic relevance in the management of acute and chronic pain, for example by increasing the production of the PC2-synthesized opioid peptides β-endorphin (for review, see Mains and Eipper, 2000) and Met-and Leu- enkephalin (Peinado et al., 2003).
We were surprised that compound 166369, which inhibits PC2 with a mixed-type mechanism at high concentrations, was found to stimulate the 87-kDa form of PC1/3 at low concentrations. It is noteworthy that this compound cannot stimulate 66-kDa PC1/3 at any concentration but instead behaves solely as an inhibitor. The difference between these two forms of PC1/3 consists of the presence of an additional 21-kDa carboxyl-terminal domain in the 87-kDa form; three groups have shown that 66-kDa PC1/3 is much more active than the 87-kDa form (Rufaut et al., 1993; Zhou and Lindberg, 1994; Boudreault et al., 1998b; Rabah et al., 2007). The carboxyl-terminal tail has been proposed as an inhibitor of PC1/3 (Jutras et al., 1997), although others have found bimodal effects (Rabah et al., 2007). We speculate that compound 166369 might bind directly to this carboxyl-terminal domain and that this would then result in enhanced catalytic efficiency of the active site, possibly mimicking the more active conformation of 66-kDa PC1/3. Once this higher affinity allosteric stimulatory site is saturated, compound 166369 could then bind directly to the active site, acting as an inhibitor, thus explaining the bimodal concentration curve. Again, further experiments will be required to demonstrate the precise mechanism and sites at which stimulators bind; however, because we lack a crystal structure for the carboxyl-terminal domain, no modeling is yet possible.
PC1/3 is the major enzyme involved in proinsulin processing; therefore, a drug that effectively stimulated PC1/3 could represent a valuable tool for increasing levels of endogenous insulin (in, for example, certain forms of diabetes in which the insulin precursor is known to be up-regulated) (Pfützner et al., 2004). If the same drug, for example compound 166369, can be shown to block proglucagon processing to glucagon, this would provide a double benefit, because glucagon acts in functional opposition to insulin, and glucagon antagonists are highly sought for purposes of glycemic control [indeed, elevated levels of glucagon seem to contribute to the progression of type 2 diabetes mellitus (Quesada et al., 2008)].
Acknowledgments
We are grateful to R. B. Mackin for providing the proglucagon-encoding bacterial vector and M. Helwig for initial help with the radioimmunoassays. We thank Pacific Telehealth and Technology (Hui, Honolulu, HI) for funding administration.
This work was supported in part by a grant from the National Institutes of Health National Institute on Drug Abuse [Grant DA050854] (to I.L.); by the State of Florida Executive Officer of the Governor's Office of Tourism, Trade and Economic Development (to K.M-M.); and by the U.S. Department of Defense, U.S. Army Medical Research and Materiel Command [Contract V549P-6073] (to A.J. and G.-S.J.), [Grant OT07-050] (to K.M.-M.).
G.-S.J. is co-corresponding author regarding the inhibitor chemistry (gjiao@pantherabio.com).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
- PC
- prohormone convertase
- 166369
- 5-(2,4-diguanidino-phenoxy)-2,4-diguanidino-cyclohexyl (4-octylphenyl)carbamate
- 166369a
- N,N′-di-tert-butoxycarbonyl-N″-{4-[2,4-di-(di-tert-butoxycarbonyl-guanidino)-phenoxy]-5-[di-(tert-butoxycarbonyl)-guanidino]-2-hydroxycyclohexyl}-guanidine
- 166550
- N-[2,4-bis(4-guanidino-phenoxy)-5-(4-guanidino-phenylamino)-cyclohexyl]-guanidine
- 166631
- N-[5-guanidino-2,4-bis-(4-guanidino-phenoxy)-cyclohexyl]-guanidine
- 166646
- 5-(2,4-diguanidino-phenoxy)-2,4-diguanidino-cyclohexyl [1,1′-biphenyl]-4-ylcarbamate
- 166691
- N-[5-(4-guanidino-naphthalen-1-ylamino)-2,4-bis-(4-guanidino-naphthalen-1-yloxy)-cyclohexyl]-guanidine
- 166811
- N-[5-guanidino-4-(4-guanidino-naphthalen-1-yloxy)-2-(4-guanidino-phenoxy)-cyclohexyl]-guanidine
- 166812
- N-{2-[2,4-diguanidino-5-(4-guanidino-phenoxy)-cyclohexyloxy]-5-guanidino-phenyl}-guanidine
- 166829
- 5-(2,4-bis(imidazolidin-2-ylideneamino)phenoxy)-2,4-bis(imidazolidin-2-ylideneamino)cyclohexanol
- 166829a
- 2,4-diamino-5-(2,4-diaminophenoxy)cyclohexanol
- 166830
- N1,N3-di(imidazolidin-2-ylidene)-4,6-bis(4 (imidazolidin-2-ylideneamino)phenoxy)cyclohexane-1,3-diamine
- 166830a
- 4,6-bis (4-aminophenoxy)cyclohexane-1,3-diamine
- 1435–6
- (2R)-4-((R)-1-cyclohexyl-3-((S)-2-(((S)-2-(cyclohexylmethyl)piperazin-1-yl)methyl)pyrrolidin-1-yl)propan-2-yl)-2-(cyclohexylmethyl)-1-(2-(4-isobutylphenyl)propyl)piperazine
- TFA
- trifluoroacetic acid
- BSA
- bovine serum albumin
- pERTKR-AMC
- pGlu-Arg-Thr-Lys-Arg-methylcoumarinamide
- PBS
- phosphate-buffered saline
- RIA
- radioimmunoassay.
Authorship Contributions
Participated in research design: Vivoli, Martínez-Mayorga, Johnson, Jiao, and Lindberg.
Conducted experiments: Vivoli and Caulfield.
Performed data analysis: Vivoli and Caulfield.
Wrote or contributed to the writing of the manuscript: Vivoli, Martínez-Mayorga, Jiao, and Lindberg.
References
- Angliker H, Wikstrom P, Shaw E, Brenner C, Fuller RS. (1993) The synthesis of inhibitors for processing proteinases and their action on the Kex2 proteinase of yeast. Biochem J 293:75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apletalina E, Appel J, Lamango NS, Houghten RA, Lindberg I. (1998) Identification of inhibitors of prohormone convertases 1 and 2 using a peptide combinatorial library. J Biol Chem 273:26589–26595 [DOI] [PubMed] [Google Scholar]
- Apletalina EV, Juliano MA, Juliano L, Lindberg I. (2000a) Structure-function analysis of the 7B2 CT peptide. Biochem Biophys Res Commun 267:940–942 [DOI] [PubMed] [Google Scholar]
- Apletalina EV, Muller L, Lindberg I. (2000b) Mutations in the catalytic domain of prohormone convertase 2 result in decreased binding to 7B2 and loss of inhibition with 7B2 C-terminal peptide. J Biol Chem 275:14667–14677 [DOI] [PubMed] [Google Scholar]
- Arnaoutova I, Smith AM, Coates LC, Sharpe JC, Dhanvantari S, Snell CR, Birch NP, Loh YP. (2003) The prohormone processing enzyme PC3 is a lipid raft-associated transmembrane protein. Biochemistry 42:10445–10455 [DOI] [PubMed] [Google Scholar]
- Basak A, Cooper S, Roberge AG, Banik UK, Chrétien M, Seidah NG. (1999) Inhibition of proprotein convertases-1, -7 and furin by diterpines of Andrographis paniculata and their succinoyl esters. Biochem J 338:107–113 [PMC free article] [PubMed] [Google Scholar]
- Basak A, Lotfipour F. (2005) Modulating furin activity with designed mini-PDX peptides: synthesis and in vitro kinetic evaluation. FEBS Lett 579:4813–4821 [DOI] [PubMed] [Google Scholar]
- Becker GL, Sielaff F, Than ME, Lindberg I, Routhier S, Day R, Lu Y, Garten W, Steinmetzer T. (2010) Potent inhibitors of furin and furin-like proprotein convertases containing decarboxylated P1 arginine mimetics. J Med Chem 53:1067–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjannet S, Mamarbachi AM, Hamelin J, Savaria D, Munzer JS, Chrétien M, Seidah NG. (1998) Residues unique to the prohormone convertase PC2 modulate its autoactivation, binding to 7B2 and enzymatic activity. FEBS Lett 428:37–42 [DOI] [PubMed] [Google Scholar]
- Bogdanovic S, Langlands B. (2005) Proteases: Technologies and Opportunities for Drug Discovery, D&MD Publications, Westborough, MA [Google Scholar]
- Bonic A, Mackin RB. (2003) Expression, purification, and PC1-mediated processing of human proglucagon, glicentin, and major proglucagon fragment. Protein Expr Purif 28:15–24 [DOI] [PubMed] [Google Scholar]
- Boudreault A, Gauthier D, Lazure C. (1998a) Proprotein convertase PC1/3-related peptides are potent slow tight- binding inhibitors of murine PC1/3 and Hfurin. J Biol Chem 273:31574–31580 [DOI] [PubMed] [Google Scholar]
- Boudreault A, Gauthier D, Rondeau N, Savaria D, Seidah NG, Chrétien M, Lazure C. (1998b) Molecular characterization, enzymatic analysis, and purification of murine proprotein convertase-1/3 (PC1/PC3) secreted from recombinant baculovirus-infected insect cells. Protein Expr Purif 14:353–366 [DOI] [PubMed] [Google Scholar]
- Brinkerhoff CJ, Podsiadlo P, Komiyama T, Fuller RS, Blum O. (2002) Protease inhibitors formed in situ from copper and tridentate chelates: a generalized approach towards metal-based pharmaceuticals. Chembiochem 3:1141–1143 [DOI] [PubMed] [Google Scholar]
- Brylinski M, Skolnick J. (2008) A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc Natl Acad Sci USA 105:129–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron A, Apletalina EV, Lindberg I. (2001) The enzymology of PC1 and PC2, in The Enzymes (Dalbey RE. ed) pp 291–331, Academic Press, San Diego [Google Scholar]
- Cameron A, Appel J, Houghten RA, Lindberg I. (2000a) Polyarginines are potent furin inhibitors. J Biol Chem 275:36741–36749 [DOI] [PubMed] [Google Scholar]
- Cameron A, Fortenberry Y, Lindberg I. (2000b) The SAAS granin exhibits structural and functional homology to 7B2 and contains a highly potent hexapeptide inhibitor of PC1. FEBS Lett 473:135–138 [DOI] [PubMed] [Google Scholar]
- Copeland RA. (2005) Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, Wiley & Sons-Interscience Hoboken, New Jersey: [PubMed] [Google Scholar]
- Cornwall GA, Cameron A, Lindberg I, Hardy DM, Cormier N, Hsia N. (2003) The cystatin-related epididymal spermatogenic protein inhibits the serine protease prohormone convertase 2. Endocrinology 144:901–908 [DOI] [PubMed] [Google Scholar]
- Dahlen JR, Jean F, Thomas G, Foster DC, Kisiel W. (1998) Inhibition of soluble recombinant furin by human proteinase inhibitor 8. J Biol Chem 273:1851–1854 [DOI] [PubMed] [Google Scholar]
- Day R, Schafer MK, Watson SJ, Chrétien M, Seidah NG. (1992) Distribution and regulation of the prohormone convertases PC1 and PC2 in the rat pituitary. Mol Endocrinol 6:485–497 [DOI] [PubMed] [Google Scholar]
- Decroly E, Vandenbranden M, Ruysschaert JM, Cogniaux J, Jacob GS, Howard SC, Marshall G, Kompelli A, Basak A, Jean F. (1994) e The convertases furin and PC1 can both cleave the human immunodeficiency virus (HIV)-1 envelope glycoprotein gp160 into gp120 (HIV-1 SU) and gp41 (HIV-I TM). J Biol Chem 269:12240–12247 [PubMed] [Google Scholar]
- Dufour EK, Denault JB, Bissonnette L, Hopkins PC, Lavigne P, Leduc R. (2001) The contribution of arginine residues within the P6–P1 region of alpha 1-antitrypsin to its reaction with furin. J Biol Chem 276:38971–38979 [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Volders K, Stanhope R, Heuschkel R, White A, Lank E, Keogh J, O'Rahilly S, Creemers JW. (2007) Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J Clin Endocrinol Metab 92:3369–3373 [DOI] [PubMed] [Google Scholar]
- Fugère M, Day R. (2005) Cutting back on pro-protein convertases: the latest approaches to pharmacological inhibition. Trends Pharmacol Sci 26:294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugère M, Limperis PC, Beaulieu-Audy V, Gagnon F, Lavigne P, Klarskov K, Leduc R, Day R. (2002) Inhibitory potency and specificity of subtilase-like pro-protein convertase (SPC) prodomains. J Biol Chem 277:7648–7656 [DOI] [PubMed] [Google Scholar]
- Furuta M, Yano H, Zhou A, Rouillé Y, Holst JJ, Carroll R, Ravazzola M, Orci L, Furuta H, Steiner DF. (1997) Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc Natl Acad Sci USA 94:6646–6651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heni M, Haupt A, Schäfer SA, Ketterer C, Thamer C, Machicao F, Stefan N, Staiger H, Häring HU, Fritsche A. (2010) Association of obesity risk SNPs in PCSK1 with insulin sensitivity and proinsulin conversion. BMC Med Genet 11:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrich S, Cameron A, Bourenkov GP, Kiefersauer R, Huber R, Lindberg I, Bode W, Than ME. (2003) The crystal structure of the proprotein processing proteinase furin explains its stringent specificity. Nat Struct Biol 10:520–526 [DOI] [PubMed] [Google Scholar]
- Henrich S, Lindberg I, Bode W, Than ME. (2005) Proprotein convertase models based on the crystal structures of furin and kexin: explanation of their specificity. J Mol Biol 345:211–227 [DOI] [PubMed] [Google Scholar]
- Hernández-Campos A, Velázquez-Martínez I, Castillo R, López-Vallejo F, Jia P, Yu Y, Giulianotti MA, Medina-Franco JL. (2010) Docking of protein kinase B inhibitors: implications in the structure-based optimization of a novel scaffold. Chem Biol Drug Des 76:269–276 [DOI] [PubMed] [Google Scholar]
- Hoshino A, Kowalska D, Jean F, Lazure C, Lindberg I. (2011) Modulation of PC1/3 activity by self-interaction and substrate binding. Endocrinology 152:1402–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38 [DOI] [PubMed] [Google Scholar]
- Jiao GS, Cregar L, Goldman ME, Millis SZ, Tang C. (2006a) Guanidinylated 2,5-dideoxystreptamine derivatives as anthrax lethal factor inhibitors. Bioorg Med Chem Lett 16:1527–1531 [DOI] [PubMed] [Google Scholar]
- Jiao GS, Cregar L, Wang J, Millis SZ, Tang C, O'Malley S, Johnson AT, Sareth S, Larson J, Thomas G. (2006b) Synthetic small molecule furin inhibitors derived from 2,5-dideoxystreptamine. Proc Natl Acad Sci USA 103:19707–19712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras I, Seidah NG, Reudelhuber TL, Brechler V. (1997) Two activation states of the prohormone convertase PC1 in the secretory pathway. J Biol Chem 272:15184–15188 [DOI] [PubMed] [Google Scholar]
- Komiyama T, Coppola JM, Larsen MJ, van Dort ME, Ross BD, Day R, Rehemtulla A, Fuller RS. (2009) Inhibition of furin/proprotein convertase-catalyzed surface and intracellular processing by small molecules. J Biol Chem 284:15729–15738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama T, VanderLugt B, Fugère M, Day R, Kaufman RJ, Fuller RS. (2003) Optimization of protease-inhibitor interactions by randomizing adventitious contacts. Proc Natl Acad Sci USA 100:8205–8210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska D, Liu J, Appel JR, Ozawa A, Nefzi A, Mackin RB, Houghten RA, Lindberg I. (2009) Synthetic small-molecule prohormone convertase 2 inhibitors. Mol Pharmacol 75:617–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd DJ, Bohan S, Gekakis N. (2006) Obesity, hyperphagia and increased metabolic efficiency in Pc1 mutant mice. Hum Mol Genet 15:1884–1893 [DOI] [PubMed] [Google Scholar]
- Mains RE, Eipper BA. (2000) Proopiomelanocortin synthesis and cell-specific processing, in Handbook of Physiology, Section 7: The Endocrine System, Volume IV: Coping with the Environment: Neural and Endocrine Mechanisms, pp 85–101, Oxford University Press, London [Google Scholar]
- Motulsky HJ, Christopoulos A. (2004) Fitting Models to Biological Data using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting, Oxford University Press, New York [Google Scholar]
- Oliva AA, Jr, Chan SJ, Steiner DF. (2000) Evolution of the prohormone convertases: identification of a homologue of PC6 in the protochordate amphioxus. Biochim Biophys Acta 1477:338–348 [DOI] [PubMed] [Google Scholar]
- Peinado JR, Li H, Johanning K, Lindberg I. (2003) Cleavage of recombinant proenkephalin and blockade mutants by prohormone convertases 1 and 2: an in vitro specificity study. J Neurochem 87:868–878 [DOI] [PubMed] [Google Scholar]
- Pfützner A, Kunt T, Hohberg C, Mondok A, Pahler S, Konrad T, Lübben G, Forst T. (2004) Fasting intact proinsulin is a highly specific predictor of insulin resistance in type 2 diabetes. Diabetes Care 27:682–687 [DOI] [PubMed] [Google Scholar]
- Podsiadlo P, Komiyama T, Fuller RS, Blum O. (2004) Furin inhibition by compounds of copper and zinc. J Biol Chem 279:36219–36227 [DOI] [PubMed] [Google Scholar]
- Qian Y, Devi LA, Mzhavia N, Munzer S, Seidah NG, Fricker LD. (2000) The C-terminal region of proSAAS is a potent inhibitor of prohormone convertase 1. J Biol Chem 275:23596–23601 [DOI] [PubMed] [Google Scholar]
- Quesada I, Tudurí E, Ripoll C, Nadal A. (2008) Physiology of the pancreatic alpha-cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol 199:5–19 [DOI] [PubMed] [Google Scholar]
- Rabah N, Gauthier D, Dikeakos JD, Reudelhuber TL, Lazure C. (2007) The C-terminal region of the proprotein convertase 1/3 (PC1/3) exerts a bimodal regulation of the enzyme activity in vitro. FEBS J 274:3482–3491 [DOI] [PubMed] [Google Scholar]
- Rhodes CJ, Thorne BA, Lincoln B, Nielsen E, Hutton JC, Thomas G. (1993) Processing of proopiomelanocortin by insulin secretory granule proinsulin processing endopeptidases. J Biol Chem 268:4267–4275 [PMC free article] [PubMed] [Google Scholar]
- Richer MJ, Keays CA, Waterhouse J, Minhas J, Hashimoto C, Jean F. (2004) The Spn4 gene of Drosophila encodes a potent furin-directed secretory pathway serpin. Proc Natl Acad Sci USA 101:10560–10565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillé Y, Bianchi M, Irminger JC, Halban PA. (1997) Role of prohormone convertase PC2 in the processing of proglucagon to glucagon. FEBS Lett 413:119–123 [DOI] [PubMed] [Google Scholar]
- Rufaut NW, Brennan SO, Hakes DJ, Dixon JE, Birch NP. (1993) Purification and characterization of the candidate prohormone-processing enzyme SPC3 produced in a mouse L cell line. J Biol Chem 268:20291–20298 [PubMed] [Google Scholar]
- Smeekens SP, Montag AG, Thomas G, Albiges-Rizo C, Carroll R, Benig M, Phillips LA, Martin S, Ohagi S, Gardner P. (1992) Proinsulin processing by the subtilisin-related proprotein convertases furin, PC2, and PC3. Proc Natl Acad Sci USA 89:8822–8826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruce MC, Potter J, Coppini DV. (2003) The pathogenesis and management of painful diabetic neuropathy: a review. Diabet Med 20:88–98 [DOI] [PubMed] [Google Scholar]
- Villemure M, Fournier A, Gauthier D, Rabah N, Wilkes BC, Lazure C. (2003) Barley serine proteinase inhibitor 2-derived cyclic peptides as potent and selective inhibitors of convertases PC1/3 and furin. Biochemistry 42:9659–9668 [DOI] [PubMed] [Google Scholar]
- Zhou A, Bloomquist BT, Mains RE. (1993) The prohormone convertases PC1 and PC2 mediate distinct endoproteolytic cleavages in a strict temporal order during proopiomelanocortin biosynthetic processing. J Biol Chem 268:1763–1769 [PubMed] [Google Scholar]
- Zhou Y, Lindberg I. (1994) Enzymatic properties of carboxyl-terminally truncated prohormone convertase 1 (PC1/SPC3) and evidence for autocatalytic conversion. J Biol Chem 269:18408–18413 [PubMed] [Google Scholar]
- Zhu X, Muller L, Mains RE, Lindberg I. (1998) Structural elements of PC2 required for interaction with its helper protein 7B2. J Biol Chem 273:1158–1164 [DOI] [PubMed] [Google Scholar]