

Abstract
Despite the potent proapoptotic effect of several antiepileptic drugs (AEDs) in developmental rodent models, little is known about the long-term impact of exposure during brain development. Clinically, this is of growing concern. To determine the behavioral consequences of such exposure, we examined phenobarbital, phenytoin, and lamotrigine for their effects on adult behaviors after administration to neonatal rats throughout the second postnatal week. AED treatment from postnatal days 7 to 13 resulted in adult deficits in spatial learning in the Morris water maze and decreased social exploration for all drugs tested. Phenobarbital exposure led to deficits in cued fear conditioning, risk assessment in the elevated plus maze, and sensorimotor gating as measured by prepulse inhibition, but it did not affect motor coordination on the rotorod task. In contrast, phenytoin and lamotrigine exposure led to impaired rotorod performance, but no deficits in sensorimotor gating. Phenytoin, but not lamotrigine or phenobarbital, increased exploration in the open field. Phenytoin and phenobarbital, but not lamotrigine, disrupted cued fear conditioning. These results indicate that AED administration during a limited sensitive postnatal period is sufficient to cause a range of behavioral deficits later in life, and the specific profile of behavioral deficits varies across drugs. The differences in the long-term outcomes associated with the three AEDs examined are not predicted by either the mechanism of AED action or the proapoptotic effect of the drugs. Our findings suggest that a history of AED therapy during development must be considered as a variable when assessing later-life cognitive and psychiatric outcomes.
Introduction
Antiepileptic drugs (AEDs) have well established safety profiles in adults, but much less is known about their safety during gestation or infancy. Because the developing brain is highly vulnerable to even transient changes in the molecular environment, AED exposure during sensitive developmental periods may alter brain maturation and adversely affect nervous system function in adulthood. This concern is underscored by accumulating preclinical and clinical evidence that the administration of AEDs during gestation or infancy can lead to later behavioral impairments.
Clinical studies have identified several striking changes after in utero exposure to AEDs, including decreased volume in key brain areas (Ikonomidou et al., 2007), as well as behavioral problems and reduced intelligence quotient (Meador et al., 2011). During the early postnatal period, phenobarbital is the first-line therapy for seizures, with phenytoin serving as a second-line alternative (Bartha et al., 2007). Because the neonatal period has the highest incidence of newly diagnosed seizures compared with any period across the lifespan, drug therapy for these seizures affects an especially large population. One of the few studies to directly examine later effects of early life exposure to phenobarbital (as a treatment for febrile seizures) found a significant association with reduced intelligence quotient (Farwell et al., 1990; Sulzbacher et al., 1999).
Behavioral outcomes caused by AED therapy cannot be revealed by clinical data alone because of confounds resulting from underlying neurological disorders. Furthermore, it is difficult to compare across drugs because clinical therapy often involves drug combinations (Kim et al., 2007a).
Rodent models are therefore ideal for evaluating the effects of exposure to different AEDs in normal subjects during defined developmental periods. AED-associated neurotoxicity during a narrow developmental window (the second postnatal week in the rat) may be sufficient to cause adverse behavioral consequences in adulthood (Forcelli et al., 2010, 2011a). In rodents, both phenobarbital and phenytoin cause neuronal apoptosis when given in therapeutically relevant doses during this specific postnatal period (Bittigau et al., 2002; Katz et al., 2007; Kim et al., 2007a,b; Forcelli et al., 2011b). This toxicity, which is shared with other AEDs (e.g., valproic acid), anesthetic agents (Jevtovic-Todorovic et al., 2003), ethanol (Ikonomidou et al., 2000), and NMDA receptor antagonists (Ikonomidou et al., 1999), is evident throughout the cortex, striatum, thalamus, and limbic system. In addition, striatal synaptic maturation is disrupted after exposure to phenobarbital, phenytoin, or lamotrigine (Forcelli 2011). Behavioral abnormalities have been reported after neonatal treatment with ethanol, anesthetics, and NMDA receptor antagonists.
The long-term effects of exposure to phenobarbital or phenytoin restricted to the second postnatal week have not been examined. Phenobarbital exposure from postnatal day 2 (P2) to P35 caused deficits in Morris water maze learning and open-field exploration in adult rats (Pereira de Vasconcelos et al., 1990; Rogel-Fuchs et al., 1992; Stefovska et al., 2008), whereas phenytoin given from P2 to P4 caused deficits in spontaneous locomotion and rotorod performance in adult mice (Ohmori et al., 1999). Phenobarbital exposure has not been examined for its effects on rotorod performance, and phenytoin exposure has not been examined for water maze learning. Neither drug has been examined for social, emotional, or somatosensory function. Thus, although data accumulated to date are sufficient to suggest that phenobarbital or phenytoin exposure in preweanling rodents has long-lasting adverse consequences for certain behaviors, a more comprehensive analysis of functional deficits after neonatal exposure to these drugs is needed.
Thus, for the present study, we selected a battery of tasks sensitive to changes in motor, cognitive, and emotional function in adult animals. Locomotor and exploratory functions were assessed by using accelerating rotorod and open-field tasks; cognitive functions were assessed by using the Morris water maze and fear-conditioning paradigms; emotionality was assessed by using the elevated plus maze and social behavior tasks; and sensorimotor gating was assessed by prepulse inhibition (PPI) of the acoustic startle response. The latter three assessments have been used to detect abnormalities that reflect aspects of human neuropsychiatric disorders (Sams-Dodd, 1999; Kamath et al., 2008). This is of particular interest because patients with a history of early life seizures exhibit a high incidence of neuropsychiatric symptoms, but it is unclear whether such symptoms are triggered by seizures, AED therapy, and/or an underlying pathology (Vestergaard et al., 2005).
We systematically evaluated and compared adult performance on the above behavioral tasks in rats that had been exposed to phenobarbital or phenytoin during the second postnatal week. For comparison, we included lamotrigine-exposed rats in our study, because unlike phenobarbital or phenytoin, lamotrigine does not induce neuronal apoptosis at therapeutically relevant doses in neonatal rats (Katz et al., 2007).
Materials and Methods
Animals.
Forty six timed pregnant Sprague-Dawley rats were obtained from Harlan (Indianapolis, IN; gestational day 14–19 upon arrival at Georgetown University). Rats were housed in a temperature-controlled (21°C) room with a 12-h light/dark cycle (lights on 6:00 AM). A total of 161 pups were used in the present studies. Upon parturition, pups were left undisturbed with their dam until P7, when female pups were culled, and male pups were weighed and numbered. Pups were treated with AEDs during the second week as described below and maintained with their dam until weaning at P21. Treatments were balanced within and across litters to avoid any litter effects, and litters were obtained across all four seasons, avoiding any seasonal variability in shipping stress. Pups were weaned into mixed-treatment cages of two to three animals for the remainder of the experiments.
Drugs and Treatments.
Sodium phenobarbital (75 mg/kg; Luminal, 5-ethyl-5-phenyl-1,3-diazinane-2,4,6-trione; Sigma-Aldrich, St. Louis, MO) and lamotrigine isethionate [20 mg/kg; Lamictal, 6-(2,3-dichlorophenyl)-1,2,4-triazine-3,5-diamine isethionate, GlaxoSmithKline, Research Triangle Park, NC] were dissolved in saline. Phenytoin (50 mg/kg; sodium diphenylhydantoin; dilantin, 5,5-diphenylimidazolidine-2,4-dione; Sigma-Aldrich) was dissolved in alkalinized saline, pH 9 to 11.
Pups were treated once daily on P7, P8, P9, P11, P12, and P13 for a total of six doses. All drugs were given intraperitoneally in a volume of 0.01 ml/g. Control animals received an equivalent volume of vehicle. Each vehicle was administered to subsets of control animals, and because no differences were detected based on vehicle, vehicle groups were collapsed. Drug doses all fell within the anticonvulsant range in neonatal rats (Kubova and Mares, 1991; Stanková et al., 1992). Furthermore, the doses of phenobarbital and phenytoin selected produce robust cell death in the developing rat brain (Bittigau et al., 2002; Katz et al., 2007; Kim et al., 2007a,b; Forcelli et al., 2011b). As a contrast, the dose of lamotrigine selected, although within the therapeutic range, does not produce cell death in the developing rat brain (Katz et al., 2007).
Behavioral Assays.
Animals were transported from the animal facility to testing rooms, where they were allowed to acclimate for a minimum of 30 min before the onset of behavioral testing. All behavioral testing was conducted and scored while blind to treatment conditions.
Accelerating Rotorod.
The accelerating rotorod is a measure of motor coordination and motor learning (Monville et al., 2006). Animals were placed on the stationary rotorod (Accuscan Instruments, Columbus, OH and IITC Life Science, Woodland Hills, CA) with a 3.2-cm-diameter drum, which accelerated from 0 to 45 rpm over the course of a 5-min test. Latency to fall was automatically recorded. Animals were tested for a total of 10 trials, with a 2-min rest period between tests.
Open Field.
Open field allows for an overall measure of spontaneous motor activity, as well as a measure of exploratory drive (animals with a greater exploratory drive may explore the center of the arena to a greater degree). Animals were placed in a Plexiglas enclosure (40 × 40 × 40 cm; TruScan Arena; Coulbourn Instruments, Allentown, PA) with 770-lux illumination over the center of the arena. Animals were allowed to explore for 20 min, during which total distance traveled, number of entries into the center zone, and time spent in the center zone were recorded by using AnyMaze software (Stoelting Co., Wood Dale, IL).
Elevated Plus Maze.
The elevated plus maze is a standard test for anxiety-like behavior in rodents. It examines the natural exploratory drive of rodents, the relative safety of the closed arms of the maze (dim and with walls), and the fear of open, unenclosed, elevated spaces. The elevated plus maze is a validated assay for anxiety-like behavior and is a commonly used screen for anxiolytic drugs. Plus maze testing was performed and scored as described previously (Forcelli and Heinrichs, 2008) in a standard gray rat elevated plus maze (50-cm arms, elevated 40 cm off the ground; Stoelting Co.). Testing was conducted under 20-lux red light. The number of arm entries and time spent in open and closed arms were recorded by using AnyMaze (Stoelting Co.).
Social Behavior.
Rats are social animals, and, as such, measures of social interaction have established face, construct, and predictive validity (Sams-Dodd, 1999; File et al., 2004). Animals were tested in a three-chamber social behavior paradigm as described previously (Billingslea, 2007). The center (“start”) chamber (15 × 10 cm; Habitest Runway; Coulbourn Instruments) was separated from the other chambers via drop doors. The left and right chambers (37 × 10 cm; Habitest Runway; Coulbourn Instruments) contained a novel conspecific adult male and a novel object, respectively. Access to the novel rat or object was limited to nose pokes through a grid with 2-cm holes, which divided the left and right chambers evenly. This paradigm, similar to that described for mice by Nadler et al. (2004), limits the initiation of social behavior to the test subject, reducing confounds. At the start of the test animals were placed in the center chamber and acclimated for 5 min, at which point the drop doors lifted and the animal was allowed to freely explore for 20 min. Time spent in each chamber was recorded by using AnyMaze software (Stoelting Co.). Social index was calculated by using the formula: (time spent in the social chamber/(time spent in social chamber + time spent in the novel object chamber)) × 100. Thus, a score of 50 would indicate no significant social preference, a score higher than 50 would indicate a social preference, and a score less than 50 would indicate a social aversion.
Prepulse Inhibition.
PPI refers to the normal reduction in startle response produced by the presentation of a weak startling stimulus before presenting the test stimulus. The reduction is considered to reflect sensorimotor gating, a function shown to be abnormal in a number of psychiatric disorders (Swerdlow et al., 2000). Testing was conducted as described previously (Kamath et al., 2008) by using SR-Lab (San Diego Instruments, San Diego, CA). Background noise (70 dB) and ventilation were provided by an electric fan. Broadband noise pulses were presented by a speaker positioned above the animal enclosure. A piezoelectric accelerometer affixed to the animal enclosure frame was used to detect and transduce motion resulting from the animals' response. Animals were placed in the Plexiglas enclosure and allowed to acclimatize to the environment for 5 min before being tested during 45 discrete trials. On the first five trials, the startle response to a 100-ms, 120-dB white noise pulse was measured to habituate the animals to the testing procedure and thus were omitted from the data analysis. On the subsequent trials, the startle pulse was either presented alone or 100 ms after the presentation of a 30-ms prepulse. The acoustic startle response to the pulse was measured after trials with prepulse intensities of 3, 6, 9, 12, 15, and 18 dB above background noise. Prepulses were varied randomly between trials, and each prepulse was presented five times; animals were randomly presented with the startle pulse alone during the other 10 trials. The average intertrial interval was 15 s (range 5–30 s). Startle magnitude was calculated as the average of the startle responses to the pulse-alone trials. PPI was calculated according to the formula: %PPI = (1 − (startle response for prepulse + pulse trials/startle response for pulse alone trials)) × 100.
Fear Conditioning.
Cued and contextual fear conditioning paradigms are standard tests of emotional memory. These behaviors rely on the integrity of amygdala, hippocampus, thalamus, and other structures. Adult male rats were conditioned as described previously (Muller et al., 1997). Animals were exposed to four pairings of a 1-kHz tone that coterminated with a 1-s, 1-mA foot shock in operant chambers (Habitest; Coulbourn Instruments). These pairings began 120 s after animals were placed into the test chamber with 60-s intertrial intervals. All animals were monitored for response to foot shock (all animals responded). Animals were tested for freezing in response to a cue (tone) 24 h later in a novel environment (altered bedding and odor) and for freezing in response to the original conditioning environment 48 h later. Stimulus presentation was controlled though an AnyMaze interface (Stoelting Co.), and freezing was measured by AnyMaze (Stoelting Co.) and verified by a well trained observer. Difference scores (to measure conditioning) were calculated as follows: cue difference score = (% time freezing in response to cue − % time freezing during test trial before cue onset), context difference score = (% time freezing during context trial − % time freezing during habituation period before conditioning on day 1).
Morris Water Maze.
Morris water maze testing was performed in a 1.5-m-diameter white water maze, as described previously (Vorhees and Williams, 2006). Water was maintained at room temperature with a depth of 32 cm. A submerged platform (11.5 cm diameter, 30.5 cm tall) was placed in a constant location in the center of the northeast quadrant of the maze. Water was made opaque by the addition of tempera paint. Morris water maze performance can be used to assess spatial learning and memory, and it critically relies on the integrity of the hippocampus. Animals received 5 days of training with the hidden platform, each day included four training sessions with a 60-s intersession interval. The start location was varied on each training trial. Latency to escape, heading error, path efficiency, thigmotactic behavior, and swim speed all were recorded by using AnyMaze Software. On the sixth day, animals were given a 60-s probe trial, in which the platform was removed and the animals were released from a novel (previously unused) start location, to ensure any navigation was based on a spatial map. Time spent in the goal quadrant was recorded.
Statistical Analysis.
Data were analyzed by using SPSS (IBM, White Plains, NY) and Prism (GraphPad Software Inc., San Diego, CA). Normally distributed data were analyzed via analysis of variance (with repeated measures as appropriate). Social behavior data and probe trials in the Morris water maze were evaluated by using a one-sample t test. Nonparametric data were analyzed by using the Kruskal-Wallis test. Post hoc tests were performed as appropriate (Bonferroni-Holm's corrected).
Testing Order.
Every animal was tested on the elevated plus maze. Subsets of animals, drawn from multiple litters were tested on the additional tasks, in order of increasing stress. Tests were performed in the following order: elevated plus maze, open field, social behavior, rotorod, fear conditioning, prepulse inhibition, and Morris water maze.
All procedures were in compliance with the American Association for Accreditation of Laboratory Animal Care standards and approved by the Georgetown University Animal Care and Use Committee. Efforts were made to minimize the number of animals used and any discomfort.
Results
Body Weight and Weight Gain.
As shown in Table 1, before treatment on P7 all groups had equivalent body weights (F3,192 = 2.464; p = 0.06). However, at the completion of treatment (on P13), the mean body weights of the phenobarbital- and phenytoin-treated groups were significantly lower than controls (F3,153 = 15.60; p < 0.0001), with the drug-treated groups exhibiting a reduced rate of weight gain (F3,151 = 32.20; p < 0.0001). In spite of these differences, the overall weight distributions were highly overlapping as reflected in the standard deviations. Lamotrigine-treated animals did not differ from controls with respect to either body weight or weight gain.
TABLE 1.
Body weight and weight gain
Data are ± S.E.M. Results in parentheses show S.D.
| Treatment | P7 Weight | P13 Weight | Weight Gain from P7 to P13 |
|---|---|---|---|
| g | |||
| Control | 14.2 ± 0.36 (2.7) | 25.7 ± 0.48 (3.6) | 11.7 ± 0.49 (3.7) |
| PB | 14.2 ± 0.28 (2.2) | 20.1 ± 0.69 (3.9)* | 4.8 ± 0.52 (2.9)* |
| PHT | 15.0 ± 0.40 (2.5) | 23.4 ± 0.64 (3.6)* | 8.5 ± 0.70 (3.8)* |
| LTG | 13.5 ± 0.40 (2.5) | 25.1 ± 0.70 (4.4) | 11.5 ± 0.50 (3.1) |
Significantly different than controls, P < 0.05.
Accelerating Rotorod.
The accelerating rotorod is a standard test of motor coordination, in which the latency to fall from the rotating rod is used as a measure of performance. With repeated trials, this task can also be used to assess motor learning as evident by increased latencies to fall (Buitrago et al., 2004). There were no differences in performance across treatments on the first trial, and all treatment groups displayed significant learning over the course of 10 repeated trials. Animals exposed to phenytoin or lamotrigine displayed significantly lower peak performance latencies (i.e., the longest latency reached by each animal) compared with controls (Fig. 1; p < 0.05; Fisher's least significant differences post hoc test).
Fig. 1.

Effect of neonatal AED exposure on adult peak performance latencies (i.e., the longest latency reached by each animal) on the accelerating rotorod. Longer latencies (higher values) indicate better performance. *, significantly reduced performance compared with controls, p < 0.05 (Fisher's least significant differences post hoc test). C, control; PB, phenobarbital; PHT, phenytoin; LTG, lamotrigine.
Open Field Locomotion.
As shown in Table 2, the experimental groups did not significantly differ in total distance traveled (F3,59 = 1.419; p = 0.25) or latency to enter the center compartment (F3,59 = 1.072; p = 0.37). Compared with controls, animals exposed to phenytoin spent more time in the central portion of the open field that was not adjacent to the walls (p < 0.05; Dunnett's multiple comparison test). When collapsing across treatment groups, center time (r = 0.221; p = 0.31) was not correlated with weight gain in the second postnatal week.
TABLE 2.
Open-field behavior
Data shown are mean ± S.E.M.
| Treatment | Total Distance | Time in Center | Latency to Enter Center |
|---|---|---|---|
| m | % | s | |
| Control | 16.4 ± 0.6 | 11.95 ± 1.2 | 25.1 ± 5.8 |
| PB | 14.87 ± 0.7 | 15.3 ± 3.5 | 20.90 ± 5.0 |
| PHT | 18.54 ± 0.9 | 20.92 ± 3.6* | 15.3 ± 6.9 |
| LTG | 17.07 ± 1.3 | 15.3 ± 5.5 | 27.76 ± 7.8 |
Significantly different than controls, P < 0.05.
Prepulse Inhibition.
For the analysis of treatment effects on PPI (Fig. 2) data were collapsed across prepulse intensities because we found no significant interaction between treatment and prepulse intensity in a two-way analysis of variance (main effect of treatment: F3,87 = 3.748, p < 0.05; prepulse intensity: F5,435 = 67.918, p < 0.0001; treatment by prepulse intensity interaction: F15,435 = 1.377, p = 0.154). Control (vehicle-treated) animals exhibited a mean of 61.6% inhibition.
Fig. 2.

Effect of neonatal AED exposure on adult PPI. A, PPI in control and AED-exposed animals, collapsed across prepulse intensity. B, baseline acoustic startle response. C, PPI as a function of prepulse intensity in control and phenobarbital-exposed animals. *, p < 0.05 (Bonferroni post hoc).
Mean PPI was significantly reduced in rats exposed to phenobarbital (p < 0.05; Dunnett's multiple comparison test) but not in those exposed to phenytoin or lamotrigine (Fig. 2A; F3,89 = 3.878). Amplitude of the acoustic startle response did not differ among treatment groups (Fig. 2B; F3,89 = 1.661; p = 0.12).
Figure 2C shows the effects of phenobarbital on PPI as a function of prepulse intensity: phenobarbital significantly attenuated PPI at prepulses 6, 9, 12, 15, and 18 dB above background (p < 0.05; Bonferroni-Holm's). When collapsed across treatment groups, PPI was correlated with neonatal weight gain (r = 0.321; p < 0.01), an effect that was exclusively driven by a correlation in the control group (r = 0.351; p < 0.05).
Elevated Plus Maze.
Control animals spent a mean of 39.2 s (of a total 300 s) in the open arms of the elevated plus maze. Time spent in the open arms was significantly greater in animals exposed to phenobarbital (F3,147 = 1.636, p = 0.18; Bonferroni's multiple comparison test, p < 0.05), but was not altered in animals exposed to phenytoin or lamotrigine (Fig. 3A). No differences between groups were found for the percentage of entries into the open arms (Fig. 3B; F3,135 = 0.26; p = 0.85) or the total arm entries (Fig. 3C; F3,148 = 2.66; p = 0.051). When collapsed across treatment groups, there was no significant correlation between time spent in the open arms and neonatal weight gain (r = 0.179; p = 0.08).
Fig. 3.
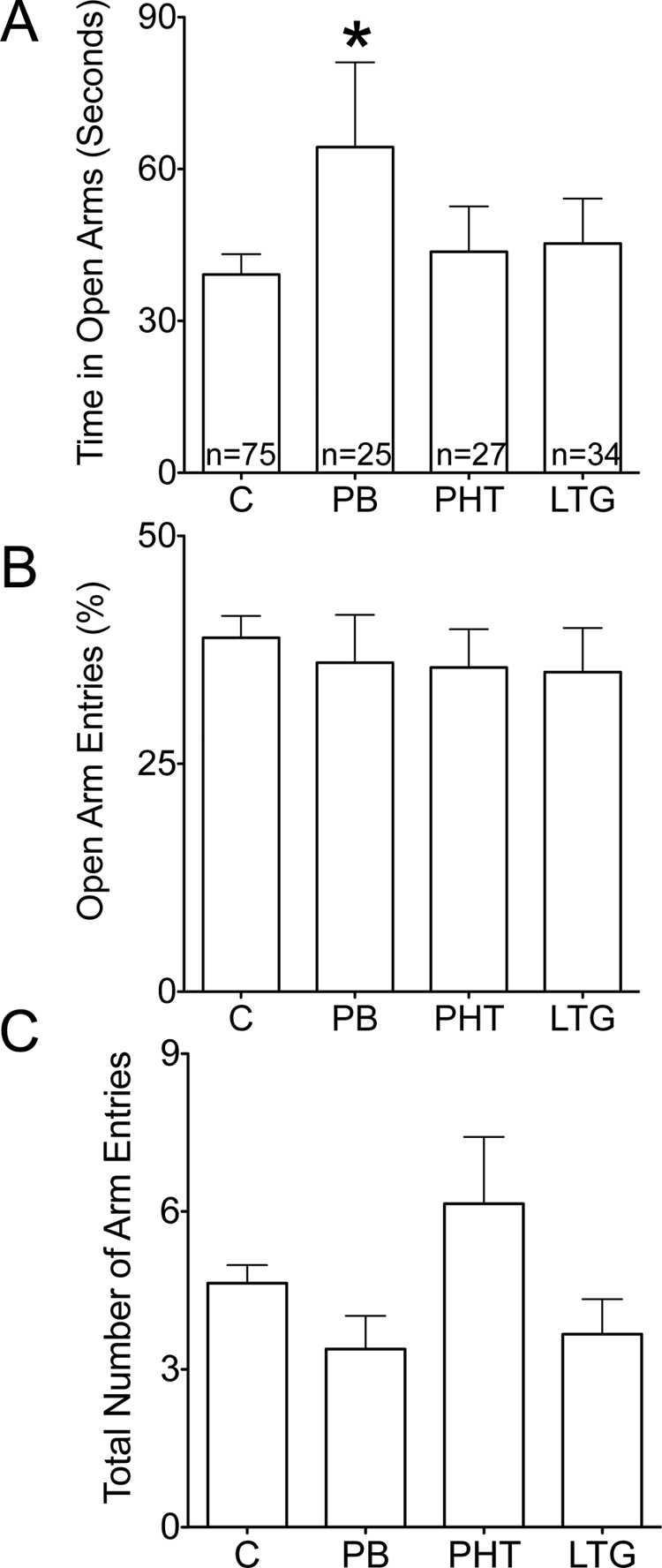
Effect of neonatal AED exposure on adult elevated plus maze. A, time spent in the open arms of the plus maze. *, p < 0.05 (Bonferroni post hoc). B, percentage of total entries that were made into the open arms. C, total number of arm entries.
Social Behavior.
Control animals exhibited a significant preference for exploration of a novel rat compared with a novel object (Fig. 4; p < 0.001). This preference was not significant in animals exposed to phenobarbital, phenytoin, or lamotrigine. Furthermore, relative time spent exploring for a novel rat was significantly lower in animals exposed to lamotrigine compared with controls (F3,42 = 4.544; p < 0.01, post hoc; Dunnett's multiple comparison test, p < 0.05). When collapsed across treatment groups, there was no significant correlation between social behavior and neonatal weight gain (r = 0.034; p = 0.83).
Fig. 4.

Effects of neonatal AED exposure on adult social exploration. Social exploration ratio was calculated as: ((time spent in the social chamber/(time spent in the social chamber + time spent in the novel object chamber)) × 100. The dotted line indicates equal preference for social and novel object chambers. *, p < 0.05 (Bonferroni post hoc). ^, no significant social preference (one-sample t test, p < 0.05).
Cued and Contextual Fear Conditioning.
As shown in Fig. 5A, cued fear conditioning in control animals was reflected by a difference score (time spent freezing during the cue minus time spent freezing during precue baseline) significantly higher than zero (p < 0.05; one-sample t test). Difference scores for phenobarbital- and phenytoin-exposed animals were significantly lower than control difference scores (F3,79 = 3.031, p < 0.05; Bonferroni post hoc, p < 0.05) and did not significantly differ from zero, indicating no conditioning (one-sample t test). Lamotrigine-exposed animals were not significantly different from controls and showed difference scores significantly higher than zero (one-sample t test; p < 0.05).
Fig. 5.

Effect of neonatal AED exposure on adult fear conditioning. A, cued fear conditioning. *, p < 0.05 (Bonferroni post hoc). ^, significant conditioning (i.e., difference score >0; one-sample t test; p < 0.05). B, contextual fear conditioning.
As shown in Fig. 5B, contextual fear conditioning was seen in all groups, reflected by difference scores significantly higher than zero (one-sample t test; p < 0.05). There were no significant differences between groups (F3,79 = 0.555; p = 0.6). When collapsed across treatment groups, fear conditioning did not correlate with neonatal body weight gain (cue: r = 0.057, p = 0.6; context: r = 0.071, p = 0.5).
Morris Water Maze.
As shown in Fig. 6 A to C, control animals learned the location of the hidden platform in the Morris water maze, as evidenced by a decrease in escape latency during training. Animals exposed to phenobarbital showed a similar learning curve, but had significantly longer escape latencies compared with controls (Fig. 6A; p < 0.05). Phenytoin-exposed (Fig. 6B) and lamotrigine-exposed (Fig. 6C) animals were not significantly different from controls during training. There was a significant main effect of training day (F4,188 = 111.673; p < 0.001), no significant main effect of treatment (F3,46 = 1.949; p = 0.14), and no significant treatment by training day interaction (F12,188 = 0.689; p = 0.72). Post hoc analysis revealed significant deficits in phenobarbital-exposed animals on the fifth training day, as well as a main effect of treatment when phenobarbital and controls were compared directly (F1,30 = 6.28; p < 0.05).
Fig. 6.

Effect of neonatal AED exposure on adult Morris water maze performance. A to C, learning curve for the 5 days of training in control and phenobarbital-exposed animals (A), control and phenytoin-exposed animals (B), and control and lamotrigine-exposed animals (C). D, time spent in the correct quadrant during the probe trial; the dotted line at 25 indicates chance level of performance. E, path efficiency across all trials. F, percentage of time spent engaged in thigmotactic behavior. *, significantly different from control, p < 0.05. G, swim speed across all trials. The post hoc values for each test are shown in the table. Bold indicate significant differences, and italics indicate trends.
On the probe trial, control and phenytoin-exposed animals displayed a significant preference for the goal quadrant (p < 0.01 and < 0.05, respectively), indicating spatial recall. Neither phenobarbital- nor lamotrigine-exposed animals displayed a significant preference for the goal quadrant (Fig. 6D; one-sample t test), indicating impaired spatial recall.
When escape route path efficiencies (Fig. 6E) were analyzed, they were found to be significantly less efficient in all AED-exposed groups compared with controls (F3,47 = 4.067; p < 0.05).
Control animals spent a mean of 21.2% of swim time along the edges of the pool (i.e., thigmotactic behavior). This measurement (Fig. 6F) was significantly greater in animals exposed to phenytoin (H = 8.133; 3 df; p < 0.05) but not in those exposed to phenobarbital or lamotrigine.
Average swim speed (Fig. 6G) over all trials did not differ between groups (F3,47 = 0.74; p = 0.5). When collapsed across groups, none of the performance measures in the Morris water maze correlated with neonatal body weight gain [percentage of time in correct quadrant during the probe trial (r = 0.218; p = 0.17); performance on day 1 (r = 0.179; p = 0.26); performance on day 2 (r = 0.077; p = 0.63); performance on day 3 (r = 0.205; p = 0.20); performance on day 4 (r = 0.02; p = 0.89); performance on day 5 (r = 0.234; p = 0.14); path efficiency (r = 0.075; p = 0.64); swim speed (r = 0.168; p = 0.29); and thigmotaxis (r = 0.007; p = 0.96)].
Discussion
Our findings support the hypothesis that AED-associated neurotoxicity during a narrow developmental window (the second postnatal week in the rat) is sufficient to cause adverse behavioral consequences in adulthood. The pattern of behavioral consequences in adulthood differed between the three AEDs we tested, as summarized in Table 3, suggesting that the drugs do not have identical mechanisms of neurodevelopmental toxicity.
TABLE 3.
Summary of behavioral findings in adult animals exposed as neonates to antiepileptic drugs
| Treatment | Elevated Plus Maze | Cued Fear Conditioning | Contextual Fear Conditioning | Prepulse Inhibition | Social Behavior | Rotorod | Morris Water Maze | Open Field |
|---|---|---|---|---|---|---|---|---|
| Phenobarbital | Decreased anxiety-like behavior | Decreased learning/memory | No change | Decreased sensorimotor gating | Decreased social exploration | No change | Impaired learning | No change |
| Phenytoin | No change | Decreased learning/memory | No change | No change | Decreased social exploration | Decreased peak performance | Impaired learning | Decreased exploration of center |
| Lamotrigine | No change | No change | No change | No change | Decreased social exploration | Decreased peak performance | Impaired learning | No change |
Our study is the first to compare the long-term effects of neonatal exposure to three commonly prescribed AEDs on a broad range of behavioral tasks relevant to human disease. Prior studies in rodents focused only on a single drug, examining only one or two behavioral assays. For example, phenobarbital exposure impaired spontaneous alternation in a T maze (Pick and Yanai, 1983; Pereira de Vasconcelos et al., 1990), water maze performance (Rogel-Fuchs et al., 1992; Stefovska et al., 2008), and rat exploratory behavior in the open field (Pereira de Vasconcelos et al., 1990). Only mouse motor function has been assessed after early life exposure to phenytoin. Simple motor behaviors (such as head elevation, pivoting, and surface righting) were impaired in pups (Hatta et al., 1999), and spontaneous locomotion and rotorod performance were impaired later in life (Ohmori et al., 1999). Long-term behavioral effects of lamotrigine have been examined in only two studies. Mikulecká et al. (2004) found that lamotrigine significantly impaired rotorod performance, and we have reported previously that lamotrigine (20 mg/kg from P7 to P13) reduced pentylenetetrazole seizure threshold in adult animals (Forcelli et al., 2011a). Here, we have shown that neonatal exposure to the AEDs results in abnormalities in motor, emotional, and social behaviors, as well as in learning and memory. Exposure to any of the three drugs adversely affected both spatial learning in the water maze and social preference. Because the dose of lamotrigine used was below the threshold for a proapoptotic action (Katz et al., 2007), these data suggest that the induction of neuronal apoptosis is not a necessary condition for adverse outcomes. Thus, long-term behavioral measures may be more sensitive indicators of neurotoxicity compared with acute histopathological analysis.
Phenobarbital and phenytoin both cause neuronal apoptosis in the neonatal rat brain. However, with the exception of a common impairment in cued fear conditioning, the deficits observed in rats exposed to each of these drugs were distinct: phenobarbital exposure was associated with abnormalities in elevated plus maze exploration and PPI, whereas phenytoin exposure was associated with abnormalities in rotorod and open-field performance. Thus, induction of neuronal apoptosis after acute treatment does not seem to be a good predictor of the full range of later behavioral outcomes after chronic treatment. It is possible that chronic treatment may, in the case of lamotrigine, lead to drug accumulation to levels that are sufficient to trigger apoptosis. The effect of repeated AED exposure on the induction of apoptosis is an open question at this point. It is unknown whether multiple exposures eliminate more neurons than an acute exposure or how/if the pattern of apoptosis might be altered with chronic administration.
Phenytoin and lamotrigine share a common mechanism of antiepileptic action, the blockade of voltage-gated sodium channels. This could account for the common mild impairment observed on rotorod performance in animals exposed to either of these drugs. However, for open-field exploration and cued fear conditioning, the effects of the two drugs were dissociated from one another. This suggests that the mechanisms underlying the toxicity may be independent of those responsible for the therapeutic action. Because these drugs belong to distinct structural classes (i.e., phenytoin is a hydantoin, lamotrigine is a phenyltriazine), generate different metabolites, and exhibit nonoverlapping sets of side effects, it is difficult to pinpoint which of the numerous differences between the drugs may account for the divergent behavioral outcomes after neonatal exposure. The fact that phenytoin has clear teratogenic actions at therapeutic doses, whereas lamotrigine does not (Ornoy, 2006), is strong evidence that the nature and severity of impairment of developmental programs is not necessarily predicted by the antiepileptic mechanism of action.
Our behavioral findings after postnatal exposure to phenytoin share some features described previously in adult mice after prenatal exposure to this drug under conditions that avoided causing physical malformations. In particular, Vorhees and colleagues observed increased open-field activity as well as deficits in maze learning (in the Morris water maze, among other tasks) in adult mice exposed to phenytoin in utero (Adams et al., 1990; Vorhees, 1994). However, the latter investigation showed increases in auditory startle, an abnormality not observed in our animals, suggesting that certain outcomes are specific to the developmental stage of exposure. Increased responsivity to sound, as well as increased activity in the open field, have also been observed in mice after in utero exposure to phenobarbital during the last week of gestation (Middaugh et al., 1981a,b). Neither of these consequences were detected in our animals after postnatal phenobarbital exposure, raising the possibility that the brain may be more vulnerable to the actions of certain drugs during the prenatal period.
Studies in nonhuman primates are an important step toward extrapolation of the rodent data to humans. Although drug-induced developmental neuronal apoptosis has been documented in monkeys after in utero or neonatal exposure to anesthetics or ethanol (Brambrink et al., 2010; Farber et al., 2010), AEDs have yet to be examined in this context. The fact that monkeys exposed to phenytoin in utero exhibited motor deficits and hyperexcitability as infants (Phillips and Lockard, 1996) suggests that nonhuman primate models are likely to detect some of the same adverse behavioral outcomes as have been observed in rodents. Studies of socio-emotional, cognitive, and motor outcomes in adult monkeys after neonatal exposure to a variety of AEDs would be of great value in extending our findings in rats to the primate brain.
The behavioral deficits we observed were unrelated to nonspecific debilitation during development or adulthood. Although there was an effect of phenobarbital and phenytoin exposure on weight gain in the neonates, this was not the case with lamotrigine exposure, which nevertheless led to impairment on tests of social, cognitive, and motor function in adulthood. Further evidence for a dissociation between neonatal weight gain and behavioral outcomes comes from the lack of correlation between weight gain during the period of drug exposure and behavioral performance on each of the individual tasks. In adulthood, there were no significant differences between body weights across treatment groups, and locomotor behavior in the open field and swim speed in the water maze were unaffected by drug exposure. This indicates that drug exposure did not induce gross motor incapacitation and motor impairment does not account for any of the observed deficits.
Our finding that phenobarbital exposure impaired PPI is consistent with a previous report that a single administration of phenobarbital on P7 disrupted PPI in adults to an extent equivalent to that seen in the neonatal ventral hippocampal lesion model of schizophrenia (Palchik et al., 2009). Damage to hippocampus as well as several other key structures in the network that supports PPI, including amygdala, thalamus, striatum, and nucleus accumbens (Bittigau et al., 2002; Forcelli et al., 2011b) may explain the PPI deficits after neonatal phenobarbital exposure. The fact that phenobarbital causes greater cell death in accumbens and amygdala than does phenytoin (Forcelli et al., 2011b) may account for the dissociation between these drugs for impairing PPI. A similar impairment in PPI has been found after neonatal exposure to NMDA receptor antagonists (Wang et al., 2001), which are also potent inducers of apoptotic cell death in the developing rat brain (Ikonomidou et al., 1999).
The abnormalities we observed in emotional and social behavior may have relevance for pediatric epilepsy, for which neuropsychiatric comorbidities are especially high (Plioplys et al., 2007). It has previously been suggested that seizures and psychiatric disorders may share an underlying brain abnormality or may be causally related. Our data suggest a third possibility: the comorbid psychiatric disorders may be iatrogenic. In support of this hypothesis, we found decreased anxiety-like behavior in the elevated plus maze in animals that had been exposed to phenobarbital and decreased social interactions in animals that had been exposed to either phenobarbital, phenytoin, or lamotrigine. These results may reflect the histological changes in limbic substrates for these behaviors (e.g., amygdala) we have reported previously (Forcelli et al., 2011b).
The time of drug exposure in the present study was limited to a restricted postnatal period (P7–P13), which corresponds to the late third trimester of gestation through infancy in humans (Dobbing and Sands, 1979). This period, characterized by a high rate of synaptogenesis and neuronal pruning, is referred to as the “brain growth spurt” (Dobbing and Sands, 1979). Our results are consistent with the one previous study that examined a behavioral outcome after drug exposure limited to this time period (Stefovska et al., 2008); Wistar rats exposed to phenobarbital (50 mg/kg) on P4, P6, P8, and P10 were impaired on water maze performance as adults.
Induction of enhanced neuronal apoptosis is one of several neurochemical changes seen in animals exposed to AEDs early in life. Early exposure (P6) to phenobarbital resulted in long-lasting changes in the cortical proteome, with long-lasting changes in the expression of proteins involved in oxidative stress, apoptosis, astroglial response, energy metabolism, and neuronal function. These changes provide at least one mechanism by which early injury can have long-lasting impacts on cortical function (Kaindl et al., 2008). Longer duration of treatment with phenobarbital resulted in reduced GABA receptor expression (Ruiz et al., 1989), increased muscarinic receptor expression in hippocampus (Rogel-Fuchs et al., 1992; Pick et al., 1993), and decreased cerebral glucose utilization (Pereira de Vasconcelos et al., 1990). In addition to these changes, hippocampal neurogenesis is impaired after early life exposure to phenobarbital (Stefovska et al., 2008). It would be particularly interesting to determine whether these changes would occur under the shorter duration of treatment we used in the present study; if the shorter duration of treatment were to be sufficient to induce these alterations, it would suggest that they may contribute to the adverse behavioral findings we detected. This is certainly compelling, because deficits in hippocampal neurogenesis have been linked to behavioral impairments in memory tasks (Saxe et al., 2006).
Our present findings demonstrate that exposure to phenobarbital, phenytoin, or lamotrigine during a narrow postnatal window, and at therapeutically relevant doses, results in long-lasting changes in emotional, social, and cognitive function. These domains of function are relevant to psychiatric conditions that are highly comorbid with epilepsy. This behavioral toxicity cannot be explained by either drug mechanism of action or the proapoptotic response to acute drug administration alone. These data suggest that analysis of the impact of multiple drugs across a battery of tests sensitive to differing domains of central nervous system function is a necessity for determining the long-term safety of AEDs for use in early life.
Acknowledgments
We thank Cameron Sweeny and Sam Paskewitz for assistance in testing animals.
This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant NS066822] (to P.A.F.); the National Institutes of Health National Institute of Mental Health [Grant MH02040] (to A.K.); an Epilepsy Foundation fellowship (to P.A.F.); and a research grant from GlaxoSmithKline (to A.K.).
The data in this article were included in the doctoral dissertation of P.A.F.: Forcelli PA (2011) Sequelae of Neonatal Antiepileptic Drug Exposure. Ph.D. thesis, Georgetown University, Washington, DC.
The data in this article have been presented previously in abstract form: Forcelli P, Ritter J, Kondratyev A, and Gale K (2008) Long-term adverse outcomes of neonatal antiepileptic drug exposure in rats, at the American Epilepsy Society Annual Meeting, 2008 Dec 5–9, Seattle, WA, American Epilepsy Society, West Hartford, CT. Forcelli PA, Kondratyev A, and Gale K (2009) Antiepileptic drug exposure in neonatal rats leads to neuropsychiatric behavioral abnormalities in adults, at the Society for Neuroscience Annual Meeting, 2009 Oct 17–21, Chicago, IL, Society for Neuroscience, Washington, DC. Forcelli P, Kozlowski R, Snyder C, Gale K, and Kondratyev A (2009) Early-life exposure to antiepileptic drugs results in behavioral and neuropsychiatric abnormalities in adult rats, at the American Epilepsy Society Annual Meeting, 2009 Dec 4–8, Boston, MA, American Epilepsy Society, West Hartford, CT. Forcelli PA, Janssen MJ, Sweeney C, Vicini S, and Gale K (2010) Neonatal exposure to phenobarbital delays maturation of synaptic transmission in striatum with behavioral consequences, at the Society for Neuroscience Annual Meeting, 2010 Nov 13–17, San Diego, CA, Society for Neuroscience, Washington, DC. Forcelli P, Janssen M, Vicini S, and Gale K (2010) Antiepileptic drugs (AEDs) in neonatal rats disrupt striatal synaptic maturation, at the American Epilepsy Society Annual Meeting, 2010 Dec 3–7, San Antonio, TX, American Epilepsy Society, West Hartford, CT.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- AED
- antiepileptic drug
- Pn
- postnatal day
- PB
- phenobarbital
- PHT
- phenytoin
- LTG
- lamotrigine
- PPI
- prepulse inhibition
- C
- control
- NMDA
- N-methyl-d-aspartate.
Authorship Contributions
Participated in research design: Forcelli, Kondratyev, and Gale.
Conducted experiments: Forcelli, Kozlowski, and Snyder.
Performed data analysis: Forcelli.
Wrote or contributed to the writing of the manuscript: Forcelli, Kondratyev, and Gale.
References
- Adams J, Vorhees CV, Middaugh LD. (1990) Developmental neurotoxicity of anticonvulsants: human and animal evidence on phenytoin. Neurotoxicol Teratol 12:203–214 [DOI] [PubMed] [Google Scholar]
- Bartha AI, Shen J, Katz KH, Mischel RE, Yap KR, Ivacko JA, Andrews EM, Ferriero DM, Ment LR, Silverstein FS. (2007) Neonatal seizures: multicenter variability in current treatment practices. Pediatr Neurol 37:85–90 [DOI] [PubMed] [Google Scholar]
- Billingslea EN. (2007) Comparisons of Behavioral Phenotypes in Multiple Methods of Serotonin Deficiency in the Rat Brain. Doctoral dissertation, Georgetown University, Washington, DC [Google Scholar]
- Bittigau P, Sifringer M, Genz K, Reith E, Pospischil D, Govindarajalu S, Dzietko M, Pesditschek S, Mai I, Dikranian K, et al. (2002) Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci U S A 99:15089–15094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Zhang X, Dissen GA, Creeley CE, Olney JW. (2010) Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology 112:834–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buitrago MM, Schulz JB, Dichgans J, Luft AR. (2004) Short- and long-term motor skill learning in an accelerated rotarod training paradigm. Neurobiol Learn Mem 81:211–216 [DOI] [PubMed] [Google Scholar]
- Dobbing J, Sands J. (1979) Comparative aspects of the brain growth spurt. Early Hum Dev 3:79–83 [DOI] [PubMed] [Google Scholar]
- Farber NB, Creeley CE, Olney JW. (2010) Alcohol-induced neuroapoptosis in the fetal macaque brain. Neurobiol Dis 40:200–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell JR, Lee YJ, Hirtz DG, Sulzbacher SI, Ellenberg JH, Nelson KB. (1990) Phenobarbital for febrile seizures–effects on intelligence and on seizure recurrence. N Engl J Med 322:364–369 [DOI] [PubMed] [Google Scholar]
- File SE, Lippa AS, Beer B, Lippa MT. (2004) Animal tests of anxiety. Curr Protoc Neurosci, Chapter 8:Unit 8.3 [DOI] [PubMed] [Google Scholar]
- Forcelli PA. (2011) Sequelae of Neonatal Antiepileptic Drug Exposure. Ph.D. thesis, Georgetown University, Washington, DC [Google Scholar]
- Forcelli PA, Gale K, Kondratyev A. (2011a) Early postnatal exposure of rats to lamotrigine, but not phenytoin, reduces seizure threshold in adulthood. Epilepsia 52:e20–e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcelli PA, Heinrichs SC. (2008) Teratogenic effects of maternal antidepressant exposure on neural substrates of drug-seeking behavior in offspring. Addict Biol 13:52–62 [DOI] [PubMed] [Google Scholar]
- Forcelli PA, Janssen MJ, Stamps LA, Sweeney C, Vicini S, Gale K. (2010) Therapeutic strategies to avoid long-term adverse outcomes of neonatal antiepileptic drug exposure. Epilepsia 51 (Suppl 3):18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcelli PA, Kim J, Kondratyev A, Gale K. (2011b) Pattern of antiepileptic drug-induced cell death in limbic regions of the neonatal rat brain. Epilepsia 52:e207–e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatta T, Ohmori H, Murakami T, Takano M, Yamashita K, Yasuda M. (1999) Neurotoxic effects of phenytoin on postnatal mouse brain development following neonatal administration. Neurotoxicol Teratol 21:21–28 [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Hörster F, Tenkova T, et al. (2000) Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 287:1056–1060 [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283:70–74 [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Scheer I, Wilhelm T, Juengling FD, Titze K, Stöver B, Lehmkuhl U, Koch S, Kassubek J. (2007) Brain morphology alterations in the basal ganglia and the hypothalamus following prenatal exposure to antiepileptic drugs. Eur J Paediatr Neurol 11:297–301 [DOI] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. (2003) Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci 23:876–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaindl AM, Koppelstaetter A, Nebrich G, Stuwe J, Sifringer M, Zabel C, Klose J, Ikonomidou C. (2008) Brief alteration of NMDA or GABAA receptor-mediated neurotransmission has long term effects on the developing cerebral cortex. Mol Cell Proteomics 7:2293–2310 [DOI] [PubMed] [Google Scholar]
- Kamath A, Al-Khairi I, Bhardwaj S, Srivastava LK. (2008) Enhanced α1 adrenergic sensitivity in sensorimotor gating deficits in neonatal ventral hippocampus-lesioned rats. Int J Neuropsychopharmacol 11:1085–1096 [DOI] [PubMed] [Google Scholar]
- Katz I, Kim J, Gale K, Kondratyev A. (2007) Effects of lamotrigine alone and in combination with MK-801, phenobarbital, or phenytoin on cell death in the neonatal rat brain. J Pharmacol Exp Ther 322:494–500 [DOI] [PubMed] [Google Scholar]
- Kim J, Kondratyev A, Gale K. (2007a) Antiepileptic drug-induced neuronal cell death in the immature brain: effects of carbamazepine, topiramate, and levetiracetam as monotherapy versus polytherapy. J Pharmacol Exp Ther 323:165–173 [DOI] [PubMed] [Google Scholar]
- Kim JS, Kondratyev A, Tomita Y, Gale K. (2007b) Neurodevelopmental impact of antiepileptic drugs and seizures in the immature brain. Epilepsia 48 (Suppl 5):19–26 [DOI] [PubMed] [Google Scholar]
- Kubova H, Mares P. (1991) Anticonvulsant effects of phenobarbital and primidone during ontogenesis in rats. Epilepsy Res 10:148–155 [DOI] [PubMed] [Google Scholar]
- Meador KJ, Baker GA, Browning N, Cohen MJ, Clayton-Smith J, Kalayjian LA, Kanner A, Liporace JD, Pennell PB, Privitera M, et al. (2011) Foetal antiepileptic drug exposure and verbal versus non-verbal abilities at three years of age. Brain 134:396–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middaugh LD, Simpson LW, Thomas TN, Zemp JW. (1981a) Prenatal maternal phenobarbital increases reactivity and retards habituation of mature offspring to environmental stimuli. Psychopharmacology (Berl) 74:349–352 [DOI] [PubMed] [Google Scholar]
- Middaugh LD, Thomas TN, Simpson LW, Zemp JW. (1981b) Effects of prenatal maternal injections of phenobarbital on brain neurotransmitters and behavior of young C57 mice. Neurobehav Toxicol Teratol 3:271–275 [PubMed] [Google Scholar]
- Mikulecká A, Kubová H, Mares P. (2004) Lamotrigine does not impair motor performance and spontaneous behavior in developing rats. Epilepsy Behav 5:464–471 [DOI] [PubMed] [Google Scholar]
- Monville C, Torres EM, Dunnett SB. (2006) Comparison of incremental and accelerating protocols of the rotarod test for the assessment of motor deficits in the 6-OHDA model. J Neurosci Methods 158:219–223 [DOI] [PubMed] [Google Scholar]
- Muller J, Corodimas KP, Fridel Z, LeDoux JE. (1997) Functional inactivation of the lateral and basal nuclei of the amygdala by muscimol infusion prevents fear conditioning to an explicit conditioned stimulus and to contextual stimuli. Behav Neurosci 111:683–691 [DOI] [PubMed] [Google Scholar]
- Nadler JJ, Moy SS, Dold G, Trang D, Simmons N, Perez A, Young NB, Barbaro RP, Piven J, Magnuson TR, et al. (2004) Automated apparatus for quantitation of social approach behaviors in mice. Genes Brain Behav 3:303–314 [DOI] [PubMed] [Google Scholar]
- Ohmori H, Ogura H, Yasuda M, Nakamura S, Hatta T, Kawano K, Michikawa T, Yamashita K, Mikoshiba K. (1999) Developmental neurotoxicity of phenytoin on granule cells and Purkinje cells in mouse cerebellum. J Neurochem 72:1497–1506 [DOI] [PubMed] [Google Scholar]
- Ornoy A. (2006) Neuroteratogens in man: an overview with special emphasis on the teratogenicity of antiepileptic drugs in pregnancy. Reprod Toxicol 22:214–226 [DOI] [PubMed] [Google Scholar]
- Palchik G, Bhardwaj SK, Gale KN, Kondratyev AD, Srivastava LK. (2009) Effects of antiepileptic drugs on neuronal survival and behavior in a developmental animal model of schizophrenia, at the Society for Neuroscience Annual Meeting; 2009 Oct 17–21; Chicago, IL Society for Neuroscience, Washington, DC [Google Scholar]
- Pereira de Vasconcelos A, Colin C, Desor D, Divry M, Nehlig A. (1990) Influence of early neonatal phenobarbital exposure on cerebral energy metabolism and behavior. Exp Neurol 108:176–187 [DOI] [PubMed] [Google Scholar]
- Phillips NK, Lockard JS. (1996) Infant monkey hyperexcitability after prenatal exposure to antiepileptic compounds. Epilepsia 37:991–999 [DOI] [PubMed] [Google Scholar]
- Pick CG, Weizman A, Fares F, Gavish M, Kanner BI, Yanai J. (1993) Hippocampal γ-aminobutyric acid and benzodiazepine receptors after early phenobarbital exposure. Brain Res Dev Brain Res 74:111–116 [DOI] [PubMed] [Google Scholar]
- Pick CG, Yanai J. (1983) Eight arm maze for mice. Int J Neurosci 21:63–66 [DOI] [PubMed] [Google Scholar]
- Plioplys S, Dunn DW, Caplan R. (2007) 10-year research update review: psychiatric problems in children with epilepsy. J Am Acad Child Adolesc Psychiatry 46:1389–1402 [DOI] [PubMed] [Google Scholar]
- Rogel-Fuchs Y, Newman ME, Trombka D, Zahalka EA, Yanai J. (1992) Hippocampal cholinergic alterations and related behavioral deficits after early exposure to phenobarbital. Brain Res Bull 29:1–6 [DOI] [PubMed] [Google Scholar]
- Ruiz G, Hamon M, Vergé D. (1989) Chronic phenytoin treatment decreases GABAA but not β-adrenoceptors in the cerebellum of young rats. Eur J Pharmacol 168:251–255 [DOI] [PubMed] [Google Scholar]
- Sams-Dodd F. (1999) Phencyclidine in the social interaction test: an animal model of schizophrenia with face and predictive validity. Rev Neurosci 10:59–90 [DOI] [PubMed] [Google Scholar]
- Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, Garcia AD, Sofroniew MV, Kandel ER, Santarelli L, et al. (2006) Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci U S A 103:17501–17506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanková L, Kubová H, Mares P. (1992) Anticonvulsant action of lamotrigine during ontogenesis in rats. Epilepsy Res 13:17–22 [DOI] [PubMed] [Google Scholar]
- Stefovska VG, Uckermann O, Czuczwar M, Smitka M, Czuczwar P, Kis J, Kaindl AM, Turski L, Turski WA, Ikonomidou C. (2008) Sedative and anticonvulsant drugs suppress postnatal neurogenesis. Ann Neurol 64:434–445 [DOI] [PubMed] [Google Scholar]
- Sulzbacher S, Farwell JR, Temkin N, Lu AS, Hirtz DG. (1999) Late cognitive effects of early treatment with phenobarbital. Clin Pediatr (Phila) 38:387–394 [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA. (2000) Animal models of deficient sensorimotor gating: what we know, what we think we know, and what we hope to know soon. Behav Pharmacol 11:185–204 [DOI] [PubMed] [Google Scholar]
- Vestergaard M, Pedersen CB, Christensen J, Madsen KM, Olsen J, Mortensen PB. (2005) Febrile seizures and risk of schizophrenia. Schizophr Res 73:343–349 [DOI] [PubMed] [Google Scholar]
- Vorhees CV. (1994) Developmental neurotoxicity induced by therapeutic and illicit drugs. Environ Health Perspect 102 (Suppl 2):145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1:848–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, McInnis J, Ross-Sanchez M, Shinnick-Gallagher P, Wiley JL, Johnson KM. (2001) Long-term behavioral and neurodegenerative effects of perinatal phencyclidine administration: implications for schizophrenia. Neuroscience 107:535–550 [DOI] [PubMed] [Google Scholar]