

Abstract
Heparanase is a heparan sulfate degrading endoglycosidase. Previous work has demonstrated that heparanase plays important roles in various biological processes including angiogenesis, wound healing and metastasis. However, the role of heparanase in the post-ischemic brain is not well defined. Transient focal cerebral ischemia in adult mice was induced by ligations of the right middle cerebral artery (MCA) and both common carotid arteries (CCAs). All mice were subjected to bromodeoxyuridine (BrdU) injection and sacrificed at different time points after stroke for immunohistochemical and Western blot analyses. Heparanase expression increased after ischemia in both cell-specific and time-dependent manners. Three to 7 days after stroke, levels of the 50-kD heparanase, basic fibroblast growth factor (FGF-2), and angiopoietin-2 (Ang-2) increased in the peri-infarct region. At early time points, heparanase expression was largely confined to proliferating vascular endothelial cells. At 14 days after ischemia, this expression had shifted to astrocytes in the same region. These data show that cerebral ischemia markedly increases heparanase levels in endothelial cells and then in astrocytes. The unique features of the heparanase upregulation imply that heparanase may play specific roles in the pathological and regenerative processes during the acute and sub-acute/chronic phase in the post-stroke brain.
Keywords: Heparanase, Cerebral ischemia, Endothelial cells, Astrocytes, Angiogenesis, FGF-2, Angiopoietin-2
1. Introduction
In the tissue repair processes, angiogenesis represents a coordinated, multicellular process that requires participation of a variety of growth factors, adhesion receptors, extracellular matrix (ECM) components, and matrix degrading enzymes (Folkman and D'Amore, 1996). Formation and reorganization of new tissues in the wound healing process are profoundly affected by the ECM microenvironment. Heparan sulfate proteoglycan (HSPG) is a major component of the ECM interacts with a variety of other ECM components and plays key roles in maintaining the structural integrity of the ECM (Goldshmidt et al., 2001). In addition to its architectural function, HSPG also provides a scaffold for storage of a range of active molecules, such as growth factors and enzymes. Heparanase, an endo-glucuronidase degrades the heparan sulfate (HS) side chains of HSPGs, is a critical mediator in tumor metastasis and angiogenesis. Heparanase is expressed in nearly all cells, including neutrophils, macrophages, endothelial cells, neurons and astrocytes (Gingis-Velitski et al., 2004; Marchetti et al., 2000; Navarro et al., 2008; Parish et al., 2001; Sasaki et al., 2004). Recent evidence suggests that heparanase modulates the expression of early growth genes such as Egr1 and Egr2 in the brain (Yan et al., 2011). At the early stages of angiogenesis, heparanase is required for the degradation of the physical barrier formed by the ECM and basement membranes (BMs). Two major pro-angiogenic molecules, basic fibroblast growth factor (bFGF or FGF-2) and vascular endothelial growth factor (VEGF), are functionally linked to the heparin-binding growth factor family (Bernfield et al., 1999; Gingis-Velitski et al., 2004; Iozzo and Murdoch, 1996). Their coordination of these molecules with heparanase in angiogenesis has been recognized recently in carcinoma and metastasis (Ilan et al., 2006; Pang and Poon, 2007), but little is known about the role of heparanase after ischemic stroke. In fact, there has been only one report of direct measurement of heparanase expression in astrocytes after a transient ischemia in the rat’s brain. This study reported a marked upregulation of heparanase in astrocytes in the peri-infarct region during the subacute phase of ischemia (from 3 to 7 days post-reperfusion) (Takahashi et al., 2007).
Astrocytes are multifunctional cells that interact with both neurons and glial cells in signaling and metabolic functions, providing a structural and nutrimental environment in the brain (Dienel and Hertz, 2005). Injury-activated astrocytes are frequently found in areas surrounding tissue lesions. An increased heparanase transcript level correlates in the hippocampus with enhanced angiogenesis following repeated hypoxia exposures (Navarro et al., 2008). So far, the information on post-stroke heparanase expression in the ischemic brain is very limited. To fill the information gap and to test the idea that heparanase expression might be associated with post-stroke pathology and regenerative activities, we examined the heparanase expression in different brain regions/cells, the time course of heparanase expression and the expression of a few key trophic factors after a focal ischemia in mice. We primarily examined the 50-kD heparanase because its higher enzymatic activity compared to the 65-kD latent protein (Gingis-Velitski et al., 2004). Our data illustrate the spatial and temporal expression of heparanase in the brain after ischemia and support the idea that heparanase may participate in the ischemic pathology and repairing process in the acute/subacute and chronic phases of ischemia.
2. Results
Ischemia-induced brain infarction and heparanase expression
Focal cerebral ischemia in mice caused a restricted infarction in the ipsilateral cortex as shown in our previous reports (Li et al., 2007). Western blotting showed that heparanase expression in the peri-infarct (penumbra) region was increased one day after ischemia and remained elevated for at least the next 13 days (Fig. 1). Increased FGF-2 and VEGF levels were identified 14 days after ischemia, while the level of Ang-2 was markedly increased beginning at day 3 after ischemia (Fig. 1). Meanwhile, we did not detect significant changes in the angiopoietin receptor Tie-2 level and there was no significant change in heparanase expression in the contralateral side of the cortex (data not shown).
Figure 1. Heparanase expression in the ischemic brain.
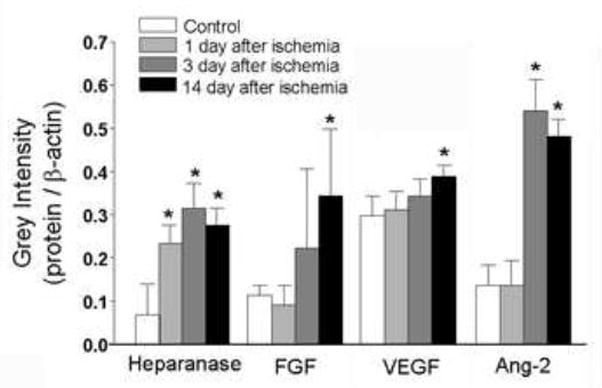
Western blotting analysis showed time-dependent changes of heparanase, VEGF, FGF-2 and Ang-2 1 to 14 days after ischemia. The bar graph summarizes increased factors in the post-ischemic cortex. Mean ± SD; n ≥ 3 assays for each test; * P<0.05 vs. controls.
Heparanase expression and cell death
We used TUNEL staining to detect DNA breaks and cell death in the ischemic cortex. Three to 7 days after ischemia, TUNEL-positive cells were detected in the ischemic core and penumbra. Noticeable heparanase staining was identified in the penumbra, but not in the ischemic core region or the contralateral cortex (Fig. 2A and 3A). In immunohistochemical images, it was clear that there was little overlap between TUNEL and heparanase staining at this time, suggesting that heparanase-positive cells were not injured or dead cells (Fig. 2).
Figure 2. Expression of heparanase in non-TUNEL-positive cells.
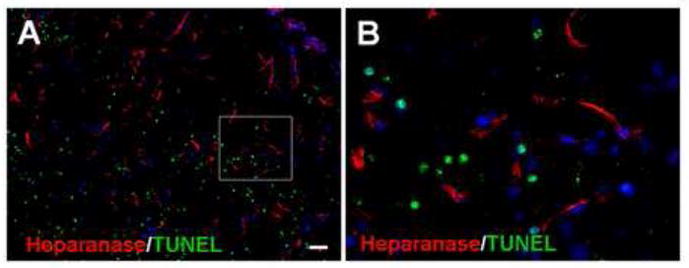
Immunostaining revealed heparanase expression (red) in the peri-infarct region 7 days after ischemia. TUNEL staining (green) was applied to detect injured and dead cells. The double staining images in A and B show that there was little overlap between heparanase and TUNEL staining, suggesting that heparanase-positive cells were not injured or dead cells. Blue is nuclei staining with Hoechst 33258. Bar: A=50μm, B=10μm.
Figure 3. Expression of heparanase in vasculature cells.
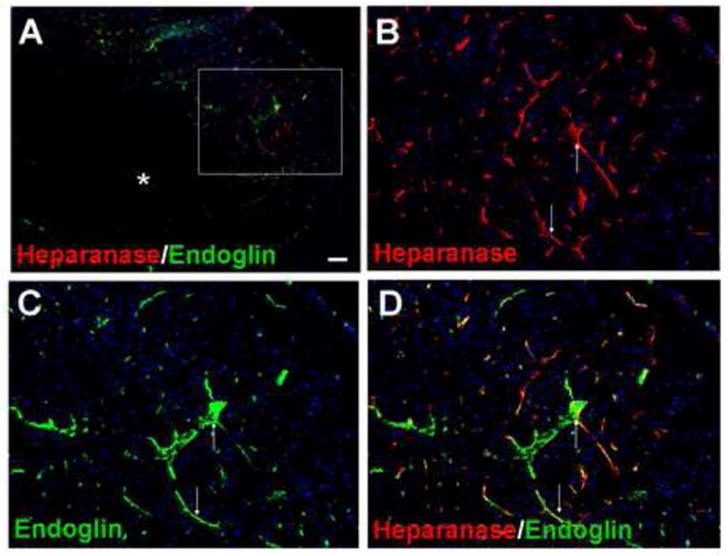
Image A shows heparanase and endoglin staining in the ischemic core (astral) and penumbra regions (up left and the frame area) 7 days after ischemia. Endoglin is a TGF- binding protein found in endothelial cells. B to D images show heparanase staining (red, B), endoglin staining (green, C) and overlay images (D), respectively. The expression of heparanase in many endoglin-positive cells supports the expression of heparanase in vasculature endothelial cells. Blue is nuclei staining with Hoechst 33258. Arrows: examples of overlapped labeling. Bar: A=100μm; B, C and D=50 μm.
Heparanase expression in blood vessels
In the penumbra, strong immunoreactivity of heparanase was observed 7 days after ischemia. Heparanase immunosignals co-localized with endoglin, an endothelial cell proliferation marker (Fig. 3). Consistent with the endoglin staining, the proliferation marker BrdU often co-localized with heparanase labeling (Fig. 4). We also found similar colocalization of heparanase with VEGF expression (Fig 5).
Figure 4. Expression of heparanase in proliferating cells.

Double fluorescent staining of heparanase and BrdU 7 days after ischemia. In the penumbra region, heparanase (green, A), BrdU (red, B) and overlay images show the overlapping of the double staining, suggesting that much of heparanase expression is mostly found in proliferating cells. Most heparanase staining shows a vessel-like morphology, while some BrdU-positive cells may be glial cells. Blue is nuclei staining with Hoechst 33258. Arrow: examples of overlaps. Bar=10μm.
Figure 5. Heparanase expression in VEGF-positive cells.
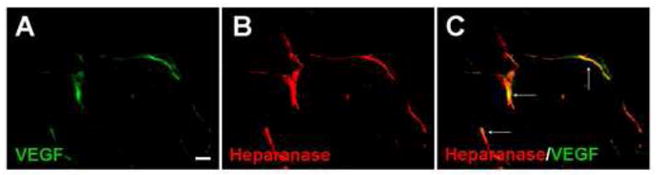
Heparanase and VEGF double staining was performed in the penumbra 7days after ischemia. VEGF (green, A) and heparanase (red, B) were seen in vessel-like cells. Image C shows overlapping of VEGF and heparanase expression (arrows). Bar=10μm.
Heparanase expression in neurons and astrocytes
Some heparanase expression was identified in NeuN-positive cells in the penumbra, consistent with a previous report that neurons express heparanase (Fig. 6A and 6B) (Navarro et al., 2008). Moreover, expression of high heparanase levels was observed in GFAP-positive astrocytes 14 days after ischemia (Fig. 6C to 6E). Interestingly, much of the heparanase expression in endoglin-positive cells had disappeared 14 days after stroke (Figs 6F to 6H).
Figure 6. Heparanase expression 14 days after ischemia.
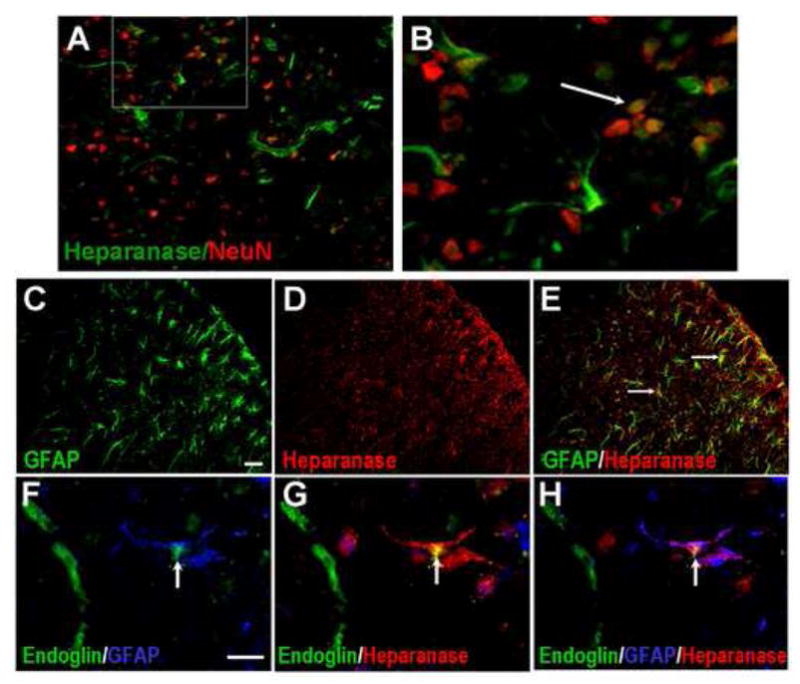
Heparanase expression in the penumbra region was examined 14 days after ischemia. A and B demonstrate heparanase expression in some NeuN positive cells. B is an enlarged image from the frame in A. The arrow points to a heparanase/NeuN double positive cells. C to E show GFAP positive astrocytes (green, C), heparanase (red) and an overlay image (E). Arrows point to some GFAP/heparanase positive cells. Many astrocytes showed heparanase expression at this time. On the other hand, F to H show that although GFAP/heparanase double stained cells were identified (arrows), endoglin-positive endothelial cells were no longer heparanase positive. Bar: C-E=50 μm, F-H=10μm.
3. Discussion
The present investigation demonstrates a time-dependent and cell-type specific upregulation of the active form (50-kD) of heparanase in the post-ischemic brain of mice. We observed a substantial increase in heparanase protein expression in the penumbra region as early as one day after ischemia. This early increase in heparanase correlated with increased angiogenic factors including Ang-2, VEGF, and FGF-2. Much of the heparanase detected was found in proliferating endothelial cells, consistent with the established role of heparanase in angiogenesis. Expression of heparanase, however, was shifted to astrocytes at later time points, which might suggest a delayed role of heparanase in tissue repair in the post-ischemic brain.
Hypoxia/ischemia-induced brain angiogenesis and pathological angiogenesis induced by tumors are thought to be regulated by similar mechanisms, involving increased expression of VEGF and heparanase (Bernaudin et al., 2002; Greenberg and Jin, 2005). In the present study, heparanase upregulation appeared to be faster and more drastic than VEGF expression. On the other hand, VEGF in the control brains already existed at a high basal level. In addition, increased levels of FGF-2 and Ang-2 were consistent with angiogenic activities. Ang-2 is a ligand of the Tie-2 receptor (Holash et al., 1999; Takahashi et al., 2007), which can initiate vessel growth in vitro and in vivo (Adamis et al., 1996; Aiello et al., 1995). Ang-2 alone can promote in vivo neovascularization (Asahara et al., 1998). Although we did not see an increase in Tie-2 expression, the increased Ang-2 level should facilitate post-ischemic angiogenesis. Supporting a role for heparanase in hypoxia-induced angiogenesis, increased heparanase transcript levels correlate in the hippocampus with enhanced angiogenesis following repeated hypoxia exposures (Navarro et al., 2008). Moreover, transgenic mice overexpressing heparanase had increased arterial thickness, cellular density, and mechanical compliance. The extent of heparanase expression within the neointima strongly correlated with the neointimal thickness following injury (Baker et al., 2009). It is likely that heparanase plays a role in the regulation of arterial structure, mechanics, and repair (Baker et al., 2009). Consistent with these observations, we demonstrate that many heparanase-expressing cells are BrdU-positive proliferating cells.
Heparanase activity has long been correlated with pathologies associated with cell invasion such as cancer metastasis, angiogenesis, and inflammation (Gingis-Velitski et al., 2004; Parish et al., 2001). Heparanase protein is expressed in the endothelium of sprouting capillaries and small vessels but not in mature quiescent blood vessels (Ilan et al., 2006). In the present investigation, an early increase in heparanase is mostly isolated to endoglin-positive and BrdU-positive cells. Like heparanase, endoglin is expressed in high levels in endothelial cells during angiogenesis in tumors and inflammation, but has lower expression in normal blood vessels (Kumar et al., 1996). The endoglin expression is associated with transforming growth factor beta (TGFβ) signaling and alterations in vascular structure (Arthur et al., 2000). We observed substantial overlaps of heparanase and endoglin staining, which is consistent with previous reports that these two factors co-localize in angiogenic cells (Kumar et al., 1996). The proposition that heparanase expression is associated with new vessel growth is also supported by the co-staining of BrdU and VEGF in these cells.
Restoration of cerebral microvascular circulation is important for functional recovery after stroke. Stroke patients with a higher density of blood vessels appear to have reduced morbidity and longer survival (Krupinski et al., 1994), as well as increased cerebral blood flow and metabolism in tissue surrounding focal brain infarcts (Cramer et al., 1997; Weiller et al., 1993). Therefore, it is expected that heparanase may promote brain regeneration after cerebral ischemia by stimulating angiogenesis during recovery phase. In tumor metastasis, on the other hand, heparanase may contribute to the destruction of both the ECM and BM of the blood brain barrier (BBB). This activity is associated with tumor cell invasion into distant organs and brain tissues (Marchetti et al., 2003; Sugahara et al., 2002; Vlodavsky and Friedmann, 2001). This effect of heparanase has been extensively studied in tumor cells but no information is available after ischemic stroke. If heparanase contributes to the deterioration of the ECM and BM of non-tumor cells during the acute phase of ischemia, the increased heparanase activity at this time can be detrimental to the brain. Although we did not seen DNA fragmentation in heparanase-positive cells three days after ischemia, this cannot rule out the possibility that the extracellular matrix and the membrane of endothelial cells are subjected to the catalytic effect of this enzyme, resulting in membrane deterioration of these cells. Confirmation of the potential pathogenic role of heparanase in BBB breakdown and consequent brain edema is an important topic and requires specific investigations.
A novel observation in this study is that the heparanase expression shows a cell type and time-dependent shift after ischemia. Heparanase protein first increases in vascular endothelial cells, but is up-regulated in astrocytes at later time points. The shift in heparanase expression from endothelial cells to astrocytes supports the report that heparanase expression may extend to astrocytes under pathological conditions as suggested during invasion of melanoma cells within the brain parenchyma (Marchetti et al., 2000). It is also consistent with the previous report in rat brain that heparanase upregulation is seen in activated astrocytes during the sub-acute phase of ischemia (from 3 to 7 days post-reperfusion) (Takahashi et al., 2007). We observed elevated heparanase levels in astrocytes at 14 days after ischemia. Several factors may explain the relatively delayed upregulation of heparanase in our study. Both the use of different ischemic insults (90-min MCA vs. 10-min CCA occlusion plus permanent MCA ligation in our study) and different animal species (rat vs. mouse) may affect the regulation of heparanase.
A previous report illustrated heparanase expression in three brain regions including the neocortex, the hippocampus, and the olfactory bulb of young adult rats (Navarro et al., 2008). The study identified heparanase expression in neurons but not in astrocytes. On the other hand, heparanase transcript was detected in neurons and astrocytes in the spinal cord of adult rats (Zhang et al., 2006). We observed heparanase expression in neurons, astrocytes, and endothelial cells. Previous investigations have suggested that heparanase plays an important role in the developing nervous system and during neural differentiation (Goldshmidt et al., 2001; Moretti et al., 2006; Navarro et al., 2008). In the injured spinal cord, pericellular heparanase may become enzymatically active and contribute to the migration of astrocytes to the site of injury (Zhang et al., 2006). Astrocytes are multifunctional cells that interact with neurons in signaling and metabolic functions, and their resistance to pathophysiological conditions can help restrict loss of tissue after an ischemic event. Reactive astrocytes, mediated by FGF-2, may promote functional recovery after cerebral ischemia (Briones et al., 2006). Our observation of the heparanase expression pattern in different cells may suggest a delayed role of heparanase in promoting tissue repair or wound healing in the post-ischemic brain.
The present investigation demonstrates a significant up-regulation of heparanase and several other angiogenic factors in the post-ischemic brain. The loss-of-function experiment was not tested in this investigation because thus far, there is no selective heparanase inhibitor available. The key role for heparanase in post-stroke angiogenesis should be confirmed once the selective blocker is available and/or using the heparanase knockout mice. Heparanase knockout (Hpse-KO) mice are fertile, exhibited a normal life span and do not have obvious pathological alterations (Zcharia et al., 2009). The lack of major abnormalities is attributed to a marked elevation in the expression of matrix metalloproteinases such as MMP2 and MMP14 (Zcharia et al., 2009). Therefore, a role for heparanase in angiogenesis may not be clearly defined using the Hpse-KO mouse because of the concurrent alterations in MMPs. A different approach to knock down heparanase may be a useful strategy to verify the post-stroke function of heparanase.
4. Materials and Methods
Animals
Young adult male SV129 mice (2–3 months old; 25–30 g; Bar Harbor, ME, USA) were used in this study. The animals were maintained at a room temperature of 22°C with a 12-hr light/dark cycle in the Animal Facility at Medical University of South Carolina, Charleston, SC. All procedures in the investigation were approved by the Institutional Animal Care and Use Committee (IACUC) and met with the standard of NIH guidelines.
Transient cerebral ischemia in mice
Focal ischemia confined to the cerebral cortex of mice was induced by ligation of the right middle cerebral artery (MCA) and temporary (10 min) occlusion of both common carotid arteries (CCAs). The mice were anesthetized by intraperitoneal (i.p.) injection of chloral hydrate 400 mg/kg. Anesthesia and surgical procedures were performed in accordance with institutional guidelines. Reperfusion of the CCAs was achieved by releasing both CCA clips after the ischemia period. Rectal temperature was monitored and maintained at 37 ° 0.3°C during ischemia and for up to 1 hr after ischemia via an electronic temperature controller linked to a heating pad and a heating lamp (Cole-Parmer, Vernon Hills, IL). Free access to food and water was allowed after recovery from anesthesia. Mice were subjected to injections of bromodeoxyuridine (BrdU; 50 mg/kg/day, i.p.), starting from three days after ischemia. Animals were sacrificed 1, 3, 7 and 14 days after the onset of ischemia for further examinations.
Immunofluorescent imaging of brain sections
The animals were anaesthetized with an intraperitoneal injection of chloral hydrate (400 mg/kg) and sacrificed for brain sectioning. Fresh frozen brains were sliced into coronal sections 14 μm thick using a cryostat vibratome (Ultapro 5000, St. Louis, MO) and fixed in 10% buffered formalin phosphate (Fisher Scientific, Pittsburgh, PA). Brain sections were then processed in an ethanol:acetic acid (2:1) solution at −20 °C for 10 min, washed 3 times in PBS solution at pH7.4, and incubated in 0.2% Triton X-100 for 5 min. Sections were blocked in 1% fish gelatin (Sigma, St. Louis, MO) in PBS for 1 hour at room temperature and incubated with antibodies including mouse anti-NeuN, rabbit anti-human heparanase, anti-GFAP, and anti-endoglin overnight at 4 °C (Chemicon international Inc., Temecula, CA), respectively. Some sections were also incubated overnight with rat anti-BrdU antibody (Abcam, Cambridge, MA). Anti-mouse Cy3 was used as secondary antibody. Results were visualized by a fluorescence microscopy (Olympus America Inc., New York, NY).
Terminal deoxynucleotidyltransferase (TdT)-mediated dUTP-biotin nick end-labeling (TUNEL)
TUNEL staining was performed using a commercial kit (DeadEndTM Fluorometric TUNEL system, Promega, Madison, WI) to assess cell death. Fresh frozen brain sections were fixed with 10% formalin for 10 min then in Ethanol:Acetone (2:1) solution for 5 min and permeabilization in 0.2% Triton-X 100 solution. The sections were then placed in equilibration buffer for 10 min. Recombinant Terminal deoxynucleotidyl Transferase (rTdT) and nucleotide mixture were added on the slide at 37°C for 60 min in the dark. Reactions were terminated by 2X SSC solution for 15 min. For neuronal cell death, cells were incubated with the NeuN antibody overnight. Anti-mouse Cy3 was used as the secondary antibody. Hoechst 33258 (Molecular Probes, Eugene, OR) was applied to stain cell nuclei. Results were visualized by a fluorescence microscopy.
Western blot analysis
Proteins in brain tissues were extracted using the modified RIPA buffer (50 mM HEPES, pH 7.3, 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM PMSF, and 10 μg/ml aprotinin). The protein concentration of each sample was determined using the bicinchoninic acid assay (Sigma, St. Louis, MO). Proteins from each sample (40 μg) were separated by SDS-PAGE in a Hoefer Mini-Gel system (Amersham Biosciences, Buckinghamshire, UK) and transferred in the Hoefer Transfer Tank to a PVDF membrane (BioRad, Hercules, CA). Membranes were blocked in 4% BSA diluted in Tris-buffered saline containing 0.1% Tween-20 (TBS-T) at room temperature for at least 2 hrs, and incubated with polyclonal rabbit anti-VEGF, anti-heparanase antibody, goat anti-ANG-2 antibody, mouse anti-FGF-2 anti-body (Chemicon International), and rabbit anti-Tie-2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA; Cell Signaling Technology, Danvers, MA), respectively. A mouse β-actin antibody (Sigma, St. Louis, MO) was used for protein loading controls. After primary antibody incubation, membranes were washed with TBS-T and incubated with alkaline phosphatase-conjugated anti-rabbit IgG antibody (Promega) for 2 hrs at room temperature. Finally, membranes were washed with TBS-T followed by three washes with TBS. Signal was detected by the addition of BCIP/NBT solution (Sigma).
Statistics analysis
The Student’s two-tailed t-test was used for comparisons of two experimental groups. Multiple comparisons were analyzed using one-way ANOVA followed by a post-hoc Tukey test for multiple pairwise examinations. Changes were identified as significant if P was less than 0.05. Mean values are reported together with the standard deviation (SD).
Highlights.
Expression of 50-kD heparanase increased in penumbra after a focal cerebral ischemia.
Most heparanase expression was confined to vascular endothelial cells and BrdU-positive cells.
Two weeks after ischemia, heparanase expression shifted to astrocytes.
The ischemia-induced heparanase increase may be associated with post-ischemic angiogenesis and tissue repair.
Acknowledgments
This work was supported by NIH grants NS057255, NS 058710, and NS062097. This work was also supported by the National Natural Science Foundation of China (30772302) and Beijing Natural Science Foundation (7082029), as well as NIH grant C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamis AP, Shima DT, Tolentino MJ, Gragoudas ES, Ferrara N, Folkman J, D'Amore PA, Miller JW. Inhibition of vascular endothelial growth factor prevents retinal ischemia-associated iris neovascularization in a nonhuman primate. Arch Ophthalmol. 1996;114:66–71. doi: 10.1001/archopht.1996.01100130062010. [DOI] [PubMed] [Google Scholar]
- Aiello LP, Pierce EA, Foley ED, Takagi H, Chen H, Riddle L, Ferrara N, King GL, Smith LE. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci U S A. 1995;92:10457–61. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur HM, Ure J, Smith AJ, Renforth G, Wilson DI, Torsney E, Charlton R, Parums DV, Jowett T, Marchuk DA, Burn J, Diamond AG. Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol. 2000;217:42–53. doi: 10.1006/dbio.1999.9534. [DOI] [PubMed] [Google Scholar]
- Asahara T, Chen D, Takahashi T, Fujikawa K, Kearney M, Magner M, Yancopoulos GD, Isner JM. Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ Res. 1998;83:233–40. doi: 10.1161/01.res.83.3.233. [DOI] [PubMed] [Google Scholar]
- Baker AB, Groothuis A, Jonas M, Ettenson DS, Shazly T, Zcharia E, Vlodavsky I, Seifert P, Edelman ER. Heparanase alters arterial structure, mechanics, and repair following endovascular stenting in mice. Circ Res. 2009;104:380–7. doi: 10.1161/CIRCRESAHA.108.180695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernaudin M, Nedelec AS, Divoux D, MacKenzie ET, Petit E, Schumann-Bard P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J Cereb Blood Flow Metab. 2002;22:393–403. doi: 10.1097/00004647-200204000-00003. [DOI] [PubMed] [Google Scholar]
- Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–77. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- Briones TL, Woods J, Wadowska M, Rogozinska M, Nguyen M. Astrocytic changes in the hippocampus and functional recovery after cerebral ischemia are facilitated by rehabilitation training. Behav Brain Res. 2006 doi: 10.1016/j.bbr.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Cramer SC, Nelles G, Benson RR, Kaplan JD, Parker RA, Kwong KK, Kennedy DN, Finklestein SP, Rosen BR. A functional MRI study of subjects recovered from hemiparetic stroke. Stroke. 1997;28:2518–27. doi: 10.1161/01.str.28.12.2518. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Hertz L. Astrocytic contributions to bioenergetics of cerebral ischemia. Glia. 2005;50:362–88. doi: 10.1002/glia.20157. [DOI] [PubMed] [Google Scholar]
- Folkman J, Klagsbrun M, Sasse J, Wadzinski M, Ingber D, Vlodavsky I. A heparin-binding angiogenic protein--basic fibroblast growth factor--is stored within basement membrane. Am J Pathol. 1988;130:393–400. [PMC free article] [PubMed] [Google Scholar]
- Folkman J, D'Amore PA. Blood vessel formation: what is its molecular basis? Cell. 1996;87:1153–5. doi: 10.1016/s0092-8674(00)81810-3. [DOI] [PubMed] [Google Scholar]
- Gingis-Velitski S, Zetser A, Flugelman MY, Vlodavsky I, Ilan N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J Biol Chem. 2004;279:23536–41. doi: 10.1074/jbc.M400554200. [DOI] [PubMed] [Google Scholar]
- Goldshmidt O, Zcharia E, Aingorn H, Guatta-Rangini Z, Atzmon R, Michal I, Pecker I, Mitrani E, Vlodavsky I. Expression pattern and secretion of human and chicken heparanase are determined by their signal peptide sequence. J Biol Chem. 2001;276:29178–87. doi: 10.1074/jbc.M102462200. [DOI] [PubMed] [Google Scholar]
- Greenberg DA, Jin K. From angiogenesis to neuropathology. Nature. 2005;438:954–9. doi: 10.1038/nature04481. [DOI] [PubMed] [Google Scholar]
- Holash J, Wiegand SJ, Yancopoulos GD. New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. Oncogene. 1999;18:5356–62. doi: 10.1038/sj.onc.1203035. [DOI] [PubMed] [Google Scholar]
- Ilan N, Elkin M, Vlodavsky I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int J Biochem Cell Biol. 2006;38:2018–39. doi: 10.1016/j.biocel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Iozzo RV, Murdoch AD. Proteoglycans of the extracellular environment: clues from the gene and protein side offer novel perspectives in molecular diversity and function. FASEB J. 1996;10:598–614. [PubMed] [Google Scholar]
- Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke. 1994;25:1794–8. doi: 10.1161/01.str.25.9.1794. [DOI] [PubMed] [Google Scholar]
- Kumar P, Wang JM, Bernabeu C. CD 105 and angiogenesis. J Pathol. 1996;178:363–6. doi: 10.1002/(SICI)1096-9896(199604)178:4<363::AID-PATH491>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu Z, Keogh CL, Yu SP, Wei L. Erythropoietin-induced neurovascular protection, angiogenesis, and cerebral blood flow restoration after focal ischemia in mice. J Cereb Blood Flow Metab. 2007;27:1043–54. doi: 10.1038/sj.jcbfm.9600417. [DOI] [PubMed] [Google Scholar]
- Marchetti D, Li J, Shen R. Astrocytes contribute to the brain-metastatic specificity of melanoma cells by producing heparanase. Cancer Res. 2000;60:4767–70. [PubMed] [Google Scholar]
- Marchetti D, Denkins Y, Reiland J, Greiter-Wilke A, Galjour J, Murry B, Blust J, Roy M. Brain-metastatic melanoma: A neurotrophic perspective. Pathology & Oncology Research. 2003;9:147–158. doi: 10.1007/BF03033729. [DOI] [PubMed] [Google Scholar]
- Moretti M, Sinnappah-Kang ND, Toller M, Curcio F, Marchetti D. HPSE-1 expression and functionality in differentiating neural cells. J Neurosci Res. 2006;83:694–701. doi: 10.1002/jnr.20753. [DOI] [PubMed] [Google Scholar]
- Navarro FP, Fares RP, Sanchez PE, Nadam J, Georges B, Moulin C, Morales A, Pequignot JM, Bezin L. Brain heparanase expression is up-regulated during postnatal development and hypoxia-induced neovascularization in adult rats. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2007.05116.x. [DOI] [PubMed] [Google Scholar]
- Pang RW, Poon RT. From molecular biology to targeted therapies for hepatocellular carcinoma: the future is now. Oncology. 2007;72(Suppl 1):30–44. doi: 10.1159/000111705. [DOI] [PubMed] [Google Scholar]
- Parish CR, Freeman C, Hulett MD. Heparanase: a key enzyme involved in cell invasion. Biochim Biophys Acta. 2001;1471:M99–108. doi: 10.1016/s0304-419x(01)00017-8. [DOI] [PubMed] [Google Scholar]
- Sasaki N, Higashi N, Taka T, Nakajima M, Irimura T. Cell surface localization of heparanase on macrophages regulates degradation of extracellular matrix heparan sulfate. J Immunol. 2004;172:3830–5. doi: 10.4049/jimmunol.172.6.3830. [DOI] [PubMed] [Google Scholar]
- Sugahara K, Okada Y, Yamada S, Toyoshima M, Dong J, Nakajima M. Structural recognition by recombinant human heparanase that plays critical roles in tumor metastasis - Hierarchical sulfate groups with differential effects and the essential target disulfated trisaccharide sequence. Journal of Biological Chemistry. 2002;277:42488–42495. doi: 10.1074/jbc.M206510200. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Matsumoto H, Kumon Y, Ohnishi T, Freeman C, Imai Y, Tanaka J. Expression of heparanase in nestin-positive reactive astrocytes in ischemic lesions of rat brain after transient middle cerebral artery occlusion. Neurosci Lett. 2007;417:250–4. doi: 10.1016/j.neulet.2007.02.075. [DOI] [PubMed] [Google Scholar]
- Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–7. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiller C, Ramsay SC, Wise RJ, Friston KJ, Frackowiak RS. Individual patterns of functional reorganization in the human cerebral cortex after capsular infarction. Ann Neurol. 1993;33:181–9. doi: 10.1002/ana.410330208. [DOI] [PubMed] [Google Scholar]
- Yan X, Jin S, Li S, Gong F, Zhang D, Zhang X, Li JP. Heparanase modulation of early growth response gene expression. Zoolog Sci. 2011;28:189–94. doi: 10.2108/zsj.28.189. [DOI] [PubMed] [Google Scholar]
- Zcharia E, Jia J, Zhang X, Baraz L, Lindahl U, Peretz T, Vlodavsky I, Li JP. Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS One. 2009;4:e5181. doi: 10.1371/journal.pone.0005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yeung MN, Liu J, Chau CH, Chan YS, Shum DK. Mapping heparanase expression in the spinal cord of adult rats. J Comp Neurol. 2006;494:345–57. doi: 10.1002/cne.20811. [DOI] [PubMed] [Google Scholar]