

Abstract
Non-technical summary
Contraction and relaxation of the heart strongly depend on calcium (Ca2+) stored in the sarcoplasmic reticulum (SR). Ca2+ stored within the SR is determined by the balance between Ca2+ uptake and Ca2+ leak that occurs mainly via Ca2+ release channels, called ryanodine receptors (RyRs). Alterations in the RyR activity can lead to enhanced SR Ca2+ leak and arrhythmias. Ca2+ tightly regulates the RyR activity from both sides of the SR (cytosolic and luminal). In this work, we studied the effects of cytosolic Ca2+ on SR Ca2+ leak in isolated ventricular myocytes. Elevation of cytosolic Ca2+ increased SR Ca2+ leak by a direct activation of RyRs. In intact myocytes, at the end of contraction and at the beginning of the relaxation phase, SR Ca2+ leak remains relatively constant due to the coordinated regulation of RyRs by cytosolic and luminal Ca2+. Thus, this dual regulation of the RyR contributes to the control of the SR Ca2+ content, preventing excessive loss of Ca2+ that could lead to pathological conditions such as cardiac arrhythmia.
Abstract
Sarcoplasmic reticulum (SR) Ca2+ leak determines SR Ca2+ content and, therefore, the amplitude of global Ca2+ transients in ventricular myocytes. However, it remains unresolved to what extent Ca2+ leak can be modulated by cytosolic [Ca2+] ([Ca2+]i). Here, we studied the effects of [Ca2+]i on SR Ca2+ leak in permeabilized rabbit ventricular myocytes. Using confocal microscopy we monitored SR Ca2+ leak as the change in [Ca2+]SR (with Fluo-5N) after complete SERCA inhibition with thapsigargin (10 μm). Increasing [Ca2+]i from 150 to 250 nm significantly increased SR Ca2+ leak over the entire range of [Ca2+]SR. This increase was associated with an augmentation of both Ca2+ spark- and non-spark-mediated Ca2+ leak. Further increasing [Ca2+]i to 350 nm led to rapid [Ca]2+]SR depletion due to the occurrence of Ca2+ waves. The augmentation of SR Ca2+ leak by high [Ca2+]i was insensitive to inhibition of Ca2+–calmodulin-dependent protein kinase II. In contrast, lowering [Ca2+]i to 50 nm markedly decreased SR Ca2+ leak rate and nearly abolished Ca2+ sparks. When the ryanodine receptor (RyR) was completely inhibited with ruthenium red (50 μm), changes in [Ca2+]i between 50 and 350 nm did not produce any significant effect on SR Ca2+ leak, indicating that [Ca2+]i alters SR Ca2+ leak solely by regulating RyR activity. In summary, [Ca2+]i in the range of 50–350 nm has a significant effect on SR Ca2+ leak rate mainly via direct regulation of RyR activity. As RyR activity depends highly on [Ca2+]i and [Ca2+]SR, SR Ca2+ leak remains relatively constant during the declining phase of the Ca2+ transient when [Ca2+]SR and [Ca2+]i change in opposite directions.
Introduction
During systole, Ca2+ influx via L-type Ca2+ channels activates ryanodine receptors (RyRs) causing global Ca2+ release that initiates contraction in cardiac muscle. This process is known as Ca2+-induced Ca2+ release (CICR; Fabiato, 1983). After CICR termination, a large portion of cytosolic Ca2+ is pumped back into the sarcoplasmic reticulum (SR) by the Ca2+-ATPase (SERCA) leading to cardiac muscle relaxation. However, RyRs are not continuously closed during diastole and spontaneous openings of RyRs generate substantial SR Ca2+ leak. By counterbalancing SR Ca2+ uptake, diastolic SR Ca2+ leak plays an important role in setting and maintaining the appropriate SR Ca2+ load in the healthy heart (Shannon et al. 2002; Zima et al. 2010). However, during the development of heart failure (HF) RyRs undergo post-translational modifications that ultimately lead to an increase in SR Ca2+ leak (Ai et al. 2005; Belevych et al. 2007; Zima et al. 2010). This excessive Ca2+ leak has been implicated in reduction of contractile force as well as in cardiac arrhythmias (George, 2008). Although the precise mechanisms of RyR modification during HF remain highly controversial (Bers et al. 2003; Lehnart & Marks, 2007), it has been shown that abnormal phosphorylation of RyR either by protein kinase A (PKA; Marx et al. 2000) or Ca2+–calmodulin-dependent kinase type II (CaMKII; Ai et al. 2005) is involved in the augmentation of SR Ca2+ leak. Additionally, oxidation of thiol groups of the RyR can contribute to an alteration of SR Ca2+ leak in HF (Terentyev et al. 2008). Failing cardiac myocytes also exhibit a slower decay of cytosolic Ca2+ transient during excitation–contraction coupling (ECC) (O'Rourke et al. 1999; Jiang et al. 2002) due, presumably, to down-regulation of SERCA activity and a subsequent impairment of SR Ca2+ reuptake (Pieske et al. 1995; Pogwizd et al. 1999). Additionally, it has been reported that in failing myocytes cytosolic [Na+] is significantly increased. This can contribute to further slowing of Ca2+ extrusion by Na+–Ca2+ exchange (NCX) (Despa et al. 2002) leading to an increase in cytosolic [Ca2+] ([Ca2+]i). High diastolic [Ca2+]i by itself can increase RyR activity and SR Ca2+ leak; however, the modulation of SR Ca2+ leak by [Ca2+]i has not been characterized in detail.
The RyR (type 2) mediates most of diastolic SR Ca2+ leak in ventricular myocytes (Neary et al. 2002; Shannon et al. 2002; Zima et al. 2010). The majority of RyRs localize in the junctional SR where they form clusters of 10–200 channels (Franzini-Armstrong et al. 1999). Each of these subcellular microdomains constitutes an SR Ca2+ release unit (Cheng & Lederer, 2008). The simultaneous activation of RyRs within the release unit generates a locally restricted increase in [Ca2+]i, or Ca2+ spark (Cheng et al. 1993; Lopez-Lopez et al. 1995). After the discovery of Ca2+ sparks, it has been suggested that the entire diastolic SR Ca2+ leak can be explained solely by these spontaneous release events (Cheng et al. 1993; Bassani & Bers, 1995). However, this concept has been recently challenged. It has been reported that in ventricular myocytes a significant portion of SR Ca2+ leak occurs as undetectable openings of single RyRs or spark-independent Ca2+ leak (Santiago et al. 2010; Zima et al. 2010; Brochet et al. 2011; Porta et al. 2011). RyR-mediated SR Ca2+ leak strongly depends on intra-SR free Ca2+ ([Ca2+]SR). At low [Ca2+]SR, SR Ca2+ leak is mainly mediated by spark-independent pathways. At higher [Ca2+]SR, however, Ca2+ sparks become a significant contributor to SR Ca2+ leak (Zima et al. 2010). In certain pathological conditions associated with SR Ca2+ overload, SR Ca2+ leak becomes exacerbated. The Ca2+ released during a spontaneous spark diffuses to neighbouring release junctions, triggers CICR and generates arrhythmogenic spontaneous Ca2+ waves (Cheng et al. 1993; Diaz et al. 1997). While the importance of [Ca2+]SR in the regulation of SR Ca2+ leak is well established, it is less clear to what extent [Ca2+]i affects the RyR-mediated Ca2+ leak (Dibb & Eisner, 2010). [Ca2+]i can regulate SR Ca2+ leak directly by binding to the high-affinity activation site on the cytosolic side of the RyR (Rousseau et al. 1986; Meissner & Henderson, 1987). Additionally, [Ca2+]i can affect SR Ca2+ leak via activation of CaMKII with subsequent phosphorylation of the RyR (Guo et al. 2006; van Oort et al. 2010).
In our previous study, we investigated the effect of [Ca2+]SR on SR Ca2+ leak (Zima et al. 2010) in conditions where [Ca2+]i was held constant. In physiological conditions, however, [Ca2+]i changes dynamically during the cardiac cycle. Therefore, it is important to understand to what extent [Ca2+]i affects SR Ca2+ leak in ventricular myocytes. In this study, we used a novel approach to directly measure SR Ca2+ leak as changes of [Ca2+]SR after complete SERCA inhibition. To measure [Ca2+]SR, we used the low-affinity Ca2+ indicator Fluo-5N entrapped within the SR. SR Ca2+ leak was studied after sarcolemma permeabilization. The advantage of this approach is that an experimental solution with known [Ca2+] can be easily introduced into the cytosol. The experimental solution also contained the high-affinity Ca2+ indicator Rhod-2 to measure Ca2+ spark and wave properties. We found that in rabbit ventricular myocytes [Ca2+]i has a pronounced effect on SR Ca2+ leak. The augmentation of SR Ca2+ leak by [Ca2+]i was attributed to an increase in both spark- and non-spark-mediated Ca2+ leak. The effect of [Ca2+]i (within the range of 50–350 nm) on SR Ca2+ leak was mainly mediated by a direct regulation of RyR activity, but not by CaMKII-dependent phosphorylation of the RyR. Part of this work has been published in abstract form (Bovo et al. 2010).
Methods
Myocyte isolation
Ventricular myocytes were isolated from New Zealand White rabbits (18 animals, 2–2.5 kg; Myrtle's Rabbitry, Thompsons Station, TN, USA). The procedure of cell isolation was approved by the Institutional Animal Care and Use Committee, and complies with US and UK regulations on animal experimentation (Drummond, 2009). Adult rabbits were anaesthetized with sodium pentobarbital (50 mg kg-1 i.v.). Following thoracotomy, hearts were quickly excised, mounted on a Langendorff apparatus and retrogradely perfused with Liberase (Roche Applied Science, Indianapolis, IN, USA) Blendzyme (Roche Applied Science, Indianapolis, IN, USA)-containing solution at 37°C according to the procedure described previously (Domeier et al. 2009). Chemicals and reagents were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise stated. All experiments were performed at room temperature (20–24°C).
Confocal microscopy
To record [Ca2+]SR and [Ca2+]i we used the low-affinity Ca2+ indicator Fluo-5N and the high-affinity Ca2+ indicator Rhod-2, respectively (both indicators were obtained from Molecular Probes/Invitrogen, Carlsbad, CA, USA). To load the SR with Ca2+ indicator, myocytes were incubated with 5 μm Fluo-5N-AM for 2.5 h at 37°C as described previously (Zima et al. 2008b; Domeier et al. 2009).
For premeabilized cell experiments, Fluo-5N-AM-loaded myocytes were permeabilized with 0.005% saponin (Zima et al. 2008a). The experimental solution containing Rhod-2 tripotassium salt (40 μm) was composed of (in mm): potassium aspartate 100; KCl 15; KH2PO4 5; MgATP 5; EGTA 0.35; CaCl2 0.067; MgCl2 0.75; phosphocreatine 10; Hepes 10; plus creatine phosphokinase 5 U ml-1; dextran (MW: 40,000) 4%, and pH 7.2 (KOH). Free [Ca2+] and [Mg2+] of this solution were 50 nm and 1 mm, respectively. Free [Ca2+] in the experimental solution was adjusted to different levels (i.e. 150, 250 and 350 nm) by adding appropriate amounts of CaCl2 (calculated using WinMAXC 2.05, Stanford University, CA, USA). In the set of experiments when effects of increased cytosolic Ca2+ buffer capacity on Ca2+ sparks and SR Ca2+ leak were studied, the fast Ca2+ buffer BAPTA (0.7 mm) was added to the experimental solution. Free [Ca2+] in this solution was kept the same as the control solution (verified with a Ca2+-sensitive electrode; Orion Research Inc.).
For intact cell experiments, Fluo-5N-AM-loaded cells were incubated at room temperature with 10 μm Rhod-2-AM for 15 min in Tyrode solution (in mm: NaCl 140; KCl 4; CaCl2 2; MgCl2 1; glucose 10; Hepes 10; pH 7.4), followed by a 20 min wash. Action potentials were induced by electrical field stimulation using a pair of platinum electrodes, which were connected to a Grass stimulator (Astro-Med. Inc., USA) set at a voltage ∼50% above the threshold for contraction. To avoid motion artifacts, the scan line was positioned along the short axis (transversal scan) in the central region of the cell where cell motion is minimal during contraction. Stimulation frequency was 0.5 Hz. Changes in [Ca2+]SR were calculated by the formula (Cannell et al. 1994): [Ca2+]SR = Kd×R/(Kd/[Ca2+]SR,diast–R+ 1), where R was the normalized Fluo-5N fluorescence (R = [F–Fmin]/[F0–Fmin]); F0 and Fmin were the fluorescence level at rest and after depletion of the SR with caffeine, respectively; Kd (Fluo-5N Ca2+ dissociation constant) was 390 μm based on in situ calibrations (Zima et al. 2010), and [Ca2+]SR,diast (diastolic [Ca2+]SR at 0.5 Hz) was 900 μm (Shannon et al. 2003). Changes in [Ca2+]i were calculated according to a similar formula (Cannell et al. 1994). Kd for Rhod-2 was 1.3 μm (calibrated in situ) and [Ca2+]i,diast (diastolic [Ca2+]i at 0.5 Hz) was 100 nm.
Changes in [Ca2+]i and [Ca2+]SR were measured with laser scanning confocal microscopy (Radiance 2000 MP, Bio-Rad, UK or LSM 410, Zeiss, Germany) equipped with a ×40 oil-immersion objective lens (NA = 1.3). Fluo-5N was excited with the 488 nm line of an argon ion laser and fluorescence was measured at 515 ± 15 nm. Rhod-2 was excited with the 543 nm line of a He–Ne laser and fluorescence was measured at wavelengths >600 nm. In experiments when Ca2+ sparks or global cytosolic Ca2+ transients were recorded simultaneously with [Ca2+]SR, images were acquired in line-scan mode (3 ms per scan; pixel size 0.12 μm). When [Ca2+]SR was recorded alone (only Fluo-5N signal), images were collected in 2-D mode (pixel size 0.2 μm) every 15 s.
Measurements of SR Ca2+ leak
SR Ca2+ leak as a function of [Ca2+]SR was measured in permeabilized myocytes according to the protocol described previously (Zima et al. 2010). Briefly, Fluo-5N was excited with minimum laser energy of an argon ion laser (to minimize dye photobleaching). To improve the signal-to-noise ratio of the low-intensity Fluo-5N signal, fluorescence was collected with an open pinhole and averaged over the entire cellular width of a line-scan or 2-D image. At the end of each experiment, minimum (Fmin) and maximum (Fmax) Fluo-5N fluorescence were estimated as we described previously (Zima et al. 2010). Fmin was measured after depletion of the SR with 10 mm caffeine in the presence of 5 mm EGTA. Fmax was measured following an increase of [Ca2+] to 10 mm in the presence of caffeine. Caffeine keeps RyRs open allowing [Ca2+] equilibration across the SR membrane (Shannon et al. 2003). The Fluo-5N signal was converted to [Ca2+] using the formula: [Ca2+]SR = Kd× (F–Fmin)/(Fmax–F), where Kd was 390 μm. SR Ca2+ leak was measured as the changes of total [Ca2+]SR ([Ca2+]SRT) over time (d[Ca2+]SRT/dt) after complete SERCA inhibition with thapsigargin (TG). [Ca2+]SRT was calculated as: [Ca2+]SRT = Bmax/(1+Kd/[Ca2+]SR) +[Ca2+]SR; where Bmax (the total concentration of SR Ca buffer) and Kd were 2700 μm and 630 μm, respectively (Shannon et al. 2000).The rate of SR Ca2+ leak (d[Ca2+]SRT/dt) was plotted as a function of [Ca2+]SR for each time point (15 s) during [Ca2+]SR decline.
Measurements of Ca2+ sparks and waves
Ca2+ sparks were detected and analysed using SparkMaster (Picht et al. 2007). To exclude false-positive events, the threshold criterion for spark detection was 3.8. At this threshold no events were detected when SR Ca2+ was emptied after simultaneous application of caffeine (10 mm) and TG (10 μm). Analysis of Ca2+ sparks included spark frequency (sparks s−1 (100 μm)−1), amplitude (ΔF/F0), full duration at half-maximal amplitude (FDHM; ms) and full width at half-maximal amplitude (FWHM; μm). F0 is the initial fluorescence recorded under steady-state conditions and ΔF = F–F0. Ca2+ waves were measured in line-scan mode and analysed in terms of amplitude, frequency and propagation velocity.
Statistics
Data are presented as mean ± SEM of n measured cells. Statistical comparisons between groups were performed with Student's t test. Differences were considered statistically significant at P < 0.05.
Results
The effect of cytosolic [Ca2+] on SR Ca2+ leak
Effects of different [Ca2+]i on SR Ca2+ leak were studied in permeabilized rabbit ventricular myocytes. SR Ca2+ leak was measured as the rate of [Ca2+]SR decline after complete SERCA inhibition with thapsigargin (TG). Figure 1A shows representative examples of [Ca2+]SR recordings in control conditions and after application of TG (10 μm). The recordings were made in different cells at [Ca2+]i of 50, 150, 250 and 350 nm. An increase of [Ca2+]i (applied at t = 0) augmented the initial [Ca2+]SR (measured before TG application). On average, initial [Ca2+]SR in the presence of 50, 150, 250 and 350 nm[Ca2+]i were 759 ± 28 μm (n = 11), 765 ± 25 μm (n = 21; NS compared to 50 nm); 828 ± 38 μm (n = 16; P > 0.05 compared to 50 nm) and 830 ± 44 μm (n = 8; P > 0.05 compared to 50 nm), respectively. These results suggest that higher [Ca2+]i has a more significant effect on SERCA-mediated Ca2+ uptake than on SR Ca2+ leak. At 350 nm[Ca2+]i, initial [Ca2+]SR frequently dropped below basal level as a result of the occurrence of Ca2+ waves.
Figure 1. SR Ca2+ leak at different [Ca2+]; values.
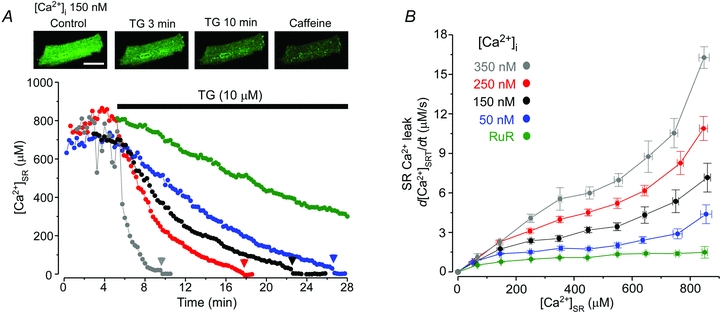
A, changes of [Ca2+]SR after inhibition of SERCA with thapsigargin (TG; 10 μm). Top, images of myocyte loaded with Fluo-5N in control conditions, after application of TG (10 μm; 3 and 10 min) and caffeine (10 mm). Calibration bar corresponds to 20 μm. Bottom, myocytes were exposed to different [Ca2+]i values: 50 nm, blue; 150 nm, black; 250 nm, red; 350 nm, grey; and 150 nm in the presence of the RyR inhibitor Ruthenium Red (RuR; 50 μm), green. Since RyR inhibition with RuR significantly increases initial [Ca2+]SR (Zima et al. 2010), for presentation purposes [Ca2+]SR decline in the presence of RuR is shown from [Ca2+]SR = 800 μm to make it comparable with the other recordings. Application of 10 mm caffeine at the end of the experiment (arrowheads) indicates complete depletion of the SR. The recordings were made from different cells. B, relationships between SR Ca2+ leak rate and [Ca2+]SR measured at different [Ca2+]i values. SR Ca2+ leak was measured after inhibition of SERCA pump with TG.
After SERCA inhibition with TG, [Ca2+]SR gradually declined until full depletion (verified as a lack of further [Ca2+]SR depletion after caffeine application; arrowheads in Fig. 1A). [Ca2+]SR after SERCA inhibition was converted to total [Ca2+]SR ([Ca2+]SRT; for details see Methods). SR Ca2+ leak rate, which was measured as changes of [Ca2+]SRT over time (d[Ca2+]SRT/dt), was plotted against the corresponding free [Ca2+]SR to obtain the relationship between SR Ca2+ leak rate and [Ca2+]SR. The leak–load relationships measured in the presence of different [Ca2+]i values are shown in Fig. 1B. SR Ca2+ leak significantly increased over the entire range of [Ca2+]SR with increasing [Ca2+]i. To test whether [Ca2+]i affected SR Ca2+ leak by activating RyRs, we studied effects of different [Ca2+]i values on SR Ca2+ leak after complete RyR inhibition with ruthenium red (RuR). Figure 1A (green symbols) shows an example of [Ca2+]SR decline in the presence of RuR (50 μm) and 150 nm[Ca2+]i. RuR significantly decreased SR Ca2+ leak, but did not prevent it (Fig. 1B). When RuR was applied at different [Ca2+]i values, the rate of the RuR-insensitive Ca2+ leak remained the same (data not shown). Thus, these results indicate that [Ca2+]i affects SR Ca2+ leak solely by modulating RyR activity.
Effects of cytosolic [Ca2+] on Ca2+ sparks and Ca2+ waves
For all [Ca2+]i values studied here, SR Ca2+ leak increased as a function of [Ca2+]SR, with a particularly steep increase at higher [Ca2+]SR (Fig. 1B). We have shown previously that the increased leak rate at higher [Ca2+]SR is attributed to higher Ca2+ spark frequency (Zima et al. 2010). Here, we studied the effects of [Ca2+]i on Ca2+ spark properties. Since [Ca2+]SR increased as a function of [Ca2+]i (Fig. 1A), we analysed the effect of changing [Ca2+]i between 50 and 250 nm in a subset of cells that had similar [Ca2+]SR. Figure 2A shows representative line-scan images of SR Ca2+ release events recorded at different [Ca2+]i values. The increase of [Ca2+]i from 50 to 250 nm had a pronounced effect on spark frequency (Fig. 2B), which increased 5-fold. Ca2+ spark amplitude decreased (Fig. 2C), whereas spark width (Fig. 2D) and duration (Fig. 2E) only slightly increased at higher [Ca2+]i. Increasing [Ca2+]i to 350 nm produced spontaneous Ca2+ waves that propagated through the cell at a constant frequency of 0.9 ± 0.2 Hz (n = 6; Fig. 2A, rightmost image). These data indicate that [Ca2+]i affects SR Ca2+ release and, therefore, spark-mediated SR Ca2+ leak. This occurs mainly via recruitment of the SR Ca2+ release units (spark frequency), but not via a profound alteration of release unit properties (spark width and duration).
Figure 2. Effects of [Ca2+]i on Ca2+ sparks and Ca2+ waves.
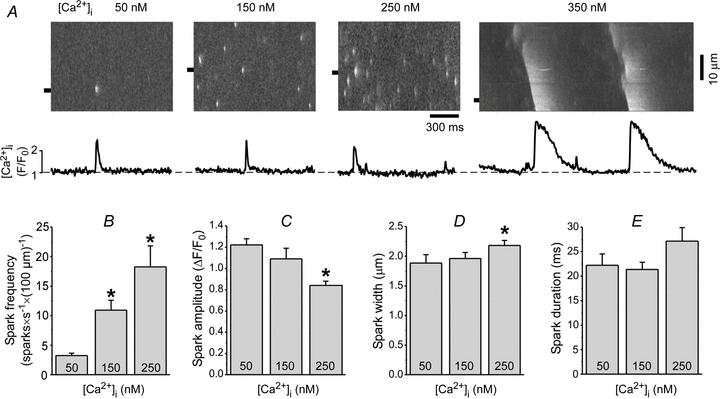
A, example line-scan images and F/F0 profiles of Ca2+ sparks and waves recorded at different [Ca2+]i values. The Ca2+ spark and wave profiles were obtained by averaging fluorescence from the 1 μm wide region marked by the black boxes. Summary data of Ca2+ spark frequency (B), amplitude (C), width (measured at half-maximal amplitude, FWHM) (D), and duration (measured at half-maximal amplitude, FDHM) (E) recorded at different [Ca]2+i values. *P < 0.05 vs. 50 nm[Ca2+]i.
Contribution of Ca2+ sparks and waves to SR Ca2+ leak at different cytosolic [Ca2+] values
We have recently shown that in rabbit ventricular myocytes RyR-mediated Ca2+ leak is composed of two main pathways: spark mediated and non-spark mediated (Zima et al. 2010). Depending on SR Ca2+ load, these two pathways contribute to a different degree to the total SR Ca2+ leak. At low [Ca2+]SR (<400 μm), Ca2+ leak occurred mostly as undetectable openings of RyRs. At high [Ca2+]SR (>600 μm), however, Ca2+ sparks became the significant pathway of SR Ca2+ leak. Here, we studied to what degree [Ca2+]i affects these two components of SR Ca2+ leak. First, we obtained RyR-dependent Ca2+ leak as a function of [Ca2+]SR (Fig. 3A) by subtracting the RuR-insensitive component of Ca2+ leak (Fig. 1B; green points) from the total SR Ca2+ leak (Fig. 1B). After subtraction, the points of SR Ca2+ leak at low [Ca2+]SR (between 50 and 450 μm) were best fitted with single Hill functions (Fig. 3B) (Zima et al. 2010). These functions were used to describe the relationship between [Ca2+]SR and non-spark-mediated Ca2+ leak for different [Ca2+]i values. Next, we estimated the spark-mediated leak by subtracting the corresponding non-spark-mediated leak (Fig. 3B) from the RyR-mediated Ca2+ leak (Fig. 3A). For different [Ca2+]i, the obtained points were best fitted with single exponential functions shown in Fig. 3C. In the case of 350 nm[Ca2+]i (grey line), SR Ca2+ leak was mainly mediated by Ca2+ waves. Figure 3D illustrates the relative contribution of spark- and non-spark-mediated Ca2+ leak to the RyR-dependent Ca2+ leak (at [Ca2+]SR = 900 μm) for different [Ca2+]i values. This analysis revealed that both components of RyR-mediated Ca2+ leak increased as a function of [Ca2+]i. While non-spark-mediated Ca2+ leak increased linearly, spark/wave-mediated leak rose exponentially with [Ca2+]i. These results indicate that cytosolic [Ca2+] significantly affects both components of RyR-mediated Ca2+ leak in ventricular myocytes.
Figure 3. Contribution of Ca2+ sparks and waves to SR Ca2+ leak.
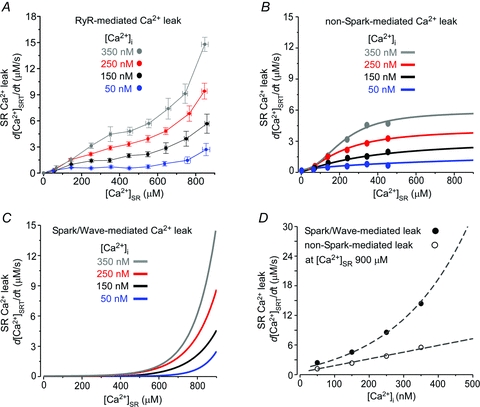
A, RyR-mediated SR Ca2+ leak as a function of [Ca2+]SR measured at different [Ca2+]i values. The RyR-mediated Ca2+ leak was obtained by subtracting SR Ca2+ leak in the presence of RuR from the total SR Ca2+ leak. B, relationships between non-spark-mediated Ca2+ leak and [Ca2+]SR obtained at different [Ca]i values. C, relationships between spark- and wave-mediated Ca2+ leak and [Ca2+]SR obtained at different [Ca]i values. D, the spark-mediated Ca2+ leak (filled circles) and the non-spark mediated Ca2+ leak (open circles) at [Ca2+]SR = 900 μm were plotted as a function of [Ca2+]i. The spark-mediated leak was fitted with an exponential function (R2 = 0.86) and the non-spark-mediated leak was fitted with a linear function (R2 = 0.91).
In the next set of experiments, we used the fast Ca2+ buffer BAPTA to eliminate Ca2+ sparks by decreasing efficacy of local CICR within RyR clusters. Addition of 0.7 mm BAPTA to the experimental solution decreased Ca2+ spark frequency by 87% (Fig. 4A and B), amplitude by 67% and width by 40%. Free [Ca2+]i of the BAPTA solution was kept the same as the control solution (250 nm). As BAPTA did not affect basal [Ca2+]SR (Fig. 4C), changes in spark properties were not due to decreased SR Ca2+ load. Therefore, by using BAPTA we were able to significantly eliminate spark-mediated Ca2+ leak without affecting single RyR activity. The analysis of SR Ca2+ leak at 250 nm[Ca2+]i in the control solution and in the presence of BAPTA (0.7 mm) (Fig. 4C) revealed that an increase of cytosolic Ca2+ buffer capacity with BAPTA significantly decreased SR Ca2+ leak, particularly at high [Ca2+]SR (>400 μm; Fig. 4D) where Ca2+ sparks significantly contribute to SR Ca2+ leak (Zima et al. 2010). Furthermore, Ca2+ leak measured in the presence of BAPTA was similar to non-spark-mediated Ca2+ leak obtained by the mathematical approach (Fig. 3B and dashed line in Fig. 4D).
Figure 4. Effects of BAPTA on Ca2+ sparks and SR Ca2+ leak.
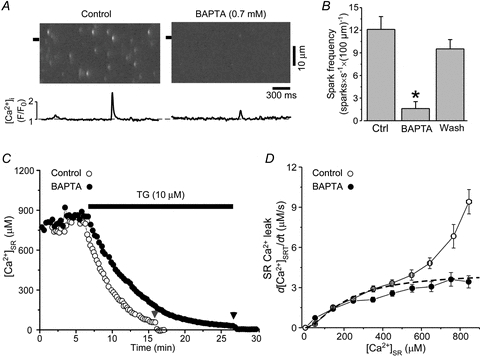
A, line-scan images and F/F0 profiles of Ca2+ sparks recorded at 250 nm[Ca]i in control and in the presence of 0.7 mm BAPTA. The Ca2+ spark profiles were obtained by averaging fluorescence from the 1 μm wide regions marked by the black boxes. B, effect of BAPTA on Ca2+ spark frequency. C, decline of [Ca2+]SR during SERCA inhibition in control solution (open circles) and in the presence of 0.7 mm BAPTA (filled circles). Free [Ca2+]i of both solutions was kept at 250 nm. D, relationships between SR Ca2+ leak and [Ca2+]SR measured at 250 nm[Ca2+]i in control (open circles) and BAPTA-containing solution (filled circles). For comparison, the dashed line indicates non-spark-mediated Ca2+ leak at 250 nm[Ca2+]i obtained by the mathematical approach (data from Fig. 3B).
Role of CaMKII in regulation of SR Ca2+ leak at high cytosolic [Ca2+]
In the following experiments, we investigated whether the augmentation of SR Ca2+ leak observed at high cytosolic [Ca2+] (>150 nm) was in part mediated by CaMKII-dependent phosphorylation of the RyR. Here we studied the effect of the specific CaMKII inhibitor autocamtide 2-related inhibitory peptide (AIP) on SR Ca2+ leak at high [Ca2+]i. Figure 5A shows a representative recording of [Ca2+]SR in the presence of AIP (5 μm), after subsequently increasing [Ca2+]i to 250 nm, as well as after application of TG (10 μm). AIP did not significantly affect initial [Ca2+]SR (recorded before TG application). Initial [Ca2+]SR was 795 ± 44 μm in control conditions and remained steady at 771 ± 66 μm (n = 10) after AIP application. In the presence of AIP, SR Ca2+ leak only changed from 7.2 ± 0.6 to 8.5 ± 1.1 μm s-1 (n = 10; [Ca2+]SR = 750 μm). Thus, inhibition of CaMKII did not have a significant effect on SR Ca2+ leak (Fig. 5B), although the results show a tendency to increase SR Ca2+ leak (∼16%) at 250 nm[Ca2+]i. Likewise, we did not observe a significant effect of AIP on SR Ca2+ leak in the presence of 350 nm[Ca2+]i. SR Ca2+ leak was 16.0 ± 0.9 μm s-1 (n = 8) in control conditions and changed to 17.8 ± 1.4 μm s-1 (n = 7) after AIP (5 μm) application. AIP also did not change the properties of Ca2+ waves (i.e. frequency, amplitude and velocity; data not shown). These results indicate that CaMKII-mediated RyR phosphorylation is not involved in the effect of [Ca2+]i (in the range from 50 to 350 nm) on SR Ca2+ leak. Thus, [Ca2+]i regulates Ca2+ leak mainly via direct action on the RyR.
Figure 5. Effects of CaMKII inhibition on SR Ca2+ leak.
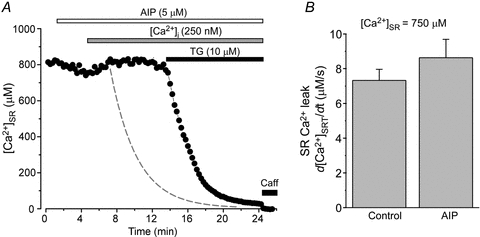
A, effect of 250 nm[Ca2+]i on decline of [Ca2+]SR during SERCA inhibition in the presence of AIP (5 μm). For comparison, the dashed line indicates the decline of [Ca2+]SR at 250 nm[Ca2+]i in the absence of CaMKII inhibition (data from Fig. 1A). B, average effect of AIP (5 μm) on SR Ca2+ leak measured at [Ca2+]SR = 750 μm.
Changes of SR Ca2+ leak during excitation–contraction coupling (ECC)
RyR-mediated Ca2+ leak is controlled by the level of Ca2+ on both sides of the SR membrane (Fig. 1B). During the cardiac cycle, cytosolic and luminal [Ca2+] dynamically change, following opposite directions ([Ca2+]SR is replenished while [Ca2+]i declines and vice versa). Thus, the decline of [Ca2+]i during late systole and early diastole would ultimately offset the stimulatory effect of [Ca2+]SR on the RyR. In the following experiments, we measured changes of [Ca2+]SR and [Ca2+]i during ECC and analysed how these changes would affect SR Ca2+ leak. [Ca2+]SR and [Ca2+]i were recorded simultaneously in intact ventricular myocytes at 0.5 Hz pacing frequency. Figure 6A shows line-scan images of Ca2+ transient (top) and corresponding [Ca2+]SR depletion (bottom) evoked by electrical field stimulation. We measured [Ca2+]SR during a SR replenishing phase at the same three [Ca2+]i values (150, 250 and 350 nm), that were studied in permeabilized cell experiments. [Ca2+]i was inversely proportional to [Ca2+]SR with a slope constant of –1100 (Fig. 6B). We estimated how changes in [Ca2+]i and [Ca2+]SR (which occur during diastole) affect SR Ca2+ leak. At 350 nm[Ca2+]i, [Ca2+]SR was 608±45 μm (n = 9; Fig. 6B) and SR Ca2+ leak was 7.9 μm s-1 (Fig. 5C, blue open circle). At 250 nm[Ca2+]i, [Ca2+]SR was replenished to 718 ± 27 μm (n = 9; Fig. 6B) and Ca2+ leak slightly decreased to 7.5 μm s-1 (Fig. 6C, red open circle). At 150 nm[Ca2+]i, [Ca2+]SR reached 819 ± 11 μm (n = 9; Fig. 6B) and SR Ca2+ leak was further reduced to 6.9 μm s-1 (Fig. 6C, black open circle). This analysis suggests that only small changes (∼13%) of SR Ca2+ leak occur during the declining phase of the Ca2+ transient. Since SR Ca2+ leak steeply depends on both [Ca2+]SR and [Ca2+]i (Fig. 1B), but [Ca2+]SR and [Ca2+]i (Fig. 6A) change in opposite directions, SR Ca2+ leak remains relatively constant during the SR replenishing phase of the cardiac cycle.
Figure 6. Estimation of SR Ca2+ leak during the declining phase of the Ca2+ transient.
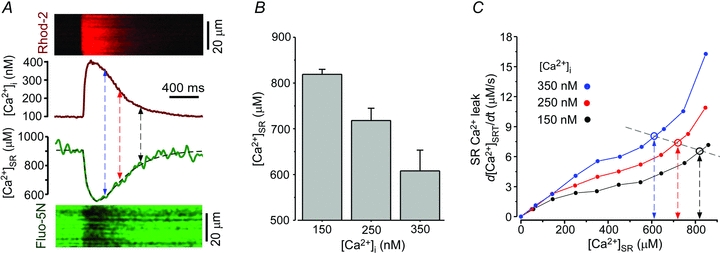
A, simultaneously recorded [Ca2+]i transient and corresponding [Ca2+]SR depletion. Top, line-scan image of Rhod-2 fluorescence and profile of [Ca2+]i. Bottom, line-scan image of Fluo-5N fluorescence and profile of [Ca2+]SR. The profiles were obtained by averaging fluorescence from the entire cellular width of line scans. B, summary data showing changes of [Ca2+]SR measured at three different [Ca2+]i values (i.e. 150, 250 and 350 nm; indicated by arrows in A). C, RyR-mediated SR Ca2+ leak as a function of [Ca2+]SR measured at different [Ca2+]i values (data were taken from Fig. 3A). For each [Ca2+]i studied, SR Ca2+ leak was extrapolated at the corresponding [Ca2+]SR, calculated from the [Ca2+]SR profile in A. The results suggest that during the declining phase of the Ca2+ transient leak remains relatively constant.
Discussion
In recent years, a significant effort has been made to understand the mechanisms that control SR Ca2+ leak in healthy and diseased hearts. A relatively small augmentation of SR Ca2+ efflux during diastole can significantly influence cardiac function by depleting the SR Ca2+ content and reducing contraction. The increased Ca2+ leak can also be pro-arrhythmic as the released Ca2+ is extruded by the electrogenic Na+–Ca2+ exchange (NCX), leading to delayed afterdepolarizations (Kass et al. 1978; Pogwizd et al. 2001). The bulk of SR Ca2+ leak is mediated by RyRs (Neary et al. 2002; Shannon et al. 2002; Zima et al. 2010), which are complexly regulated by Ca2+ from both cytosolic and luminal sides of the channel (Fill & Copello, 2002; Meissner, 2004). Since Fabiato's work (Fabiato, 1985a,b), [Ca2+]i has been considered a crucial factor in the activation of global SR Ca2+ release during systole. However, it is less clear to what extent dynamic changes of [Ca2+]i during diastole affects RyR activity and SR Ca2+ leak. Here we directly measured Ca2+ leak as the rate of [Ca2+]SR decline after SERCA inhibition in permeabilized rabbit ventricular myocytes. By introducing different [Ca2+]i levels into the cytosol, we found that: (1) [Ca2+]i increases SR Ca2+ leak primarily by activating the RyR-mediated leak pathway; (2) with increasing [Ca2+]i, Ca2+ sparks and waves become significant pathways of Ca2+ leak; (3) within the range of 50–350 nm[Ca2+]i, the effect on SR Ca2+ leak is predominantly mediated by direct activation of the RyR and not by CaMKII-mediated phosphorylation of the channel; and (4) during late systole and early diastole (when [Ca2+]SR and [Ca2+]i change in opposite directions) SR Ca2+ leak remains relatively constant as a result of strong dependence from both [Ca2+]i and [Ca2+]SR.
Our results show that [Ca2+]i affects SR Ca2+ leak entirely by regulating RyR activity. We found that in the presence of the potent RyR inhibitor RuR, the residual SR Ca2+ leak was insensitive to changes in [Ca2+]i. High [Ca2+]i can facilitate SR Ca2+ leak by increasing RyR open probability. However, it also decreases the SR Ca2+ gradient and therefore the driving force for SR Ca2+ leak. Our results show that despite decrease of the SR Ca2+ gradient (by changing [Ca2+]i from 50 to 350 nm), RyR-mediated Ca2+ leak increased more than 5 times (measured at the same [Ca2+]SR; Figs 1B and 3A). This finding suggests that SR Ca2+ leak is not a simple diffusion of Ca2+ from the SR, but rather a well-regulated process. We have previously shown that RyR-mediated Ca2+ leak can occur as spontaneous Ca2+ sparks, but also as undetectable openings of RyRs (non-spark-mediated Ca2+ leak; Zima et al. 2010). The augmentation of SR Ca2+ leak by cytosolic Ca2+ was attributed to an increase of both components of SR Ca2+ leak (Fig. 3B and C). These two pathways, however, were differently regulated by [Ca2+]i. While non-spark-mediated Ca2+ leak linearly depended on [Ca2+]i (Fig. 3D), spark-mediated Ca2+ leak rose steeply at higher [Ca2+]i. The latter was mainly mediated by an increase of Ca2+ spark frequency, since other spark properties (width and duration) were only minimally affected by [Ca2+]i (Fig. 2). A similar effect of [Ca2+]i on spark frequency was observed in permeabilized rat ventricular myocytes (Lukyanenko & Gyorke, 1999).
The analysis of the spark properties also shows that the amplitude proportionally decreased with an increase of [Ca2+]i, indicating that this spark parameter is mainly determined by SR Ca2+ release flux and, therefore, by the SR Ca2+ gradient. However, despite the Ca2+ flux during individual release events at 350 nm[Ca2+]i being smaller than at [Ca2+]i < 350 nm, it is sufficient to increase [Ca2+]i next to neighbouring junctions to activate local CICR and trigger Ca2+ waves. When [Ca2+]SR was depleted to ∼500 μm (during SERCA inhibition) Ca2+ waves ceased completely (data not shown), suggesting that the [Ca2+] on both sides of the RyR plays an important role in the generation of Ca2+ waves. It appears that high [Ca2+]i increases the sensitivity of release units to CICR and elevated [Ca2+]SR maintains SR Ca2+ flux strong enough to produce Ca2+ waves via a ‘fire–diffuse–fire’ mechanism (Keizer & Smith, 1998). We found that the transformation of SR Ca2+ release from localized sparks to propagating waves almost doubled RyR-mediated Ca2+ leak (Fig. 3C), suggesting that Ca2+ waves represent spontaneous SR Ca2+ release events in their extreme form.
It has been suggested that an increase of SR Ca2+ leak by high [Ca2+]i is mediated partially by CaMKII-dependent phosphorylation of RyRs (Guo et al. 2006; van Oort et al. 2010). Using the selective CaMKII inhibitor AIP, we found that the augmentation of SR Ca2+ leak at high [Ca2+]i was insensitive to CaMKII inhibition. In contrast to previous studies, AIP treatment showed some tendency to accelerate SR Ca2+ leak (Fig. 5). The discrepancy with previously published work can be explained by the fact that CaMKII was not activated under our experimental conditions. It has been shown that activation of endogenous CaMKII requires much higher [Ca2+]i (∼500 nm), phosphatase inhibition (e.g. with okadaic acid) and calmodulin (Guo et al. 2006). In our experiments none of these conditions were applied. The small acceleration of SR Ca2+ leak observed in the presence of AIP can be the result of a potential direct interaction of the peptide with the RyR. Thus, in the range of 50–350 nm[Ca2+]i regulates SR Ca2+ leak mainly via a CaMKII-independent mechanism.
Another important finding of this study is that SR Ca2+ leak is highly sensitive to both [Ca2+]i and [Ca2+]SR (Fig. 1B). It remains unresolved whether luminal and cytosolic Ca2+ regulate the RyR via entirely independent mechanisms or via a well coordinated process. It has been shown that the RyR can be activated independently by Ca2+ from the luminal or cytosolic sides of the channel (Sitsapesan & Williams, 1994; Gyorke & Gyorke, 1998; Qin et al. 2008). However, luminal [Ca2+] can also regulate RyR by acting on the cytosolic Ca2+ activation site of the channel via a ‘feed-through’ mechanism (Laver, 2007). The combination of all these regulatory mechanisms contributes to the fine control of the RyR. Furthermore, our data suggest that [Ca2+]SR regulation provides an important feedback mechanism to counter the positive feedback of high [Ca2+]i during the declining phase of the Ca2+ transient, whereas at the end of diastole, the high SR Ca2+ leak driving force determined by high [Ca2+]SR is well equilibrated by the low [Ca2+]i. The balance established by this dual regulation results in a SR Ca2+ leak rate that remains relatively constant during the decline of the Ca2+ transient when [Ca2+]SR and [Ca2+]i change in opposite directions. However, alterations in the activity of RyR during diastole can affect the basal Ca2+ leak rate and, therefore, the amount of Ca2+ stored in the SR. This would lead to altered systolic Ca2+ transients, potential generation of arrhythmias and contractile dysfunctions. There are, however, significant differences between animal species regarding the role of RyR modification and its effect on SR Ca2+ release and ECC. In rat ventricular myocytes, for example, RyR sensitization with low doses of caffeine produced only a transient positive inotropic effect (Trafford et al. 2000). However, studies conducted on ventricular myocytes from larger animals (e.g. dogs and rabbits) (Belevych et al. 2007; Domeier et al. 2009) showed that caffeine led to significant depletion of [Ca2+]SR and a decrease of Ca2+ transient amplitude. The differences between rats and rabbits are probably due to the differences in expression of SERCA and NCX (Bers, 2001). In contrast to rats, rabbits have less SERCA but increased NCX expression. This translates functionally into less SR Ca2+ reuptake by SERCA, more Ca2+ extrusion from the cell via NCX, and greater net cellular Ca2+ loss.
The coordinated regulation of RyR by luminal and cytosolic Ca2+ provides a fundamental mechanism that contributes to setting the appropriate SR Ca2+ load during diastole and therefore preventing the onset of pathological conditions (e.g. SR Ca2+ overload). This is an important step towards the understanding of the RyR regulation. In intact cells, however, additional mechanisms are probably involved. For instance, our estimation did not account for a potential SR Ca2+ release refractoriness which can occur during ECC in intact cells. In permeabilized cells, the potential contribution of Ca2+-dependent inactivation (Fabiato, 1985b) in offsetting the SR Ca2+ leak at high [Ca2+]i can be ruled out based on several previous findings. First, Ca2+-dependent inactivation has not been reported in permeabilized cell experiments when SR Ca2+ release was measured at [Ca2+]i >10 μm (Stevens et al. 2009). Second, the study of cardiac RyR in lipid bilayers has shown that RyR inactivation occurs at significantly higher [Ca2+]i (>0.5 mm) (Xu et al. 1996). Thus, the 13% difference between the leak rates at the different points of [Ca2+]i and corresponding [Ca2+]SR during the declining phase of the Ca2+ transient (Fig. 6C), might be explained by the fact that our estimations obtained in permeabilized cells are not accounting for all the regulatory mechanisms that play a role in setting the RyR activity in the intact cellular environment (e.g. use-dependent inactivation).
In conclusion, this study is the first that we know of to characterize the effect of [Ca2+]i on SR Ca2+ leak in permeabilized ventricular myocytes. We found that [Ca2+]i plays a critical role in controlling SR Ca2+ leak. The increase of [Ca2+]i and RyR sensitivity to [Ca2+]i in heart failure or other pathological conditions, can lead to the alteration of diastolic SR Ca2+ leak and to the generation of arrhythmogenic events. These findings are of great importance because they explain a new regulatory mechanism of SR Ca2+ leak that is critical to maintain the physiological homeostasis of Ca2+ in the heart.
Acknowledgments
This work was supported by National Institutes of Health Grants HL62231, HL80101 and HL101235, the Leducq Foundation (to L.A.B.) and the McCormick Foundation (to A.V.Z.). The authors also would like to thank Drs Seth L. Robia and Joshua Maxwell for critical reading of the manuscript.
Glossary
Abbreviation
- AIP
autocamtide 2-related inhibitory peptide
- [Ca2+]i
cytosolic free calcium concentration
- CaMKII
Ca2+–calmodulin-dependent kinase type II
- CICR
Ca2+-induced Ca2+ release
- ECC
excitation–contraction coupling
- FDHM
full duration at half-maximum
- FWHM
full width at half-maximum
- HF
heart failure
- NCX
Na+–Ca2+ exchange
- PLB
phospholamban
- RuR
ruthenium red
- RyR
ryanodine receptor
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+-ATPase
- SR
sarcoplasmic reticulum
- [Ca2+]SR
sarcoplasmic reticulum free calcium concentration
- TG
thapsigargin
Author contributions
E.B., S.R.M., A.V.Z. and L.A.B. contributed to the conception and design of the study, interpretation of data and writing of the manuscript. E.B., S.R.M. and A.V.Z. performed the experimental work and analysis of results. All authors have approved the version to be published.
References
- Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- Bassani RA, Bers DM. Rate of diastolic Ca release from the sarcoplasmic reticulum of intact rabbit and rat ventricular myocytes. Biophys J. 1995;68:2015–2022. doi: 10.1016/S0006-3495(95)80378-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Gyorke S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J. 2007;93:4083–4092. doi: 10.1529/biophysj.107.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht: Kluwer Academic Publishers; 2001. [Google Scholar]
- Bers DM, Eisner DA, Valdivia HH. Sarcoplasmic reticulum Ca2+ and heart failure: roles of diastolic leak and Ca2+ transport. Circ Res. 2003;93:487–490. doi: 10.1161/01.RES.0000091871.54907.6B. [DOI] [PubMed] [Google Scholar]
- Bovo E, Blatter LA, Zima AV. Regulation of sarcoplasmic reticulum calcium leak by cytosolic calcium in rabbit ventricular myocytes. Biophys J. 2010;98:102a. doi: 10.1113/jphysiol.2011.214171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochet DX, Xie W, Yang D, Cheng H, Lederer WJ. Quarky calcium release in the heart. Circ Res. 2011;108:210–218. doi: 10.1161/CIRCRESAHA.110.231258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys J. 1994;67:1942–1956. doi: 10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- Diaz ME, Trafford AW, O'Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol. 1997;501:3–16. doi: 10.1111/j.1469-7793.1997.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibb K, Eisner D. A small leak may sink a great ship but what does it do to the heart? J Physiol. 2010;588:4849. doi: 10.1113/jphysiol.2010.200840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domeier TL, Blatter LA, Zima AV. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol. 2009;587:5197–5209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol Cell Physiol. 1983;245:C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Rapid ionic modifications during the aequorin-detected calcium transient in a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985a;85:189–246. doi: 10.1085/jgp.85.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985b;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CH. Sarcoplasmic reticulum Ca2+ leak in heart failure: mere observation or functional relevance? Cardiovasc Res. 2008;77:302–314. doi: 10.1093/cvr/cvm006. [DOI] [PubMed] [Google Scholar]
- Guo T, Zhang T, Mestril R, Bers DM. Ca2+/calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res. 2002;91:1015–1022. doi: 10.1161/01.res.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- Kass RS, Lederer WJ, Tsien RW, Weingart R. Role of calcium ions in transient inward currents and aftercontractions induced by strophanthidin in cardiac Purkinje fibres. J Physiol. 1978;281:187–208. doi: 10.1113/jphysiol.1978.sp012416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keizer J, Smith GD. Spark-to-wave transition: saltatory transmission of calcium waves in cardiac myocytes. Biophys Chem. 1998;72:87–100. doi: 10.1016/s0301-4622(98)00125-2. [DOI] [PubMed] [Google Scholar]
- Laver DR. Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Biophys J. 2007;92:3541–3555. doi: 10.1529/biophysj.106.099028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart S, Marks AR. Regulation of ryanodine receptors in the heart. Circ Res. 2007;101:746–749. doi: 10.1161/CIRCRESAHA.107.162479. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 1995;268:1042–1045. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- Lukyanenko V, Gyorke S. Ca2+ sparks and Ca2+ waves in saponin-permeabilized rat ventricular myocytes. J Physiol. 1999;521:575–585. doi: 10.1111/j.1469-7793.1999.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Meissner G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium. 2004;35:621–628. doi: 10.1016/j.ceca.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Meissner G, Henderson JS. Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. J Biol Chem. 1987;262:3065–3073. [PubMed] [Google Scholar]
- Neary P, Duncan AM, Cobbe SM, Smith GL. Assessment of sarcoplasmic reticulum Ca2+ flux pathways in cardiomyocytes from rabbits with infarct-induced left-ventricular dysfunction. Pflugers Arch. 2002;444:360–371. doi: 10.1007/s00424-002-0794-0. [DOI] [PubMed] [Google Scholar]
- O'Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res. 1999;84:562–570. doi: 10.1161/01.res.84.5.562. [DOI] [PubMed] [Google Scholar]
- Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster – automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol. 2007;293:C1073–C1081. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- Pieske B, Kretschmann B, Meyer M, Holubarsch C, Weirich J, Posival H, et al. Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation. 1995;92:1169–1178. doi: 10.1161/01.cir.92.5.1169. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- Porta M, Zima AV, Nani A, Diaz-Sylvester PL, Copello JA, Ramos-Franco J, et al. Single ryanodine receptor channel basis of caffeine's action on Ca2+ sparks. Biophys J. 2011;100:931–938. doi: 10.1016/j.bpj.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Valle G, Nani A, Nori A, Rizzi N, Priori SG, et al. Luminal Ca2+ regulation of single cardiac ryanodine receptors: insights provided by calsequestrin and its mutants. J Gen Physiol. 2008;131:325–334. doi: 10.1085/jgp.200709907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau E, Smith JS, Henderson JS, Meissner G. Single channel and 45Ca2+ flux measurements of the cardiac sarcoplasmic reticulum calcium channel. Biophys J. 1986;50:1009–1014. doi: 10.1016/S0006-3495(86)83543-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago DJ, Curran JW, Bers DM, Lederer WJ, Stern MD, Rios E, Shannon TR. Ca sparks do not explain all ryanodine receptor-mediated SR Ca leak in mouse ventricular myocytes. Biophys J. 2010;98:2111–2120. doi: 10.1016/j.bpj.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS, Bers DM. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys J. 2000;78:322–333. doi: 10.1016/S0006-3495(00)76595-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Guo T, Bers DM. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res. 2003;93:40–45. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- Sitsapesan R, Williams AJ. Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca2+-release channel by luminal Ca2+ J Membr Biol. 1994;137:215–226. doi: 10.1007/BF00232590. [DOI] [PubMed] [Google Scholar]
- Stevens SC, Terentyev D, Kalyanasundaram A, Periasamy M, Gyorke S. Intra-sarcoplasmic reticulum Ca2+ oscillations are driven by dynamic regulation of ryanodine receptor function by luminal Ca2+ in cardiomyocytes. J Physiol. 2009;587:4863–4872. doi: 10.1113/jphysiol.2009.175547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafford AW, Sibbring GC, Diaz ME, Eisner DA. The effects of low concentrations of caffeine on spontaneous Ca release in isolated rat ventricular myocytes. Cell Calcium. 2000;28:269–276. doi: 10.1054/ceca.2000.0156. [DOI] [PubMed] [Google Scholar]
- van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Mann G, Meissner G. Regulation of cardiac Ca2+ release channel (ryanodine receptor) by Ca2+, H+, Mg2+, and adenine nucleotides under normal and simulated ischemic conditions. Circ Res. 1996;79:1100–1109. doi: 10.1161/01.res.79.6.1100. [DOI] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Picht E, Bers DM, Blatter LA. Partial inhibition of sarcoplasmic reticulum Ca release evokes long-lasting Ca release events in ventricular myocytes: role of luminal Ca in termination of Ca release. Biophys J. 2008a;94:1867–1879. doi: 10.1529/biophysj.107.114694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Picht E, Bers DM, Blatter LA. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res. 2008b;103:e105–e115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]