

Abstract
Non-technical summary
Myocardial stretch increases force in two phases. The first one is immediate and attributed to an increase in myofilament Ca2+ responsiveness (Frank–Starling mechanism). The second phase gradually develops and is known as slow force response (SFR) or Anrep effect due to an increase in intracellular Ca2+ transient. We previously showed that Ca2+ entry through reverse Na+/Ca2+ exchange underlies the SFR, as the final step of an autocrine/paracrine loop involving release of angiotensin II/endothelin, transactivation of the epidermal growth factor receptor, increased mitochondrial oxidative stress and a Na+/H+ exchanger (NHE-1) activation-mediated rise in Na+. In the present study we show that mineralocorticoid receptor activation is a necessary step between endothelin and epidermal growth factor receptor activation in the stretch-triggered reactive oxygen species-mediated NHE-1 activation leading to the SFR.
Abstract
The increase in myocardial reactive oxygen species after epidermal growth factor receptor transactivation is a crucial step in the autocrine/paracrine angiotensin II/endothelin receptor activation leading to the slow force response to stretch (SFR). Since experimental evidence suggests a link between angiotensin II or its AT1 receptor and the mineralocorticoid receptor (MR), and MR transactivates the epidermal growth factor receptor, we thought to determine whether MR activation participates in the SFR development in rat myocardium. We show here that MR activation is necessary to promote reactive oxygen species formation by a physiological concentration of angiotensin II (1 nmol l-1), since an increase in superoxide anion formation of ∼50% of basal was suppressed by blocking MR with spironolactone or eplerenone. This effect was also suppressed by blocking AT1, endothelin (type A) or epidermal growth factor receptors, by inhibiting NADPH oxydase or by targeting mitochondria, and was unaffected by glucocorticoid receptor inhibition. All interventions except AT1 receptor blockade blunted the increase in superoxide anion promoted by an equipotent dose of endothelin-1 (1 nmol l-1) confirming that endothelin receptors activation is downstream of AT1. Similarly, an increase in superoxide anion promoted by an equipotent dose of aldosterone (10 nmol l-1) was blocked by spironolactone or eplerenone, by preventing epidermal growth factor receptor transactivation, but not by inhibiting glucocorticoid receptors or protein synthesis, suggesting non-genomic MR effects. Combination of aldosterone plus endothelin-1 did not increase superoxide anion formation more than each agonist separately. We found that aldosterone increased phosphorylation of the redox-sensitive kinases ERK1/2-p90RSK and the NHE-1, effects that were eliminated by eplerenone or by preventing epidermal growth factor receptor transactivation. Finally, we provide evidence that the SFR is suppressed by MR blockade, by preventing epidermal growth factor receptor transactivation or by scavenging reactive oxygen species, but it is unaffected by glucocorticoid receptor blockade or protein synthesis inhibition. Our results suggest that MR activation is a necessary step in the stretch-triggered reactive oxygen species-mediated activation of redox-sensitive kinases upstream NHE-1.
Introduction
After stretch, the force developed by cardiac muscle increases in two phases. The first phase (Frank–Starling mechanism) occurs immediately and is attributed to an increase in myofilament calcium responsiveness. The second phase, known as the slow force response (SFR), occurs gradually over the next 10–15 min. The SFR is due to a progressive increase in the calcium transient amplitude (Allen & Kurihara, 1982) that results from an autocrine/paracrine mechanism involving release of angiotensin II (Ang II) and endothelin (ET) (Alvarez et al. 1999; Perez et al. 2001). This phase is thought to be the in vitro manifestation of the Anrep effect, first described in 1912 (von Anrep, 1912).
An important step in the chain of events leading to SFR generation is the increased production of mitochondrial reactive oxygen species (ROS) (Caldiz et al. 2007; Villa-Abrille et al. 2010). Conversely, suppression of mitochondrial ROS production blunts the generation of SFR (Caldiz et al. 2007; Zhang et al. 2009). SFR generation is induced by the autocrine/paracrine actions of Ang II/ET, which are known to have stimulatory effects on NADPH oxidase (NOX) activity (Sugden & Clerk, 2006). We reported previously that a necessary step for inducing mitochondrial ROS release is transactivation of the epidermal growth factor receptor (EGFR) after the effect of Ang II/ET (Villa-Abrille et al. 2010).
H2O2, the product of superoxide anion (O2·) dismutation, is a well-known activator of the cardiac Na+/H+ exchanger (NHE-1) through redox-sensitive kinases like the extracellular signal-regulated protein kinases (ERK1/2) and the p90 ribosomal S6 kinase (p90RSK) (Sabri et al. 1998; Wei et al. 2001; Rothstein et al. 2002). Enhanced NHE-1 activity via phosphorylation brings sodium into cardiomyocytes after stretch, changing the reverse potential of the Na+/Ca2+ exchanger and driving its reverse mode of operation (Perez et al. 2001). The critical role played by NHE-1 activation in the Anrep phenomenon was demonstrated pharmacologically with NHE-1 inhibitors (for review see Cingolani et al. 2011), and also by specific NHE-1 silencing following direct intramyocardial injection of small interfering RNA into the rat left ventricular wall (Perez et al. 2011).
Recently, a link between Ang II or its receptor AT1 and the mineralocorticoid receptor (MR) was reported (Lemarie et al. 2008, 2009; Grossmann & Gekle, 2009). Although still somewhat controversial, aldosterone (ALD), which is known to be regulated by Ang II, appears to be synthesized and/or released by cardiac muscle (Silvestre et al. 1998, 1999; Takeda et al. 2000a; Gomez-Sanchez et al. 2004; Chai & Danser, 2006).
Furthermore, stretching cardiac muscle induces SFR development, in which Ang II and EGFR transactivation are crucial steps. Therefore, we hypothesized that if crosstalk between Ang II and the MR exists during ROS production, and ROS is crucial for SFR development, inhibition of the MR would blunt the SFR. In the present study, our objective was to prove that MR activation is involved in the signalling pathway leading to the Anrep effect.
Methods
Animals
Experiments were carried out with 4- to 5-month-old male Wistar rats, in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and with the guidelines from the Animal Welfare Committee of La Plata School of Medicine. Animals were provided by our own Faculty Animal Facility.
Four- to five-month-old Wistar rats (n = 36) were anaesthetized by intraperitoneal injection of sodium pentobarbitone (35 mg (kg body weight)-1). The chests were opened to excise the heart when deep anaesthesia was reached, verified by the loss of corneal reflex and appearance of slow deep diaphragmatic breathing. Each heart was used to provide papillary muscles as well as cardiac tissue slices. Immediately after excised, each heart was perfused through the aorta with Krebs–Ringer buffer pH 7.4 to eliminate the blood and two papillary muscles as well as 8–10 cardiac tissue slices from the left ventricle (1 × 5 mm) were dissected.
Determination of the SFR to stretch in isolated papillary muscles
Papillary muscles from the left ventricle were used to assess the SFR to stretch. Briefly, the muscles were mounted in a perfusion chamber placed on the stage of an inverted microscope (Olympus) and superfused with a CO2/HCO3--buffered solution containing (mmol l-1): NaCl 128.3, KCl 4.5, CaCl2 1.35, NaHCO3 20.23, MgSO4 1.05, glucose 11.0 and equilibrated with 5% CO2–95% O2 (pH ∼7.40). The possible participation of cathecholamines released by the nerve endings was prevented by adrenergic receptors blockade with 1.0 μmol l-1 prazosin plus 1.0 μmol l-1 atenolol. The muscles were paced at 0.2 Hz at a voltage 10% over threshold maintained at 30°C, and isometric contractions were recorded. Cross-sectional area (calculated as 0.75 of the product of thickness by width) was used to normalize force records obtained with a silicon strain gauge (model AEM 801, Sensonor, Kronex Technologies Corporation, California, USA). The slack length of each muscle was determined after mounting, and then the muscles were progressively stretched to the length at which they developed maximal twitch force (Lmax). After a few minutes at Lmax they were shortened to a length that approximated 98% of Lmax and was referred to as L98. Then, the muscles were shortened to 92% of Lmax (L92) and maintained at this length until the beginning of the experimental protocol, when they were abruptly stretched from L92 to L98. The drugs were added to the perfusate 20 min before stretch, and during this period, none of them changed developed force (DF) by more than 3–4%.
Measurement of O2
Cardiac slices from the left ventricle (1 × 5 mm) were dissected and kept at 4°C until assayed. Assay buffer consisted of Krebs buffer with the following composition (in mm): 118.3 NaCl, 4.7 KCl, 1.35 CaCl2, 1.2 MgSO4, 1 K2HPO4, 25 NaHCO3, 11 glucose, 20 Hepes (pH 7.4 after 1.5 h aeration with 95% O2–5% CO2 at 37°C). We used the lucigenin-enhaced chemiluminescence to measure O2· by rat cardiac tissue in Krebs-Hepes buffer with 5 μm lucigenin as previously described (Caldiz et al. 2007). Briefly, cardiac slices were incubated in assay buffer in the presence of Ang II, ET-1, ALD or ALD+ET-1 during 30 min in a metabolic incubator under 95% O2–5% CO2 at 37°C before measuring O2·. Inhibitors or receptor blockers were added to the buffer 5 min before Ang II, ET-1, ALD or ALD+ET-1. Chemiluminescence in arbitrary units was recorded with a luminometer (Chameleon; Hidex, Finland) for 30 s each with 4.5 min interval for 30 min. Since we have previously reported that superoxide production stabilized after 15 min (Caldiz et al. 2007), the averaged data obtained at this time are presented. The lucigenin-containing assay buffer with tissue slices minus background and responses to the different drugs assayed were reported. O2· production was normalized to milligrams dry weight tissue per minute. It has been reported that the method with lucigenin-enhanced chemiluminescence that we used does not allow detection of basal O2· production (Dikalov et al. 2007). However, control tissue slices without any pharmacological intervention produced an increase in O2· that was different from the background. This O2· production was not abolished by the NADPH oxidase inhibitor apocinyn (Apo) but it was abrogated with the ROS scavenger 2-mercapto-propionyl-glycine (MPG, not shown). This could be explained with the finding that NOX 4 is not inhibited by Apo (Ago et al. 2011). Therefore, the increase in O2· production was expressed as delta (Δ) percentage of basal value after 15 min. Although we induced and measured mitochondrial O2· formation, O2· is quite unstable and rapidly converted into H2O2 by several isoforms of superoxidase dismutases (SOD), like EC-SOD, a membrane-bound extracellular SOD, Mn SOD in the mitochondrial matrix and Cu-Zn-SOD in the insterticial space and cytosol. The activation of the redox sensitive intracellular signals probably occurs after O2· dismutation.
Immunoanalysis by Western blot, and determination of ERK1/2, p90RSK, and NHE-1 phosphorylation
Cardiac tissue slices were homogenized in lysis buffer (300 μmol l-1 sacarose, 1 μmol l-1 DTT, 4 μmol l-1 EGTA, protease inhibitors cocktail (Complete Mini Roche), 20 μmol l-1 Tris-HCl, pH 7.4) at the end of the experimental protocols. After a brief centrifugation the supernatant was kept and protein concentration determined by the Bradford method. Samples were denatured and equal amounts of protein were subjected to PAGE and electrotransferred to PVDF membranes. Membranes were then blocked with non-fat dry milk and incubated overnight with either anti-phospho-ERK1/2 or anti-ERK 2 or anti-P-p90RSK polyclonal antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). For NHE-1 phosphorylation determination samples were immunoprecipitated using a NHE-1 polyclonal antibody (Chemicon, Temecula, CA, USA) and then subjected to PAGE, electrotransferred and incubated with an anti-14-3-3 binding motif antibody (Cell Signaling Technology, Inc., Danvers, MA, USA). Previous reports have shown that the regulatory Ser703 of the NHE-1 lies within a sequence which creates upon phosphorylation a binding motif for 14-3-3 proteins (Takahashi et al. 1999; Lehoux et al. 2001). Thus, the anti-P-14-3-3 binding motif antibody when probed with immunoprecipitated NHE-1 represents a useful tool to estimate NHE-1 phosphorylation at Ser703 (Snabaitis et al. 2006).
Total ERK-2, p90RSK and NHE-1 as loading controls were assayed. Peroxidase-conjugated anti-rabbit or anti-mouse IgG (Santa Cruz Biotechnology) were used as secondary antibodies and bands were visualized using the ECL-Plus chemiluminescence detection system (Amersham). Immunoreactive band signals were analysed by densitometry (Scion Image).
Statistics
Data are expressed as means ± SEM. Differences between groups were assessed by one-way ANOVA followed by Student–Newman–Keuls test. P < 0.05 was considered significant.
Results
As an initial approach to determining whether MR activation participates in the chain of events leading to the SFR, and based on the crucial role of ROS in the signalling pathway leading to NHE-1 activation, we decided to explore whether MR blockade interferes with Ang II/ET-1-induced mitochondrial ROS formation. At the physiological concentration of 1 nmol l-1, Ang II increased the basal rate of O2· production by approximately 50%. This effect was abrogated by the MR antagonists spironolactone (Sp) and eplerenone (Ep), as shown in Fig. 1A. The Ang II effect was also blunted by blocking the AT1 and ET (ETA) receptors, by NOX inhibition with apocynin, or by inhibiting the mitochondrial ATP-sensitive potassium channels (5HD or glibenclamide) or its complex I with rotenone (Fig. 1A and B). However, this effect was unaffected by glucocorticoid receptor inhibition with RU-486 (Fig. 1B). These data indicate that at this physiological concentration, Ang II increases myocardial ROS production entirely through ETA receptor activation, and that this increase is NOX dependent and of mitochondrial origin.
Figure 1. Superoxide anion production induced by angiotensin II.
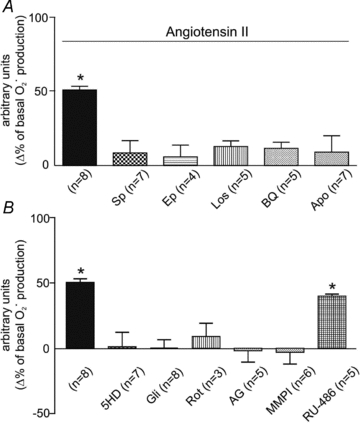
A, MR blockade with spirolactone (Sp, 10 μmol l-1) or eplerenone (Ep, 10 μmol l-1) abrogated the effect of 1 nmol l-1 Ang II on the basal rate of O2· production. This effect was also blunted by the AT1 and ETA receptor antagonists losartan (Los, 1 μmol l-1) and BQ123 (BQ, 10 μmol l-1), respectively, and by NOX inhibition with apocynin (Apo, 300 μmol l-1). B, Ang II-induced O2· formation was also blunted by targeting mitochondria with 5HD (100 μmol l-1), glibenclamide (Gli, 50 μmol l-1), or rotenone (Rot, 10 μmol l-1), and by preventing EGFR activation either by EGFR blockade with AG1478 (AG, 1 μmol l-1) or by inhibiting the metalloproteinase involved in EGFR transactivation with MMPI (3 μmol l-1). Glucocorticoid receptor inhibition with Ru-486 (10 μmol l-1) did not influence the effect of Ang II. For ease of comparison, the effect of Ang II alone on the basal rate of O2· production was repeated in both panels (filled black bar). *P < 0.05 vs. basal O2· production.
In light of recent reports (Anderson et al. 2004; Duquesnes et al. 2009) and our finding that EGFR transactivation is an intermediate step in SFR development in adult cat myocardium (Villa-Abrille et al. 2010), we next explored the consequences of preventing EGFR transactivation on the ROS-promoting effects of Ang II in rat myocardium. We found that the Ang II-mediated increase in O2· formation was blunted by direct EGFR blockade (AG1478), and by preventing EGFR transactivation with an inhibitor of metalloproteinases (MMPI) (Fig. 1B).
Consistent with the notion that the effects of Ang II are mediated by endogenous ET, the effect of exogenous ET-1, at a dose (1 nmol l-1) that mimicked the effect of 1 nmol l-1 Ang II on basal O2· production, was inhibited by all of the pharmacological interventions (except for AT1 receptor blockade) that effectively inhibited Ang II (Fig. 2). These results confirm previous experiments in cat papillary muscles showing that ET is downstream of AT1 receptors in the chain of events leading to increased O2· formation, and they exclude the possibility of bidirectional crosstalk between Ang II and ET.
Figure 2. Superoxide anion production induced by endothelin-1.
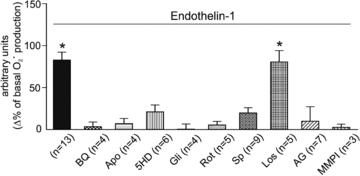
The effect of ET-1 at a concentration (1 nmol l-1) that mimicked the effect of Ang II on the basal rate of O2· production was inhibited by all of the pharmacological interventions that were effective for Ang II except AT1 receptor blockade. These results are consistent with the notion that ET is downstream of AT1 receptors in the chain of events leading to increased O2· formation, and they exclude the possibility of bidirectional crosstalk between the two peptides. *P < 0.05 vs. basal O2· production.
Interestingly, similar results were observed when an equipotent dose of ALD (10 nmol l-1) was used to induce an increase in O2· production. ALD at this concentration increased myocardial O2· production by ∼65% of basal production (Fig. 3A). As expected, the effect of ALD was suppressed by the addition of Sp or Ep, but not by inhibition of glucocorticoid receptors or by preventing protein synthesis with cycloheximide. This suggests that MR activation had a non-genomic effect that was induced by ALD, and it excludes the possibility of glucocorticoid receptor activation. On the other hand, as shown for Ang II and ET-1, the ALD-mediated increase in ROS formation was abrogated by NOX inhibition with apocynin and by preventing EGFR transactivation with MMPI or EGFR blockade (Fig. 3A). These results support the idea that receptor transactivation occurs from the activated MR to EGFR, and that it requires metalloproteinase activation. Interestingly, combined ALD and ET did not promote any further increase in O2· production (Fig. 3B). The increased O2· production was suppressed by MR blockade but unaffected by ETA receptor blockade, supporting the notion that the sequence of events leading to increased ROS formation occurs in the ETA-to-MR direction, and not the other way around.
Figure 3. Superoxide anion production induced by aldosterone.
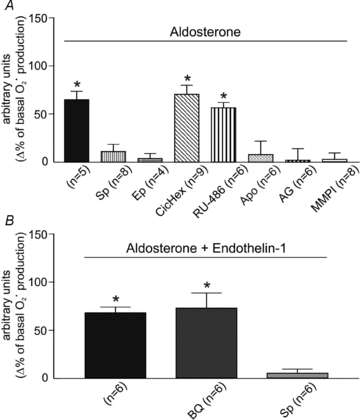
A, the effect of aldosterone at a concentration (10 nmol l-1) that mimicked the effect of Ang II and ET on the basal rate of O2· production was suppressed by spirolactone (Sp) and eplerenone (Ep), but not by the glucocorticoid receptor inhibitor Ru-486 or by preventing protein synthesis with cycloheximide (CicHex, 7 μmol l-1). This demonstrates that MR activation has non-genomic consequences and excludes the possibility of glucocorticoid receptor activation. On the other hand, as shown for Ang II and ET, the ALD-mediated increase in ROS formation was prevented by NOX inhibition (Apo) and by preventing EGFR activation (AG and MMPI). This suggests that transactivation occurs in the direction of activated MR to EGFR, and that metalloproteinase activation downstream of MR is crucial for EGFR transactivation. B, the combination of ALD and ET did not promote any further increase in O2· production. Under this condition, O2· production was abrogated by spironolactone (Sp), but unaffected by ETA receptor blockade with BQ123 (BQ), indicating that the only possible sequence of events is from ETA to MR. *P < 0.05 vs. basal O2· production.
The crucial role of myocardial ROS formation in SFR development appears to be the activation of redox-sensitive kinases upstream of NHE-1, such as ERK1/2 and p90RSK (Caldiz et al. 2007; Villa-Abrille et al. 2010). In agreement with this, 10 nmol l-1 ALD induced an increase in ERK1/2 and p90RSK phosphorylation that was accompanied by an increase in NHE-1 phosphorylation (detected by a phosphospecific antibody that recognizes the phospho-Ser703 in the 14-3-3 protein binding motif of the carboxyl tail of the exchanger) (Fig. 4). These effects were suppressed by the addition of Ep or by preventing metalloproteinase activation with MMPI.
Figure 4. ERK1/2, p90RSK and NHE-1 phosphorylation after aldosterone stimulation.
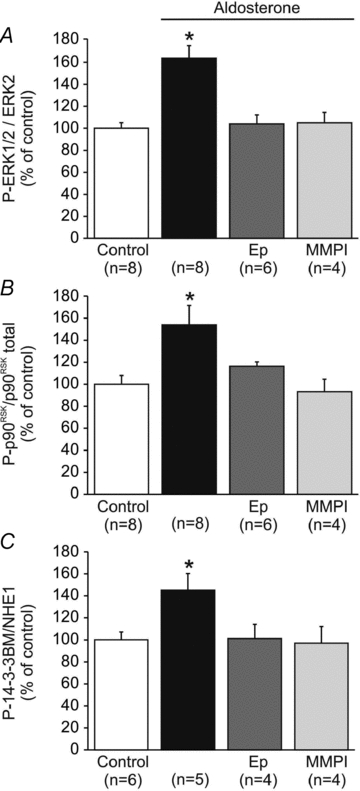
Aldosterone (10 nmol l-1) induced an increase in ERK1/2 (A) and p90RSK (B) phosphorylation that was accompanied by an increase in NHE-1 phophorylation (C). These effects were, as expected, suppressed not only by MR blockade with eplerenone (Ep), but also by preventing metalloproteinase activation with MMPI. These results support the presence of MR–EGFR crosstalk. *P < 0.05 vs. control.
In light of these results showing the signalling pathway activated by ALD, we decided to specifically explore whether MR activation was involved in the SFR in rat myocardium.
Figure 5A–C shows that two different MR blockers, Sp and Ep, completely suppressed the SFR, which was, however, unaffected by glucocorticoid receptor inhibition or by preventing protein synthesis with cycloheximide. Interestingly, we also found that, as reported previously in cat papillary muscles (Villa-Abrille et al. 2010), the SFR required EGFR transactivation, since it was blunted either by direct EGFR inhibition with AG1478 or by cancelling transactivation with the metalloproteinase inhibitor MMPI (Fig. 5D). Furthermore, the SFR was suppressed by the ROS scavenger MPG, providing additional support to the notion that ROS formation is a key factor in the autocrine/paracrine chain of events leading to the Anrep effect (Fig. 5D).
Figure 5. SFR and MR activation.
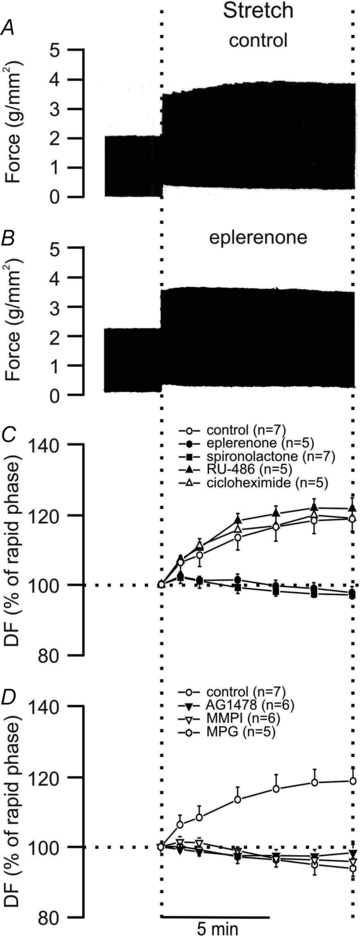
A, typical force record from rat papillary muscle subjected to an increase in length from 92% to 98% of Lmax. The biphasic force response to stretch can be observed. B, same as A but from a muscle pre-treated with the MR blocker eplerenone, demonstrating that prevention of MR activation after stretch eliminated the SFR. C, averaged results of the SFR expressed as percentages of the initial rapid phase. MR blockade, not only by eplerenone but also by spironolactone, completely suppressed the SFR. However, the SFR was unaffected by the glucocorticoid receptor inhibitor Ru-486 or the protein synthesis inhibitor cycloheximide. D, as reported previously in cat myocardium (Villa-Abrille et al. 2010) the SFR required EGFR transactivation, since it was blunted either by direct EGFR inhibition (AG1478) or by blocking transactivation with MMPI. Furthermore, the SFR was suppressed by the ROS scavenger MPG, supporting the notion that ROS formation is a key factor in the chain of events leading to the Anrep effect.
Discussion
To the best of our knowledge, the data presented here constitute the first demonstration that the Anrep effect, reflected in vitro by the SFR, can be blunted by MR inhibition. In previous works, we (Villa-Abrille et al. 2006, 2010; Caldiz et al. 2007; De Giusti et al. 2008) and others (Sand et al. 2003; Anderson et al. 2004; Zhang et al. 2009) proposed a crucial role for mitochondrial ROS formation in the chain of events triggered by stretch and leading to NHE-1 activation to explain the SFR development. However, the possibility of MR activation as an intermediate step between ETA receptors activation and EGFR transactivation was never considered. Indeed, we are not aware of previous reports supporting that myocardial stretch induces MR activation. We present evidence that the Ang II/ET-1-induced mitochondrial ROS formation was inhibited by the corresponding receptor antagonists and by MR antagonists, while the ALD effect was suppressed by its specific receptor antagonists or by preventing EGFR activation (but not by ETA receptor blockade). Thus, the present results suggest that MR activation is downstream of the AT1/ETA receptors and upstream of EGFR in the chain of intracellular signals. This indicates that EGFR transactivation does not occur directly via G protein coupled receptor (GPCR), as we previously suggested (Villa-Abrille et al. 2010). Instead, it occurs via MR. Figure 6 depicts our proposed hypothesis, in which transactivation takes place after metalloproteinase activation following MR activation. In adult rat cardiac myocytes, Rude et al. (2005) showed that ALD stimulates ROS generation and kinases, although neither the sequence of events nor transactivation and NHE-1 activation were explored. The participation of ET in many cardiovascular actions classically attributed to Ang II is well known (Ito et al. 1993; Rajagopalan et al. 1997; Liang & Gardner, 1998; Ortiz et al. 2001; Aiello et al. 2002; Perez et al. 2003; Cingolani et al. 2006; Villa-Abrille et al. 2006). Supporting a role for ET participation in the cardiovascular actions of Ang II, previous works demonstrated an increase in preproET-1 mRNA after the addition of 1 nmol l-1 Ang II to isolated cardiac myocytes (Cingolani et al. 2006; Villa-Abrille et al. 2006), and an increase in ET-3 mRNA after stretching isolated papillary muscles (Ennis et al. 2005), perhaps as an initial step leading to increased ET-1 (Tamamori et al. 1996). These findings may reflect increased ET synthesis secondary to its release after myocardial stretch, most likely in order to replenish intracellular pools. Similarly, Anderson et al. (2004) reported stretch-induced ET-1 release followed by NOX activation in cultured neonatal rat cardiomyocytes. Interestingly, they also reported stretch-induced EGFR transactivation in the same preparation, a finding that was later confirmed by Duquesnes et al. (2009). EGFR transactivation appears to be carried out by metalloproteinase-dependent cleavage of proheparin binding epidermal growth factor (pro-HB-EGF), which promotes ectoshedding of HB-EGF (Anderson et al. 2004; Krieg et al. 2004). HB-EGF is a well-known ligand of EGFR, and binding of HB-EGF to EGFR leads to activation of the receptor by tyrosine autophosphorylation. However, the exact metalloproteinase responsible for the pro-HB-EGF cleavage induced by MR activation is not known. In the present study, we used the hydroxomate-based metalloproteinase inhibitor MMPI, which is considered to be a broad-spectrum inhibitor (Krieg et al. 2004; Rude et al. 2005).
Figure 6. Proposed chain of events triggered by stretch.
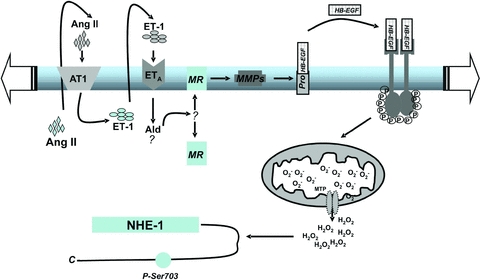
MR activation appears to be located downstream of Ang II/ET release and upstream of EGFR activation in the chain of intracellular signals leading to the SFR. Thus, EGFR transactivation does not originate from GPCR, as suggested previously by us (Villa-Abrille et al. 2010) but from the MR to EGFR. Although our own data and those of Anderson et al. (2004), Duquesnes et al. (2009) and Krieg et al. (2004) appear to support the proposed intracellular signalling pathway described in our study, the data would also be consistent with both EGFR transactivation and MR being required, although acting separately.
The fact that EGFR transactivation can be triggered by MR activation was recognized previously by other authors (Grossmann & Gekle, 2009; Huang et al. 2009; Grossmann et al. 2010). Once EGFR is phosphorylated, the downstream signals increase mitochondrial O2· production. We found that the increase in O2· formation induced by Ang II, ET, or ALD was apocynin sensitive. At present, we do not know which NOX isoform is activated, whether NOX activation occurs at only one or at more than one step in this signalling pathway, or even if trafficking of NOX-dependent ROS signalling within the endosomal compartment is required for redox-dependent signal transduction (Oakley et al. 2009).
A new finding of the present study is that myocardial stretch, in addition to triggering hypertrophic signals such as Ang II/ET, also activates MRs, which are known to induce both hypertrophy and fibrosis (Rude et al. 2005; Zia et al. 2010; Creemers & Pinto, 2011).
We did not explore how the MR becomes activated after myocardial stretch. Actually, our data do not allow concluding that stretch in fact activates the MR. In this regard, either an active MR or an increased activation of the MR is a conceivably valid hypothesis to explain our findings. We may speculate that stretch induces the release or synthesis of ALD by the heart and activates cytosolic or sarcolemmal receptors. This is supported by the finding that ALD is present in heart perfusate and homogenate (Silvestre et al. 1998, 1999; Takeda et al. 2000a), although its origin is disputed (Gomez-Sanchez et al. 2004; Chai & Danser, 2006). Previous reports demonstrated the presence in the heart not only of ALD synthase, a partial processing enzyme that hydroxylates 11-deoxycorticosterone to produce cortiscosterone, 18-hydrocortisone and ALD, but also steroidogenic acute regulatory protein (StAR), which is a crucial factor in the rate-limiting step of ALD biosynthesis (Silvestre et al. 1998, 1999; Takeda et al. 2000a, b; 2001; Casal et al. 2003; Gomez-Sanchez et al. 2004; Chai & Danser, 2006). However, Gomez-Sanchez et al. (2004) and Chai & Danser (2006) have both suggested that there may not be enough of the enzymes in the heart to catalyse the reactions. The simplistic assumption that stretch promotes ALD release/formation with consequent cytosolic or sarcolemmal MR activation cannot be verified without measuring the tissue content of ALD and/or ALD–MR binding in the different cellular fractions before and after myocardial stretch. Interestingly, it was reported that at least a small fraction of classic MR is located in the cell plasma membrane (Grossmann et al. 2010).
Among the putative mechanisms of MR activation after stretch, several ALD-independent pathways should be considered: (1) glucocorticoid-mediated MR activation, especially under conditions of enhanced ROS production, as reported by Mihailidou et al. (2009); (2) direct MR phosphorylation independent of its own ligand, as proposed by Kato et al. (1995) for the oestrogen receptor; and (3) specific changes in MR conformation induced by strain, as proposed by Zou et al. (2004) to explain AT1 receptor activation by mechanical stretch.
Experiments performed by Allen & Kurihara (1982), and later confirmed by other authors including us (Kentish & Wrzosek, 1998; Alvarez et al. 1999), demonstrated that the SFR is solely due to a progressive increase in the calcium transient, without changes in myofilament Ca2+ sensitivity. However, recent studies proposed a certain degree of increase in myofilament Ca2+ sensitivity during the SFR in human normal atrium (Kockskamper et al. 2008a,b) and in ventricle from end stage heart failure (Kockskamper et al. 2008b).
In summary, activated MRs are required for the effect of stretch on cardiac force, and the mechanism involved may be an autocrine/paracrine effect of ALD, or an ALD-independent pathway, such as glucocorticoid-mediated activation, direct receptor phosphorylation, or specific strain-induced conformational changes of the MR. Our proposal that, in addition to its redox-triggering effect, MR activation is one of the earliest intracellular signals following myocardial stretch provides a reasonable explanation of how oxidative stress and metalloproteinase activation may interact to promote the development of myocardial fibrosis and hypertrophy. Finally, from a clinical perspective, our findings suggest that prevention of oxidative stress should be considered as a potential key factor for the salutary effects of ALD antagonism in humans.
Acknowledgments
This work was supported in part by grants PICT 25475 and 01031 from Agencia Nacional de Promoción Científica of Argentina to H.E.C. and N.G.P., respectively, and PIP 1386 from Consejo Nacional de Investigaciones Científicas y Técnicas of Argentina to N.G.P. We specially thank Gador SA, Buenos Aires, Argentina for kindly providing eplerenone. R. G. Díaz is a Fellow of Agencia Nacional de Promoción Científicas y Tecnológica (ANPCyT), Argentina. G. E. Chiappe de Cingolani, I. L. Ennis, H. E. Cingolani and N. G. Pérez are Established Investigators of Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina.
Glossary
Abbreviations
- ALD
aldosterone
- Ang II
angiotensin II
- AT1
angiotensin type 1 receptor
- Apo
apocinyn
- DF
developed force
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- Ep
eplerenone
- ERK1/2
extracellular signal-regulated protein kinases
- ET
endothelin
- ETA
endothelin type A receptor
- GPCR
G protein coupled receptor
- pro-HB-EGF
proheparin binding-epidermal growth factor
- MMPI
metalloproteinase inhibitor
- MPG
2-mercapto-propionyl-glycine
- MR
mineralocorticoid receptor
- NHE-1
Na+/H+ exchanger-1
- NOX
NAPDH oxidase
- O2·
superoxide anion
- p90RSK
p90 ribosomal S6 kinase
- ROS
reactive oxygen species
- SFR
slow force response
- SOD
superoxidase dismutase
- Sp
spironolactone
- StAR
steroidogenic acute regulatory protein
Author contributions
I.L.E., H.E.C. and N.G.P. have contributed to conception and design of the experiments, collection, analysis and interpretation of data, and drafting the article and revising it critically for important intellectual content. C.I.C., R.G.D., M.B.N. and G.E.C. have contributed to collection, analysis and interpretation of data, and drafting the article and revising it critically for important intellectual content. All authors have read and approved the final version of the manuscript.
References
- Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2011;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello EA, Villa-Abrille MC, Cingolani HE. Autocrine stimulation of cardiac Na+-Ca2+ exchanger currents by endogenous endothelin released by angiotensin II. Circ Res. 2002;90:374–376. doi: 10.1161/hh0402.105373. [DOI] [PubMed] [Google Scholar]
- Alvarez BV, Perez NG, Ennis IL, Camilion de Hurtado MC, Cingolani HE. Mechanisms underlying the increase in force and Ca2+ transient that follow stretch of cardiac muscle: a possible explanation of the Anrep effect. Circ Res. 1999;85:716–722. doi: 10.1161/01.res.85.8.716. [DOI] [PubMed] [Google Scholar]
- Allen DG, Kurihara S. The effects of muscle length on intracellular calcium transients in mammalian cardiac muscle. J Physiol. 1982;327:79–94. doi: 10.1113/jphysiol.1982.sp014221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson HD, Wang F, Gardner DG. Role of the epidermal growth factor receptor in signaling strain-dependent activation of the brain natriuretic peptide gene. J Biol Chem. 2004;279:9287–9297. doi: 10.1074/jbc.M309227200. [DOI] [PubMed] [Google Scholar]
- Caldiz CI, Garciarena CD, Dulce RA, Novaretto LP, Yeves AM, Ennis IL, Cingolani HE, Chiappe de Cingolani G, Perez NG. Mitochondrial reactive oxygen species activate the slow force response to stretch in feline myocardium. J Physiol. 2007;584:895–905. doi: 10.1113/jphysiol.2007.141689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casal AJ, Silvestre JS, Delcayre C, Capponi AM. Expression and modulation of steroidogenic acute regulatory protein messenger ribonucleic acid in rat cardiocytes and after myocardial infarction. Endocrinology. 2003;144:1861–1868. doi: 10.1210/en.2002-220943. [DOI] [PubMed] [Google Scholar]
- Cingolani HE, Ennis IL, Aiello EA, Perez NG. Role of autocrine/paracrine mechanisms in response to myocardial strain. Pflugers Arch. 2011;462:29–38. doi: 10.1007/s00424-011-0930-9. [DOI] [PubMed] [Google Scholar]
- Cingolani HE, Villa-Abrille MC, Cornelli M, Nolly A, Ennis IL, Garciarena C, Suburo AM, Torbidoni V, Correa MV, Camilionde Hurtado MC, Aiello EA. The positive inotropic effect of angiotensin II: role of endothelin-1 and reactive oxygen species. Hypertension. 2006;47:727–734. doi: 10.1161/01.HYP.0000208302.62399.68. [DOI] [PubMed] [Google Scholar]
- Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res. 2011;89:265–272. doi: 10.1093/cvr/cvq308. [DOI] [PubMed] [Google Scholar]
- Chai W, Danser AH. Why are mineralocorticoid receptor antagonists cardioprotective? Naunyn Schmiedebergs Arch Pharmacol. 2006;374:153–162. doi: 10.1007/s00210-006-0107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Giusti VC, Correa MV, Villa-Abrille MC, Beltrano C, Yeves AM, de Cingolani GE, Cingolani HE, Aiello EA. The positive inotropic effect of endothelin-1 is mediated by mitochondrial reactive oxygen species. Life Sci. 2008;83:264–271. doi: 10.1016/j.lfs.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquesnes N, Vincent F, Morel E, Lezoualc'h F, Crozatier B. The EGF receptor activates ERK but not JNK Ras-dependently in basal conditions but ERK and JNK activation pathways are predominantly Ras-independent during cardiomyocyte stretch. Int J Biochem Cell Biol. 2009;41:1173–1181. doi: 10.1016/j.biocel.2008.09.032. [DOI] [PubMed] [Google Scholar]
- Ennis IL, Garciarena CD, Perez NG, Dulce RA, Camilion de Hurtado MC, Cingolani HE. Endothelin isoforms and the response to myocardial stretch. Am J Physiol Heart Circ Physiol. 2005;288:H2925–2930. doi: 10.1152/ajpheart.01202.2004. [DOI] [PubMed] [Google Scholar]
- Gomez-Sanchez EP, Ahmad N, Romero DG, Gomez-Sanchez CE. Origin of aldosterone in the rat heart. Endocrinology. 2004;145:4796–4802. doi: 10.1210/en.2004-0295. [DOI] [PubMed] [Google Scholar]
- Grossmann C, Gekle M. New aspects of rapid aldosterone signaling. Mol Cell Endocrinol. 2009;308:53–62. doi: 10.1016/j.mce.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Grossmann C, Husse B, Mildenberger S, Schreier B, Schuman K, Gekle M. Colocalization of mineralocorticoid and EGF receptor at the plasma membrane. Biochim Biophys Acta. 2010;1803:584–590. doi: 10.1016/j.bbamcr.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Huang S, Zhang A, Ding G, Chen R. Aldosterone-induced mesangial cell proliferation is mediated by EGF receptor transactivation. Am J Physiol Renal Physiol. 2009;296:F1323–1333. doi: 10.1152/ajprenal.90428.2008. [DOI] [PubMed] [Google Scholar]
- Ito H, Hirata Y, Adachi S, Tanaka M, Tsujino M, Koike A, Nogami A, Murumo F, Hiroe M. Endothelin-1 is an autocrine/paracrine factor in the mechanism of angiotensin II-induced hypertrophy in cultured rat cardiomyocytes. J Clin Invest. 1993;92:398–403. doi: 10.1172/JCI116579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- Kentish JC, Wrzosek A. Changes in force and cytosolic Ca2+ concentration after length changes in isolated rat ventricular trabeculae. J Physiol. 1998;506:431–444. doi: 10.1111/j.1469-7793.1998.431bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockskamper J, Khafaga M, Grimm M, Elgner A, Walther S, Kockskamper A, von Lewinski D, Post H, Grossmann M, Dorge H, Gottlieb PA, Sachs F, Eschenhagen T, Schondube FA, Pieske B. Angiotensin II and myosin light-chain phosphorylation contribute to the stretch-induced slow force response in human atrial myocardium. Cardiovasc Res. 2008a;79:642–651. doi: 10.1093/cvr/cvn126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockskamper J, von Lewinski D, Khafaga M, Elgner A, Grimm M, Eschenhagen T, Gottlieb PA, Sachs F, Pieske B. The slow force response to stretch in atrial and ventricular myocardium from human heart: functional relevance and subcellular mechanisms. Prog Biophys Mol Biol. 2008b;97:250–267. doi: 10.1016/j.pbiomolbio.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg T, Cui L, Qin Q, Cohen MV, Downey JM. Mitochondrial ROS generation following acetylcholine-induced EGF receptor transactivation requires metalloproteinase cleavage of proHB-EGF. J Mol Cell Cardiol. 2004;36:435–443. doi: 10.1016/j.yjmcc.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Lehoux S, Abe J, Florian JA, Berk BC. 14-3-3 Binding to Na+/H+ exchanger isoform-1 is associated with serum-dependent activation of Na+/H+ exchange. J Biol Chem. 2001;276:15794–15800. doi: 10.1074/jbc.M100410200. [DOI] [PubMed] [Google Scholar]
- Lemarie CA, Paradis P, Schiffrin EL. New insights on signaling cascades induced by cross-talk between angiotensin II and aldosterone. J Mol Med. 2008;86:673–678. doi: 10.1007/s00109-008-0323-5. [DOI] [PubMed] [Google Scholar]
- Lemarie CA, Simeone SM, Nikonova A, Ebrahimian T, Deschenes ME, Coffman TM, Paradis P, Schiffrin EL. Aldosterone-induced activation of signaling pathways requires activity of angiotensin type 1a receptors. Circ Res. 2009;105:852–859. doi: 10.1161/CIRCRESAHA.109.196576. [DOI] [PubMed] [Google Scholar]
- Liang F, Gardner DG. Autocrine/paracrine determinants of strain-activated brain natriuretic peptide gene expression in cultured cardiac myocytes. J Biol Chem. 1998;273:14612–14619. doi: 10.1074/jbc.273.23.14612. [DOI] [PubMed] [Google Scholar]
- Mihailidou AS, Loan Le TY, Mardini M, Funder JW. Glucocorticoids activate cardiac mineralocorticoid receptors during experimental myocardial infarction. Hypertension. 2009;54:1306–1312. doi: 10.1161/HYPERTENSIONAHA.109.136242. [DOI] [PubMed] [Google Scholar]
- Oakley FD, Abbott D, Li Q, Engelhardt JF. Signaling components of redox active endosomes: the redoxosomes. Antioxid Redox Signal. 2009;11:1313–1333. doi: 10.1089/ars.2008.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz MC, Sanabria E, Manriquez MC, Romero JC, Juncos LA. Role of endothelin and isoprostanes in slow pressor responses to angiotensin II. Hypertension. 2001;37:505–510. doi: 10.1161/01.hyp.37.2.505. [DOI] [PubMed] [Google Scholar]
- Perez NG, de Hurtado MC, Cingolani HE. Reverse mode of the Na+-Ca2+ exchange after myocardial stretch: underlying mechanism of the slow force response. Circ Res. 2001;88:376–382. doi: 10.1161/01.res.88.4.376. [DOI] [PubMed] [Google Scholar]
- Perez NG, Nolly MB, Roldan MC, Villa-Abrille MC, Cingolani E, Portiansky EL, Alvarez BV, Ennis IL, Cingolani HE. Silencing of NHE-1 blunts the slow force response to myocardial stretch. J Appl Physiol. 2011;111:874–880. doi: 10.1152/japplphysiol.01344.2010. [DOI] [PubMed] [Google Scholar]
- Perez NG, Villa-Abrille MC, Aiello EA, Dulce RA, Cingolani HE, Camilion de Hurtado MC. A low dose of angiotensin II increases inotropism through activation of reverse Na+/Ca2+ exchange by endothelin release. Cardiovasc Res. 2003;60:589–597. doi: 10.1016/j.cardiores.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Laursen JB, Borthayre A, Kurz S, Keiser J, Haleen S, Giaid A, Harrison DG. Role for endothelin-1 in angiotensin II-mediated hypertension. Hypertension. 1997;30:29–34. doi: 10.1161/01.hyp.30.1.29. [DOI] [PubMed] [Google Scholar]
- Rothstein EC, Byron KL, Reed RE, Fliegel L, Lucchesi PA. H2O2-induced Ca2+ overload in NRVM involves ERK1/2 MAP kinases: role for an NHE-1-dependent pathway. Am J Physiol Heart Circ Physiol. 2002;283:H598–605. doi: 10.1152/ajpheart.00198.2002. [DOI] [PubMed] [Google Scholar]
- Rude MK, Duhaney TA, Kuster GM, Judge S, Heo J, Colucci WS, Siwik DA, Sam F. Aldosterone stimulates matrix metalloproteinases and reactive oxygen species in adult rat ventricular cardiomyocytes. Hypertension. 2005;46:555–561. doi: 10.1161/01.HYP.0000176236.55322.18. [DOI] [PubMed] [Google Scholar]
- Sabri A, Byron KL, Samarel AM, Bell J, Lucchesi PA. Hydrogen peroxide activates mitogen-activated protein kinases and Na+-H+ exchange in neonatal rat cardiac myocytes. Circ Res. 1998;82:1053–1062. doi: 10.1161/01.res.82.10.1053. [DOI] [PubMed] [Google Scholar]
- Sand C, Peters SL, Pfaffendorf M, van Zwieten PA. The influence of endogenously generated reactive oxygen species on the inotropic and chronotropic effects of adrenoceptor and ET-receptor stimulation. Naunyn Schmiedebergs Arch Pharmacol. 2003;367:635–639. doi: 10.1007/s00210-003-0745-0. [DOI] [PubMed] [Google Scholar]
- Silvestre JS, Heymes C, Oubenaissa A, Robert V, Aupetit-Faisant B, Carayon A, Swynghedauw B, Delcayre C. Activation of cardiac aldosterone production in rat myocardial infarction: effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation. 1999;99:2694–2701. doi: 10.1161/01.cir.99.20.2694. [DOI] [PubMed] [Google Scholar]
- Silvestre JS, Robert V, Heymes C, Aupetit-Faisant B, Mouas C, Moalic JM, Swynghedauw B, Delcayre C. Myocardial production of aldosterone and corticosterone in the rat. Physiological regulation. J Biol Chem. 1998;273:4883–4891. doi: 10.1074/jbc.273.9.4883. [DOI] [PubMed] [Google Scholar]
- Snabaitis AK, D'Mello R, Dashnyam S, Avkiran M. A novel role for protein phosphatase 2A in receptor-mediated regulation of the cardiac sarcolemmal Na+/H+ exchanger NHE1. J Biol Chem. 2006;281:20252–20262. doi: 10.1074/jbc.M600268200. [DOI] [PubMed] [Google Scholar]
- Sugden PH, Clerk A. Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxid Redox Signal. 2006;8:2111–2124. doi: 10.1089/ars.2006.8.2111. [DOI] [PubMed] [Google Scholar]
- Takahashi E, Abe J, Gallis B, Aebersold R, Spring DJ, Krebs EG, Berk BC. p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J Biol Chem. 1999;274:20206–20214. doi: 10.1074/jbc.274.29.20206. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Yoneda T, Demura M, Furukawa K, Miyamori I, Mabuchi H. Effects of high sodium intake on cardiovascular aldosterone synthesis in stroke-prone spontaneously hypertensive rats. J Hypertens. 2001;19:635–639. doi: 10.1097/00004872-200103001-00017. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Yoneda T, Demura M, Miyamori I, Mabuchi H. Cardiac aldosterone production in genetically hypertensive rats. Hypertension. 2000a;36:495–500. doi: 10.1161/01.hyp.36.4.495. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Yoneda T, Demura M, Miyamori I, Mabuchi H. Sodium-induced cardiac aldosterone synthesis causes cardiac hypertrophy. Endocrinology. 2000b;141:1901–1904. doi: 10.1210/endo.141.5.7529. [DOI] [PubMed] [Google Scholar]
- Tamamori M, Ito H, Adachi S, Akimoto H, Marumo F, Hiroe M. Endothelin-3 induces hypertrophy of cardiomyocytes by the endogenous endothelin-1-mediated mechanism. J Clin Invest. 1996;97:366–372. doi: 10.1172/JCI118424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Abrille MC, Caldiz CI, Ennis IL, Nolly MB, Casarini MJ, Chiappe de Cingolani GE, Cingolani HE, Perez NG. The Anrep effect requires transactivation of the epidermal growth factor receptor. J Physiol. 2010;588:1579–1590. doi: 10.1113/jphysiol.2009.186619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Abrille MC, Cingolani HE, Garciarena CD, Ennis IL, Aiello EA. [Angiotensin II-induced endothelin-1 release in cardiac myocytes] Medicina. 2006;66:229–236. [PubMed] [Google Scholar]
- von Anrep G. On the part played by the suprarenals in the normal vascular reactions of the body. J Physiol. 1912;45:307–317. doi: 10.1113/jphysiol.1912.sp001553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Rothstein EC, Fliegel L, Dell'Italia LJ, Lucchesi PA. Differential MAP kinase activation and Na+/H+ exchanger phosphorylation by H2O2 in rat cardiac myocytes. Am J Physiol Cell Physiol. 2001;281:C1542–1550. doi: 10.1152/ajpcell.2001.281.5.C1542. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Dingle L, Hall R, Casadei B. The role of nitric oxide and reactive oxygen species in the positive inotropic response to mechanical stretch in the mammalian myocardium. Biochim Biophys Acta. 2009;1787:811–817. doi: 10.1016/j.bbabio.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zia AA, Kamalov G, Newman KP, McGee JE, Bhattacharya SK, Ahokas RA, Sun Y, Gerling IC, Weber KT. From aldosteronism to oxidative stress: the role of excessive intracellular calcium accumulation. Hypertens Res. 2010;33:1091–1101. doi: 10.1038/hr.2010.159. [DOI] [PubMed] [Google Scholar]
- Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]