

Abstract
Non-technical summary
Each heartbeat is accompanied by the coordinated release of calcium ions into cardiac cells through ryanodine receptors, which span the membrane of the sarcoplasmic reticulum. We show that an intra-sarcoplasmic reticulum protein, junctin, interacts with ryanodine receptor channels and appears to activate them when calcium inside the sarcoplasmic reticulum is low. Conversely, junctin appears to act as an inhibitor of ryanodine receptors when calcium inside the sarcoplasmic reticulum is high. Knowledge of how junctin interacts with ryanodine receptors helps us to understand how calcium within the sarcoplasmic reticulum helps to regulate ryanodine receptor activity in normal hearts and also helps us to understand why junctin is decreased in patients diagnosed with certain forms of heart failure.
Abstract
Junctin, a 26 kDa intra-sarcoplasmic reticulum (SR) protein, forms a quaternary complex with triadin, calsequestrin and the ryanodine receptor (RyR) at the junctional SR membrane. The physiological role for junctin in the luminal regulation of RyR Ca2+ release remains unresolved, but it appears to be essential for proper cardiac function since ablation of junctin results in increased ventricular automaticity. Given that the junctin levels are severely reduced in human failing hearts, we performed an in-depth study of the mechanisms affecting intracellular Ca2+ homeostasis in junctin-deficient cardiomyocytes. In concurrence with sparks, JCN-KO cardiomyocytes display increased Ca2+ transient amplitude, resulting from increased SR [Ca2+] ([Ca2+]SR). Junctin ablation appears to affect how RyRs ‘sense’ SR Ca2+ load, resulting in decreased diastolic SR Ca2+ leak despite an elevated [Ca2+]SR. Surprisingly, the β-adrenergic enhancement of [Ca2+]SR reverses the decrease in RyR activity and leads to spontaneous Ca2+ release, evidenced by the development of spontaneous aftercontractions. Single channel recordings of RyRs from WT and JCN-KO cardiac SR indicate that the absence of junctin produces a dual effect on the normally linear response of RyRs to luminal [Ca2+]: at low luminal [Ca2+] (<1 mmol l−1), junctin-devoid RyR channels are less responsive to luminal [Ca2+]; conversely, high luminal [Ca2+] turns them hypersensitive to this form of channel modulation. Thus, junctin produces complex effects on Ca2+ sparks, transients, and leak, but the luminal [Ca2+]-dependent dual response of junctin-devoid RyRs demonstrates that junctin normally acts as an activator of RyR channels at low luminal [Ca2+], and as an inhibitor at high luminal [Ca2+]. Because the crossover occurs at a [Ca2+]SR that is close to that present in resting cells, it is possible that the activator-inhibitor role of junctin may be exerted under periods of prevalent parasympathetic and sympathetic activity, respectively.
Introduction
Cardiac ryanodine receptors (RyR2s) are the Ca2+ release channels of sarcoplasmic reticulum (SR), which function to provide the majority of Ca2+ for contraction of the heart. RyR2s are exquisitely regulated by a myriad of cytosolic factors (Fill & Copello, 2002; Lanner et al. 2010; Capes et al. 2011). Nevertheless, because small elevations in SR Ca2+ load cause steep increases in Ca2+ release (Shannon et al. 2003, 2005), luminal (intra-SR) Ca2+ has taken a prominent role in the control of Ca2+ release and cardiac performance. A quaternary protein complex composed of the RyR2, the luminal Ca2+ buffering protein calsequestrin (Csq2), and the SR transmembrane spanning proteins triadin-1 and junctin (Zhang et al. 1997) are believed to be involved in determining the RyR2 response to alterations in SR Ca2+ load.
Junctin is a 26 kDa SR membrane-spanning protein with a 165 amino acid luminal domain containing KEKE motifs, which are repeated sequences of 13 amino acids or longer starting and ending with Lys or Glu and where >60% of the residues are Lys or Glu/Asp. These KEKE motifs are presumed to be involved in interactions between junctin and Csq2, RyR2, and triadin-1 (Jones et al. 1995). The current model of RyR2 regulation by intraluminal proteins places junctin in direct contact with luminal and/or membrane-spanning segments of the RyR2 and also as an anchoring protein for Csq2 (Zhang et al. 1997; Wang et al. 1998; Györke et al. 2004). However, whether junctin serves as direct ligand of RyR2 or as transducer of Csq2 signals as the latter binds Ca2+ is less established. Recent experiments have shown that cardiac-specific overexpression of junctin results in alterations in cellular morphology including a tighter packing of Csq2 in the proximity of the junctional SR membrane and a larger surface association of the SR with the T-tubular region (Zhang et al. 2001; Tijskens et al. 2003). Junctin overexpression also had profound effects on Ca2+ handling in mouse cardiomyocytes, which led to the development of impaired cellular relaxation and cardiac hypertrophy (Kirchhefer et al. 2003, 2006). Likewise, ablation of the murine junctin gene severely disrupts normal cardiac performance, leading to the development of delayed after-depolarization (DAD)-induced arrhythmias with an increased incidence of sudden death under conditions of physiological stress (Yuan et al. 2007). These studies emphasize the physiological importance of junctin in the maintenance of normal cardiac function.
Junctin depletion (>90%) appears to be among the most consistent findings in several forms of human heart failure (HF), a multifactorial syndrome characterized by pathological chamber remodelling and ventricular dysfunction (Gergs et al. 2007; Pritchard & Kranias, 2009). Unfortunately, a multitude of cellular constituents and signalling pathways are also affected in HF, making it difficult to dissect the contribution of this junctin deficiency to the Ca2+ mishandling and contractile dysfunction that is prominent in the advance stages of this syndrome. In the current study, we utilized the junctin knockout mouse (JCN-KO) to comprehensively determine how cellular Ca2+ homeostasis is affected by the ablation of junctin. Our approach is valid because unlike in HF, or in animal models of junctin overexpression (Kirchhefer et al. 2003, 2006) and triadin ablation (Chopra et al. 2009), the JCN-KO mouse does not undergo structural remodelling or alterations in the density of the most critical proteins of E-C coupling (Yuan et al. 2007). By determining the effects of junctin ablation on single reconstituted RyR2s, Ca2+ sparks and intracellular Ca2+ transients, and SR Ca2+ load under various physiological conditions, we determined that junctin modulates RyR2 through at least two functionally interacting sites, and that junctin may both attenuate and increase RyR2 activity depending on SR Ca2+ loads that are likely to be within the range controlled by β-adrenergic stimulation of ventricular myocytes. Importantly, normal levels of junctin are obligatorily needed to optimize luminal regulation of Ca2+ release and to protect against cardiac arrhythmias. Our results not only expand upon the initial characterization of the JCN-KO mouse (Yuan et al. 2007), but importantly, they may explain the cellular etiology of the DAD-induced arrhythmias in this mouse model.
Methods
Generation of junctin knockout mice
JCN-KO mice were generated as previously described (Yuan et al. 2007). Three- to seven-month-old male and female JCN-KO mice and wild-type littermates were used in this study, and all animal experiments were performed with the approval of the Animal Care and Use Committee at the University of Wisconsin–Madison and in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (publication No. 85-23, revised 1985).
Mouse ventricular cell isolation
Adult mice were treated with 0.075 ml heparin by peritoneal injection and were subjected to cervical dislocation. The heart was quickly excised and placed in ice-cold Tyrode solution containing (in mmol l−1): 130 NaCl, 5.4 KCl, 0.4 NaH2PO4, 0.5 MgCl2, 25 Hepes, 22 glucose, 0.01 μg ml−1 insulin, (pH 7.4 with NaOH). All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise noted. The aorta was cannulated, attached to a Langendorf perfusion apparatus, and perfused with warm (37°C), oxygenated Tyrode solution supplemented with 0.1 mmol l−1 EGTA for 2 min. Mouse ventricular cardiomyocytes were subsequently isolated using modifications of an enzymatic perfusion method (Wolska & Solaro, 1996), as described in Benkusky et al. (2007).
Ca2+ spark measurements
Intact isolated cardiomyocytes were loaded with 10 μmol l−1 Fluo-4 AM (Molecular Probes/Invitrogen, Carlsbad, CA, USA) for 20 min at room temperature and subsequently washed with storage solution. Cardiomyocytes were initially paced at 0.2 Hz for 10 s to ensure cell viability and preload the SR. Following this brief period of stimulation, spontaneous Ca2+ sparks were recorded in quiescent cells, which were continuously perfused with Tyrode solution supplemented with 1 mmol l−1 CaCl2 in the absence or presence of the indicated concentration of Iso. Solutions were perfused onto the cells with a microperfusion system containing a three-barrelled pipette (Warner Instruments, Hamden, CT, USA). All experiments were conducted at room temperature (21–23°C). Ca2+ sparks were visualized using the C-Apochromat 40× water immersion objective of a Zeiss LSM 510 confocal microscope. Fluo-4 was excited with an argon laser at 488 nm, with emitted fluorescence measured at >505 nm. All images were collected in line-scan mode (1.92 ms per line, 0.1–0.3 μm per pixel, with the scan line oriented along the longitudinal axis of the myocytes). All line scan images were processed and analysed using IDL software (Research Systems Inc., v.5.5, Boulder, CO, USA) with modifications custom-written by A. M. Gómez. Images were corrected for background fluorescence, and the fluorescence values (F) were then normalized to the basal fluorescence (F0). Using an automated detection method (Cheng et al. 1999), Ca2+ sparks were detected in regions where the Ca2+ fluorescence increased by four times the standard deviation of the image.
Measurements of intracellular Ca2+ transients and SR Ca2+ load
Intact isolated cardiomyocytes were placed in Tyrode solution supplemented with 1.8 mmol l−1 CaCl2. Cells were loaded with 10 μmol l−1 Fluo-3 AM (Molecular Probes) for 20 min at room temperature, and were then resuspended in the same solution without dye. During image acquisition, cells were continuously perfused with Tyrode solution. Confocal images (using the Zeiss LSM 510 microscope) were acquired in the line-scan mode at 1.92 ms per line as detailed in Ca2+ spark measurements (see above). Fluo-3 AM was excited using the 488 nm line of an argon ion laser. Emission was collected at >505 nm. The line scan was selected parallel to the longitudinal cell axis in order to measure the associated cell shortening. Cells were field-stimulated at 0.5, 1, 2, and 3 Hz (at 40 V) through two platinum electrodes placed along the bath and connected to an external high voltage stimulator, and line scans of steady-state [Ca2+]i transients were collected. Stimulation was then stopped, and Tyrode solution containing the indicated concentration of Iso was perfused onto the cells for 2 min. The entire pacing protocol was then repeated in the presence of Iso. In another set of experiments, cells were field-stimulated at up to four different pacing frequencies (0.5, 1, 2, and 3 Hz) either in the absence or presence of Iso, and caffeine (10 mmol l−1) was rapidly perfused to estimate the SR Ca2+ load after each pacing frequency. Alternatively, some cells were placed in the Tyrode solution, and without initially field-stimulating them, 10 mmol l−1 caffeine was immediately perfused in order to estimate the SR Ca2+ load in a quiescent population of cells. Experiments were carried out at room temperature (21–23°C). All confocal line scans were corrected for the background fluorescence. The fluorescence values (F) were then normalized to the basal fluorescence (from a region of the image before each electrical stimulation, F0) to obtain the fluorescence ratio (F/F0).
Measurements of diastolic SR Ca2+ leak and cellular aftercontractions
Mouse ventricular cardiomyocytes were isolated and loaded with Ca2+ indicator as described above, and SR Ca2+ leak was measured using a protocol described previously (Shannon et al. 2003). Briefly, resting [Ca2+]i was measured using Fluo-3 AM fluorescence in the presence and absence of SR Ca2+ leak. Cardiomyocytes were stimulated at least 20 times at 0.5, 1, 2, or 3 Hz in normal Tyrode solution supplemented with 1.8 mmol l−1 Ca2+ to bring the SR Ca2+ content to steady-state. After the last pulse, the perfusion was switched to a 0 Ca2+/0 Na+ solution containing (in mmol l−1): 130 LiCl, 5.4 KCl, 0.4 KH2PO4, 0.5 MgCl2, 25 Hepes, 10 EGTA-K, 22 glucose, 0.01 μg ml−1 insulin (pH 7.4 with KOH). Under these conditions, Na+–Ca2+ exchange, the primary Ca2+ influx and efflux mechanism at rest, was blocked so that little or no Ca2+ left the cell. The 0 Ca2+/0 Na+ solution was perfused for 1 min, and the [Ca2+]i was measured as the diastolic fluorescence signal (F0). After 1 min, the perfusate was switched to 0 Ca2+/0 Na+ solution supplemented with 1 mmol l−1 tetracaine, in order to block RyR2 Ca2+ leak (Overend et al. 1997). After 1 min, the F0 in the presence of tetracaine was measured, and the SR leak was determined by the following equation: SR Ca2+ leak = F0(0Ca/0Na)–F0(Tetra). The entire protocol was then repeated in the presence of 1 μmol l−1 Iso supplemented in each of the three perfusion solutions. For the measurement of spontaneous aftercontractions, cardiomyocytes were perfused with Tyrode solution containing 1.8 mmol l−1 Ca2+ and 1 μmol l−1 Iso for 2 min. Subsequently, cells were paced at 3 Hz for 2–3 s, and the pacing was then stopped to allow for the recording of spontaneous aftercontractions. Confocal line-scan images were taken for up to ∼20 s. All experiments were carried out at room temperature (21–23°C), and fluorescence measurements were obtained using IDL software as described above.
Preparation of SR-enriched mouse microsomes and planar bilayer recordings of RyR2
Mouse cardiac homogenates and SR-enriched microsomes were obtained as described previously (Benkusky et al. 2007). WT and JCN-KO single RyR2 channels were reconstituted into planar lipid bilayers, also as previously described (Farrell et al. 2003; Altschafl et al. 2007). Briefly, the trans (700 μl) and the cis (700 μl) chambers (corresponding to the luminal and cytosolic side of the channel, respectively) contained symmetrical 300 mmol l−1 CsCHSO3 and 20 mmol l−1 Na-Hepes (pH 7.2). A phospholipid bilayer of phosphatidylethanolamine and phosphatidylserine (1:1 dissolved in 20 mg ml−1 n-decane) was ‘painted’ with a glass rod across an aperture of ∼200 μm diameter in a delrin cup. The trans chamber was the voltage control side connected to the head stage of a 200A Axopatch amplifier (Molecular Devices, Sunnyvale, CA, USA), while the cis side was held at virtual ground. All recordings were performed in the presence of nominal Ca2+ (3–5 μmol l−1, as determined by a Ca2+ electrode) bathing the cis chamber, and CaCl2 was then added in step increments to the trans chamber to raise free [Ca2+] to 0.01–10 mmol l−1. Channel activity was filtered with an 8-pole low pass Bessel filter set at 1.5 kHz and digitized at a rate of 4 kHz using a Digidata 1200 AD/DA interface. Data acquisition and analysis were performed with Molecular Devices (Axon Instruments) hardware and software (pCLAMP 8).
Immunoblotting
Samples run in NuPAGE (Invitrogen Corp., Carlsbad, CA, USA) Novex 4–12% Bis-Tris gel electrophoresis in MES SDS running buffer were transferred to nitrocellulose membranes. The membranes were incubated with either of the following primary antibodies: goat polyclonal anti-glutathione S-transferase (GST), horseradish peroxidase (HRP) conjugate (1:5000; Amersham Biosciences Europe GmbH, Freiburg, Germany), or murine monoclonal anti-maltose binding protein (MBP) covalently linked to HRP (1:2000; New England Biolabs Inc., Ipswich, MA, USA). Protein signals were detected using electrogenerated chemiluminescence (ECL) reagents according to the manufacturer's protocol (Amersham Biosciences Europe Gmbh). The intensities of the bands of interest from at least three different experiments were quantified with the use of ImageJ software (v. 1.33a).
Construction of recombinant proteins
For the generation of human RyR2 and junctin constructs, total RNA was extracted from human, post-mortem, non-failing left ventricular heart tissue using Trizol (Invitrogen Corp.), and reverse transcribed by Superscript II RNAse H reverse transcriptase (Invitrogen Corp.). PCR products were digested with EcoRI and SalI (Takara Bio Inc., Shiga, Japan) and subcloned into EcoRI/SalI cloning sites of either pGEX-5x-1 or pMAL-c2X vector (New England Biolabs). The following primer sets were used for amplification: RyR2-A (4520–4553 amino acid (aa) residues), 5′-GAA TTC TAT AAG GTC TCC ACT TCT TCT CAC-3′, 5′-GTC GAC TCT ATG GGA GCT GCT GTC CAG-3′; RyR2-B (4789–4818 aa), 5′-GAA TTC TTC CGA AAA TTC TAC AAT AAA-3′, 5′-GTC GAC ATA CAT GTG GAA CAT ATA GCA-3′; RyR2-C (4819–4846 aa), 5′-GAA TTC GTT GGA GTT CGT GCT GGA GGA-3′, 5′-GTC GAC AAA GAT GAT TCG ATA GAT CTC-3′; RyR2-D (4789–4846 aa), 5′-GAA TTC TTC CGA AAA TTC TAC AAT AAA-3′, 5′-GTC GAC AAA GAT GAT TCG ATA GAT CTC-3′; junctin-A (47–77 aa) and junctin iso(1) (the exon 2A containing junctin isoform, 47–92 aa), 5′-GAA TTC GAT CTT GTT GAC TAT GAG GAA G-3′, 5′-GTC GAC TAA AAC TTT GGC ATC ATC CAC-3′; junctin-B (78–161 aa), 5′-GAA TTC TTA GAA GGA CCC AGT GGG GTA-3′, 5′-GTC GAC ACC CTT AGG AGA CTC CTT CCT-3′; junctin-C (160–210 aa), 5′-GAA TTC AAG GGT AAA AAG GAC AGA GAA A-3′, 5′-GTC GAC TTA GCC GTT TCT TTT CTG GGT-3′; junctin-D (78–210 aa), 5′-GAA TTC TTA GAA GGA CCC AGT GGG GTA-3′, 5′-GTC GAC TTA GCC GTT TCT TTT CTG GGT-3′. Following preparation of these DNA fragments and digestion with EcoRI and SalI, they were subcloned in the pGEX-5x-1 and pMAL-c2X vectors. The obtained clones were verified by direct sequencing. GST and MBP recombinant polypeptides were generated following induction with 0.5 mmol l−1 isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h, in BL21 Star (DE3) (Invitrogen Corp.) and library efficient DH5a E. coli strains, respectively, according to the manufacturer's instructions. Recombinant polypeptides were purified by affinity chromatography by either Glutathione Sepharose 4B for GST fusion proteins or amylose resin for MBP fusion proteins.
Blot overlay assays
Briefly, ∼2.5 μg of bacterially expressed, affinity-purified MBP, MBP–junctin iso(1) (47–92 aa), MBP–junctin-A (47–77 aa), MBP–junctin-B (78–161 aa), MBP–junctin-C (160–210 aa), and MBP–junctin-D (78–210 aa), as well as GST, GST–RyR2-A (4520–4553 aa), GST–RyR2-B (4789–4818 aa), GST–RyR2-C (4819–4846 aa), and GST–RyR2-D (4789–4846 aa) fusion proteins were separated on different gels by NuPAGE Novex 4–12% Bis-Tris gel electrophoresis system and transferred to nitrocellulose membranes. Non-specific sites on the nitrocellulose membranes were blocked in buffer A (50 mmol l−1 Tris, pH 7.2, 140 mmol l−1 NaCl, 0.1% Tween 20, 5% non-fat skimmed milk, 2 mmol l−1 DTT, 0.5% Nonidet P-40) plus protease inhibitors for 1 h at 25°C. Then the membranes of MBP–junctin analysed constructs were overlaid with 3 μg ml−1 of either GST–RyR2-A, -B, -C, or -D fusion proteins, while the membranes of GST–RyR2 constructs were overlaid with 3 μg ml−1 of either MBP–junctin iso(1), MBP–junctin-A, -B, -C, or -D fusion proteins in the same buffer in the presence of 1 mmol l−1 ATP for 2 h at 25°C. Blots were washed 5× (15 min each) with buffer A and once with buffer B (1× phosphate-buffered saline (PBS), pH 7.2, 10 mmol l−1 NaN3, and 0.1% Tween 20). Following blocking of non-specific reactive sites in buffer C (1× PBS, pH 7.2, 10 mmol l−1 NaN3, 0.1% Tween 20, and 3% non-fat skimmed milk), membranes were probed with goat anti-GST HRP conjugate or rabbit anti-MBP HRP conjugate antibody, respectively, diluted in buffer C. Immunoreactive bands were visualized using ECL reagents (Amersham Biosciences).
Statistical analysis
Statistical analysis was performed using either the Origin (v. 7.5, OriginLab Corp., Northampton, MA, USA) or SigmaStat software (v. 3.0, RockWare Inc., Golden, CO, USA). Data are presented as the mean ± SEM. Data were compared using Student's unpaired t test or a one-way analysis of variance (with secondary comparisons made using a Student–Newman–Keuls test). Differences between sample means were considered significant if P < 0.05 unless indicated otherwise.
Results
Human failing hearts appear to be ‘junctin-deficient’ (Gergs et al. 2007), but the alteration of density and function of multiple cellular components precludes direct determination of junctin's role in this syndrome. Our first characterization of the JCN-KO model used here revealed that junctin deficiency leads to intracellular Ca2+ mishandling and increased ventricular automaticity (Yuan et al. 2007), most likely the underlying mechanisms of the premature death observed in these mice. In this study, we designed an in-depth approach to shed light on the molecular and cellular basis of the increased ventricular automaticity resulting from junctin deficiency and to understand the normal role of junctin under basal and adrenergically stimulated conditions. Towards these efforts, measurements of Ca2+ sparks and Ca2+ transients were reassessed, but with the notable differences outlined below.
Ca2+ sparks in intact WT and JCN-KO cardiomyocytes
Ca2+ sparks result from spontaneous and coordinated openings of a cluster of RyRs gating in situ (Cheng & Lederer, 2008), and thus examination of individual Ca2+ spark parameters reveals localized RyR activity in its resting state. In a previous study, we showed that Ca2+ sparks in permeabilized myocytes of JCN-KO mice were altered with respect to WT (Yuan et al. 2007), but those measurements were done in an artificially controlled intracellular milieu that precluded characterization of Ca2+ signalling under the influence of β-adrenergic stimulation and under a more physiological setting. Figure 1A and B shows representative line scan images and three-dimensional Ca2+ spark reconstructions obtained from intact WT and JCN-KO cardiomyocytes under basal conditions. Ca2+ spark frequency is significantly reduced (Fig. 1C) but Ca2+ spark amplitude is higher in intact JCN-KO compared with WT cardiomyocytes (Fig. 1D). Moreover, the temporal properties of the Ca2+ sparks are different: the time-to-peak (TTP) of the Ca2+ release event is faster (Fig. 1G) and the full duration at half-maximal amplitude (FDHM) is significantly reduced in JCN-KO cardiomyocytes (Fig. 1E). On the other hand, the full width at half-maximal amplitude (FWHM) is the same for both groups of cells indicating that there is no change in the spatial spread of the Ca2+ sparks (Fig. 1F). JCN-KO cardiomyocytes thus produce fewer but brighter Ca2+ sparks that reach their maximal peak quicker and decay faster than sparks from WT cardiomyocytes.
Figure 1. JCN-KO cardiomyocytes produce fewer but brighter Ca2+ sparks than WT cardiomyocytes.

A and B, representative confocal line-scan images and three-dimensional Ca2+ sparks recorded in intact WT and JCN-KO cardiomyocytes, respectively. C–G, bar graphs depicting Ca2+ spark frequency measured as the number of events per unit time and length, the maximal Ca2+ spark amplitude (F/F0), the average of full duration at half-maximal amplitude (FDHM), the average of full width at half-maximal amplitude (FWHM), and the time-to-peak (TTP) of Ca2+ spark amplitude in WT (blue bars) and JCN-KO (aqua bars) cells, respectively. ***P < 0.001. n = 1622 (WT) and 2743 (JCN-KO) Ca2+ sparks.
We next perfused isoproterenol (Iso) onto intact cardiomyocytes to directly evaluate the effect of β-adrenergic stimulation on Ca2+ sparks. Previous studies have postulated that β-adrenergic agonists and activation of PKA are associated with increased SR Ca2+ load and RyR2 phosphorylation, resulting in increased Ca2+ spark frequency (Li et al. 2002; Benkusky et al. 2007). Figure 2A and B shows representative line scan images and three-dimensional Ca2+ spark reconstructions obtained from intact WT and JCN-KO cardiomyocytes 2 min after the perfusion of 300 nmol l−1 Iso, which significantly increases the frequency of Ca2+ sparks in JCN-KO cardiomyocytes and to a lesser extent in WT cardiomyocytes (Fig. 2C). This differential effect of Iso cancels the disparity in Ca2+ spark frequency between JCN-KO and WT cardiomyocytes seen in the absence of Iso (Fig. 1C). Iso also increased the amplitude of Ca2+ sparks in both WT and JCN-KO cardiomyocytes; however, at variance with the Ca2+ spark frequency, the spark amplitude was still greater in JCN-KO cells after β-adrenergic stimulation (Fig. 2D). Interestingly, Iso-treated JCN-KO cardiomyocytes exhibited Ca2+ sparks of longer duration with greater spatial spread (Fig. 2E and F) as opposed to exhibiting Ca2+ sparks of shorter duration with equal spatial spread, as seen in the absence of Iso. The abbreviated TTP of the Ca2+ sparks in JCN-KO cardiomyocytes was not further shortened as a result of Iso perfusion (Fig. 2G). Overall then, Iso perfusion increased Ca2+ spark frequency and amplitude in both JCN-KO and WT cells, as expected from increases in SR Ca2+ load and release caused by β-adrenergic stimulation, but JCN-KO cells exhibited greater response to Iso, thus blurring the differences seen in unstimulated cells.
Figure 2. Isoproterenol perfusion attenuates the decreased Ca2+ spark frequency in JCN-KO cardiomyocytes.

A and B, representative confocal line-scan images and three-dimensional Ca2+ sparks following the perfusion of 300 nmol l−1 Iso onto intact WT and JCN-KO cardiomyocytes, respectively. C–G, bar graphs depicting Ca2+ spark frequency, amplitude, FDHM, FWHM and TTP, respectively, in Iso-perfused WT and JCN-KO cells. ***P < 0.001. n = 3837 (WT) and 2157 (JCN-KO) Ca2+ sparks.
Intracellular Ca2+ transients and SR load in WT and JCN-KO cardiomyocytes
The Ca2+ spark results with Iso indicate that β-adrenergic stimulation is a critical factor to determine the phenotype of the JCN-KO cardiomyocytes. We thus measured field-stimulated [Ca2+]i transients in WT and JCN-KO cells at four pacing frequencies and in the absence and presence of Iso. JCN-KO cardiomyocytes displayed higher [Ca2+]i transient amplitudes than WT cardiomyocytes, both in the absence (Fig. 3A) and in the presence (Fig. 3B) of Iso, but the difference declined with increasing pacing frequencies. Also, in the absence of Iso, the rate of decay of the [Ca2+]i transient (τ) was similar for both groups at every pacing frequency (Fig. 3C); however, Iso accelerated τ in JCN-KO cardiomyocytes significantly more than in WT cardiomyocytes (Fig. 3D). Thus, the general trend for JCN-KO cardiomyocytes was to exhibit [Ca2+]i transients of higher amplitude but shorter duration.
Figure 3. JCN-KO cardiomyocytes display a significant increase in the amplitude of the intracellular Ca2+ transient and an isoproterenol-induced faster rate of decay.
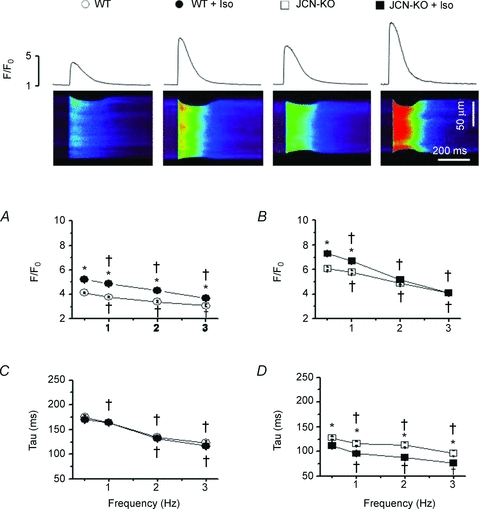
Representative confocal line scan images of Ca2+ transients elicited from cardiomyocytes field-stimulated at 0.5 Hz with the associated Ca2+ transient profiles (top panel). Line graphs depict the relationship between stimulation frequency and the amplitude of the Ca2+ transient (F/F0) (A and B) and the monoexponential decay of the Ca2+ transient (tau) (C and D). Open and filled symbols represent WT and JCN-KO cardiomyocytes, respectively. Circles and squares represent cardiomyocytes before and after 1 μmol l−1 Iso perfusion, respectively. †P < 0.05 vs. 0.5 Hz. *P < 0.05 JCN-KO vs. WT. n≥ 36 transients from ≥5 mouse hearts.
To determine whether the augmented Ca2+ spark and transient amplitudes in JCN-KO cardiomyocytes correlate with an increase in SR Ca2+ content, we measured the size of the caffeine-releasable Ca2+ pool at various pacing frequencies, similar to above. Figure 4 shows that indeed, caffeine-elicited Ca2+ transients were modestly but statistically higher in JCN-KO cardiomyocytes than in WT cardiomyocytes (F/F0 = 5.10 ± 0.18 and 5.67 ± 0.16 for WT and JCN-KO cardiomyocytes, respectively, n = 15 cells, P < 0.05).
Figure 4. JCN-KO cardiomyocytes have a higher SR Ca2+ content than WT cardiomyocytes.

A and B, representative line scan images and amplitude profiles of caffeine-induced Ca2+ transients elicited from WT and JCN-KO cardiomyocytes, respectively. C, bar graph depicting the amplitude (F/F0-caffeine) of the caffeine-elicited Ca2+ transients. *P < 0.05. n = 53 (WT) and 38 (JCN-KO) caffeine-induced Ca2+ transients.
SR Ca2+ leak measurements in WT and JCN-KO cardiomyocytes
The reduction of Ca2+ spark frequency and resulting enhanced SR Ca2+ load suggest that there may be a decrease in diastolic SR Ca2+ leak in JCN-KO cells. Figure 5A shows SR Ca2+ leak (calculated as described in Methods) for WT and JCN-KO cardiomyocytes at four different pacing frequencies, in the absence of Iso. SR Ca2+ leak increased with stimulation frequency for both WT and JCN-KO cardiomyocytes, but the latter displayed reduced SR Ca2+ leak at all pacing frequencies. The average SR leak (determined by summing all data points and dividing by the number of points) was 6.57 ± 0.53 for WT and 4.85 ± 0.59 for JCN-KO cells (Fig. 5C). Surprisingly, when the exact same protocol was repeated in the continuous presence of 1 μmol l−1 Iso to stimulate the β-adrenergic pathway, JCN-KO cardiomyocytes exhibited higher SR Ca2+ leak than WT cardiomyocytes (Fig. 5B). The relationship between pacing frequency and SR leak was slightly flattened compared with cells without Iso, but JCN-KO cells had augmented leak at all pacing frequencies. Again, the average SR Ca2+ leak calculated from all data points shows that Iso-treated JCN-KO cells had significantly higher SR leak than WT cells (8.38 ± 0.87 and 5.38 ± 0.70, respectively, Fig. 5D). Thus, under non-stimulated conditions, SR Ca2+ leak in JCN-KO cells is depressed with respect to control, but becomes higher under β-adrenergic stimulation. The functional implications of this crossover in SR leak for JCN-KO cells are discussed below.
Figure 5. The higher SR Ca2+ content in JCN-KO cardiomyocytes is potentially caused by a decrease in SR Ca2+ leak, which is reversed in the presence of isoproterenol.

A and B, line graphs depicting the relationship between stimulation frequency and SR Ca2+ leak in the absence and presence of 1 μmol l−1 Iso, respectively. C and D, bar graphs depicting the combined SR Ca2+ leak in the absence and presence of 1 μmol l−1 Iso, respectively. *P < 0.05. n≥ 6 measurements per pacing frequency from 4 hearts. E, bar graphs depicting the percentage of WT (n = 5 of 87) and JCN-KO (n = 32 of 66) cardiomyocytes that developed spontaneous aftercontractions. Data were analysed using a Chi2 test. *P < 0.05. F and G, WT and JCN-KO cardiomyocytes were paced for 2–3 s at 3 Hz in the presence of 1 μmol l−1 Iso, then pacing was stopped to image spontaneous Ca2+ release events. Representative Ca2+ transient profiles (top traces), line scan images (middle images), and cell shortening profiles (bottom traces) from a WT and JCN-KO cardiomyocyte.
Spontaneous aftercontractions in JCN-KO cells
The disproportionate increase in SR Ca2+ leak caused by β-adrenergic stimulation in JCN-KO cardiomyocytes suggests that junctin ablation may lead to a higher propensity for spontaneous Ca2+ release, which is greatly influenced by diastolic SR Ca2+ leak. We tested this possibility by subjecting isolated cardiomyocytes to pacing at 3 Hz and in the continuous presence of 1 μmol l−1 Iso. This protocol rarely caused spontaneous SR Ca2+ release in WT cardiomyocytes (Fig. 5F). In fact, spontaneous Ca2+ release and associated aftercontractions were only observed in ∼6% (n = 5 of 87 cells) of WT cardiomyocytes (Fig. 5E). Conversely, aftercontractions were detected in 50% of JCN-KO cardiomyocytes (n = 32 of 66 cells, Fig. 5E), indicating that these cells do indeed have a greater propensity for spontaneous Ca2+ release events that develop into aftercontractions (Yuan et al. 2007). Although we observed the propagation of some waves in the presence of Iso in both WT and JCN-KO cardiomyocytes, we defined spontaneous aftercontractions as unsolicited, global Ca2+-induced Ca2+ release (CICR) events that produced discrete cellular contractions, as demonstrated by the two final Ca2+ release events in the JCN-KO cardiomyocyte (Fig. 5G). In the first characterization of the JCN-KO mice, we found that over 50% of JCN-KO mice experience sudden death before reaching 12 months of age (Yuan et al. 2007). The differential effect of Iso in Ca2+ sparks and leak observed in this study (Figs 2 and 5), plus the development of β-adrenergic-elicited aftercontractions (Fig. 5G and Yuan et al. 2007) may explain the increased incidence of sudden death in JCN-KO mice.
Single channel recordings reveal direct effect of junctin on RyR2 activity
We reconstituted SR-enriched microsomes from hearts of JCN-KO and control littermates in planar lipid bilayers and recorded single RyR2 channel activity. A side-by-side comparison between these two groups allowed us to attribute differences in RyR activity to the selective absence of junctin, since other components of the Ca2+ release complex (triadin-1, FKBP12.6, HRC, Csq2, RyR2 itself) are unaffected in the JCN-KO mouse (Yuan et al. 2007). Channels were reconstituted in a nominally Ca2+-free solution, but ‘contaminant’ Ca2+ (∼5 μmol l−1 free [Ca2+]) served to activate RyR2s (Farrell et al. 2003; Altschafl et al. 2007). Upon incorporation of channels in the bilayer, cytosolic[Ca2+] was lowered to 100 nmol l−1 with a Ca:EGTA mixture, and luminal[Ca2+] was elevated to the concentrations indicated in Fig. 6 by bolus addition of CaCl2 from a 100-fold stock.
Figure 6. Effect of junctin removal on single channel RyR2 activity is dependent on the luminal Ca2+ concentration.
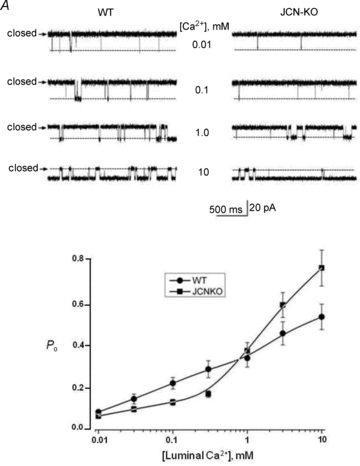
A, single RyR channels recorded from WT and JCN-KO cardiac microsomes reconstituted into a planar lipid bilayer with 0.01–10 mmol l−1 Ca2+ bathing the trans (luminal) side of the channel. B, data points depict the relationship between luminal [Ca2+] and the open probability (Po) of the RyR2 channel as a function of luminal [Ca2+]. Circles and squares represent WT and JCN-KO single channel data, respectively. Data points are joined by a line with no theoretical function and correspond to the mean ± SEM for n = 8 and 6 channels (WT and JCN-KO, respectively).
At low luminal [Ca2+] (<1 mmol l−1), WT channels consistently displayed higher activity than JCN-KO channels. For example, increasing luminal [Ca2+] from 0.01 to 0.1 mmol l−1 increased RyR2 Po from 0.062 ± 0.009 to 0.221 ± 0.023 (n = 8) in WT channels, i.e. an increase of 3.57-fold, but only from 0.053 ± 0.008 to 0.093 ± 0.011 (n = 6) in JCN-KO channels, i.e. an increase of 1.75-fold (P = 0.05). Conversely, at high luminal [Ca2+], JCN-KO channels were hyperactive with respect to their WT controls. The difference was especially prominent at the highest luminal [Ca2+], i.e. 10 mmol l−1, but could also be noted at 3 mmol l−1[Ca2+]. Increasing luminal [Ca2+] from 0.01 to 10 mmol l−1 increased RyR2 Po to 0.778 ± 0.118 in JCN-KO channels, an increase of 14.7-fold, but only to 0.481 ± 0.098 in WT channels, or an increase of 7.75-fold (P = 0.05). Thus, the absence of junctin has direct effects on RyR2 activity, with an apparent dual mode of regulation: at low luminal [Ca2+], junctin-devoid RyR2 channels have lower activity than WT channels, and at high luminal [Ca2+], JCN-KO channels are hyperactive. The crossover occurs at ∼1 mmol l−1 luminal [Ca2+], approximately the calculated intra-SR [free Ca2+] of ventricular cells at rest (Shannon & Bers, 1997).
Identification of the minimal interacting domains between junctin and RyR2
The dual mode of junctin regulation of RyR activity strongly suggests that junctin and the RyR2 channel are capable of multiple protein–protein interactions. To determine the minimal interacting domains between junctin and RyR2, we generated a series of constructs for their SR lumen-projected domains, which were fused to maltose binding protein (MBP) and glutathione-S-transferase (GST), respectively (Fig. 7A). The MBP–junctin and GST–RyR2 fusion peptides were expressed in E. coli and affinity-purified. Equal amounts of the aforementioned MBP–junctin peptides, with respective molecular masses of ∼47.6, ∼45.9, ∼50.4, ∼50.3 and ∼58.0 kDa (Fig. 7B), along with control MBP protein (42.5 kDa), were subjected to electrophoresis and transferred to nitrocellulose membranes (see Methods). The membranes were overlaid with affinity-purified GST–RyR2 constructs and immunoprobed with anti-GST. GST–RyR2-A bound specifically and efficiently MBP–junctin iso(1) and MBP–junctin-A, but not any of the other MBP–junctin fusion peptides or control MBP protein, while GST–RyR2-D bound specifically and efficiently MBP–junctin-D (Fig. 7B). In a parallel set of experiments, equal amounts of the aforementioned GST–RyR2 peptides, with respective molecular weights of ∼29.7, ∼29.0, ∼29.7 and ∼32.7 kDa (Fig. 7C), along with control GST protein (26.0 kDa) were subjected to electrophoresis and transferred to nitrocellulose membranes. The membranes were overlaid with affinity-purified MBP–junctin constructs and immunoprobed with anti-MBP. MBP–junctin iso(1) and MBP–junctin-A bound specifically and efficiently GST–RyR2-A, but not any of the other GST–RyR2 fusion peptides or control GST protein, while MBP–junctin-D bound specifically and efficiently GST–RyR2-D (Fig. 7C). Interestingly, smaller fragments of junctin KEKE-like domain do not bind GST–RyR2-D, indicating that the full length of this domain is required for this interaction. Thus, the results obtained separately with junctin and RyR2 constructs confirm each other and indicate that two distinct domains of junctin interact with the RyR2.
Figure 7. Junctin and RyR2 interacting domains.

A, diagram depicting the junctin and RyR2 interacting domains probed in this study. B, MBP–junctin fusion peptides separated by electrophoresis were transferred to membranes and blot-overlaid with GST–RyR2-A and GST–RyR2-D. C, GST–RyR2 fusion peptides separated by electrophoresis were transferred to membrane and blot-overlaid with MBP–junctin iso(1), MBP–junctin-A and MBP–junctin-D.
Discussion
There is consensus that junctin, a 26 kDa transmembrane protein of the junctional SR, is a structural component of the Ca2+ release machinery that, either alone or in concert with Csq2 and triadin-1 (Jones et al. 1995; Zhang et al. 1997), plays an important role in controlling RyR activity and Ca2+ release in cardiac cells. This notion has been reaffirmed by studies in which acute (Gergs et al. 2007) or chronic (Kirchhefer et al. 2003, 2006) overexpression of junctin leads to decreased Ca2+ transients and contractility of cardiac cells, and in others where genetic ablation of junctin enhances contractility but predisposes mice to delayed after-depolarizations (DADs) and premature death (Yuan et al. 2007). However, the precise mechanism(s) by which junctin exerts control of luminal Ca2+ sensitivity of RyRs and its role in cardiac dysfunction remain unclear. In this study, we compared the activity of RyRs in WT and Junctin −/− (JCN-KO) mice at several levels of integration, encompassing single channel recordings, Ca2+ sparks and global Ca2+ transients, to elucidate some of the most outstanding functional attributes of junctin in cardiac cells. The inferences are appropriate since the JCN-KO mouse, in direct contrast to the Triadin−/− mouse, does not modify the expression of other key structural components of the Ca2+ release unit, including the RyR, Csq2, triadin-1, FKBP12.6, HRC and the DHPR (Yuan et al. 2007). By comparing the activity of RyRs in these various molecular and cellular settings and in the presence of increasing luminal [Ca2+] (by direct addition or by β-adrenergic stimulation), we were able to unveil novel mechanisms of junctin function that may explain the increased propensity for arrhythmias of the JCN-KO mice, and the downregulation of junctin as a potential compensatory event in HF (Gergs et al. 2007).
Junctin as a modulator of RyR2 Ca2+ release: direct or indirect role?
An outstanding question regarding the role of junctin in Ca2+ release is whether it functions as a direct modifier of RyR activity via protein–protein interactions, or whether it limits its role to anchor Csq2 to the Ca2+ release unit and thus indirectly alters RyR2 activity. Triadin-1, which shares ∼60% homology with junctin and also links Csq2 to RyR2s by the same structural KEKE motifs, has been shown to be capable of sensitizing RyRs to luminal Ca2+ by directly interacting with the channel protein (Györke et al. 2004). However, in the Triadin−/− mice, dramatic alterations to the junctional SR architecture (50% reduction in the number of contacts between SR and T-tubules; Chopra et al. 2009) are likely to contribute to its decreased E-C coupling efficiency, making it difficult to discern any potential role of triadin-1 as a direct ligand of RyR2s. Two sets of data led us to hypothesize that, despite their high homology and similar structural role, junctin acts distinctively from triadin-1 in cellular settings (Dulhunty et al. 2009) and is likely to exert a direct role on RyR2 regulation. First, major structural rearrangement of the junctional SR, a hallmark of the Triadin−/− mouse, appears unlikely in the JCN-KO mice, as their membrane capacitance (an index of sarcolemmal and T-tubule surface area) and density of major E-C coupling proteins are both indistinguishable from WT mice (Yuan et al. 2007). This preservation of the Ca2+ release unit architecture limits the role of altered Ca2+ gradients as modifiers of E-C coupling efficiency in the JCN-KO mouse. Further, it also underscores the value of the second set of data that supports a direct effect of junctin on modulating RyR2 activity: the recording of single RyR2 channels in the absence and the presence of junctin (Fig. 6).
Side-by-side recording of RyR2 channels from WT and JCN-KO cardiac SR indicates that the absence of junctin produces a dual effect on the normally linear response of RyR2 to luminal [Ca2+]. At low luminal [Ca2+] (<1 mmol l−1), junctin-devoid RyR2 channels are less responsive to luminal [Ca2+] than their WT counterparts; conversely, high luminal [Ca2+] turns JCN-KO channels hypersensitive to this form of channel modulation. At face value, these results indicate a multifaceted regulation of RyR2 by junctin and suggest multiple junctin-RyR2 interacting sites. In support of this, biochemical experiments using junctin and RyR2 constructs (Fig. 7) indicate that two distinct domains of junctin, one containing the classical KEKE motif, bind to two different domains of the RyR2 channel, confirming multiple protein–protein interactions and providing the structural basis for a dual role of junctin in regulation of RyR2 activity.
It is possible that some of the effects of junctin ablation on RyR2 mentioned above may be compounded by Csq2, which binds to the RyR2–junctin–triadin complex in a Ca2+-dependent manner and indirectly regulates RyR2 activity (Wang et al. 1998; Györke et al. 2004; Wei et al. 2009). Although earlier studies suggested that both Csq1 (Wang et al. 1998) and Csq2 (Györke et al. 2004), the skeletal and cardiac forms of this protein, respectively, dissociate from junctin/triadin at high [Ca2+] (>3–5 mmol l−1), a more recent report (Wei et al. 2009) found that low luminal [Ca2+] (∼0.5–10 μmol l−1) may also dissociate most of Csq2 from the RyR2–junctin–triadin complex. Our experimental conditions did not allow us to ascertain with confidence whether our reconstituted channels lacked Csq2, but since we started the titration of RyR2 activity at low [Ca2+] (10 μmol l−1, Fig. 6), it is possible that Csq2 was absent from the beginning of the experiments and played no significant role in the RyR2 response to increasing luminal [Ca2+]. This would reinforce the notion that the difference in Po between WT and JCN-KO channels is indeed due to direct effects of junctin on RyR2, but the possibility that Csq2 acted in tandem with junctin cannot be discarded.
Effect of junctin removal on Ca2+ sparks and its relation to Po of RyR2 channels
In cellular settings, the luminal [Ca2+]-dependent biphasic response of RyR2 to junctin would translate into complex effects on Ca2+ sparks and [Ca2+]i transients, but the results obtained in single channel experiments guided interpretation of these two phenomena. Under non-stimulated (Iso-free) resting conditions, JCN-KO cardiomyocytes exhibited fewer and more intense Ca2+ sparks than WT cardiomyocytes (Fig. 1). These results bode well with the single channel data that indicated that junctin-devoid RyR2 channels are less active than WT under low luminal [Ca2+] (Fig. 6). However, probably as a result of the restrained RyR2 activity, the SR Ca2+ content was higher in the JCN-KO mice (Fig. 4), forcing more Ca2+ out of the SR whenever a Ca2+ spark was produced (higher intensity). Conversely, under β-adrenergic stimulation and when SR Ca2+ content increased even more, Ca2+ spark frequency in JCN-KO cardiomyocytes was practically identical to that of WT cardiomyocytes even though intensity continued to be higher (Fig. 2). This suggested that the sensitivity of JCN-KO RyR2 channels to high luminal Ca2+ was increased, again fitting well with the single channel data that indicated that the Po of junctin-devoid RyR2 channels was actually higher than WT at luminal [Ca2+] > 1 mmol l−1 (Fig. 6). Thus, not surprisingly, the frequency and intensity of the Ca2+ sparks appeared directly related to the Po of RyR2 channels and the SR Ca2+ content, respectively (Cheng & Lederer, 2008), both of which were altered in the JCN-KO cardiomyocytes.
[Ca2+]i transients in JCN-KO cardiomyocytes
The amplitude and kinetics of [Ca2+]i transients are more complexly regulated, as expected from the multitude of molecules involved in this process, but an overriding mechanism controlling their amplitude in this study appeared to be the SR Ca2+ content. Our hypothesis to explain changes in [Ca2+]i transients in the JCN-KO mouse is as follows: in the absence of Iso stimulation and thus, under relatively low luminal [Ca2+], the amplitude of the [Ca2+]i transient was higher in the JCN-KO cardiomyocytes because their SR Ca2+ content was higher. Under constant pacing, the incoming Ca2+ ions from L-type Ca2+ current (ICa) and CICR were such powerful stimuli to open RyRs that they both overrode the lower Po of junctin-devoid RyRs demonstrated in single channel recordings. On the other hand, under β-adrenergic stimulation and higher pacing frequency (2–3 Hz), conditions that have both been shown to increase the SR Ca2+ content (Bers, 2002), the amplitude of the [Ca2+]i transients in WT and JCN-KO cardiomyocytes was higher compared with [Ca2+]i transients in the absence of Iso; however, there was no difference in [Ca2+]i transient amplitude between WT and JCN-KO cells, presumably because a ‘ceiling’ of luminal [Ca2+] control was reached.
Biphasic effect of junctin removal on SR Ca2+ leak and its relation to DADs
In the above scenario, the changes in Po of RyR2 attributed to the absence of junctin appeared weighed down by CICR and the luminal mechanisms that control Ca2+ release; accordingly, the modulation of RyR2 activity by junctin may be more conspicuous in the absence of ICa (during diastole) and in determining SR Ca2+ leak. The results of Fig. 5 showed that in the absence of Iso, JCN-KO cardiomyocytes exhibited lower SR Ca2+ leak than WT, but under conditions of higher luminal Ca2+ content induced by β-adrenergic stimulation, JCN-KO cells had higher SR Ca2+ leak than WT. The exact luminal [Ca2+] at which the crossover occurred could not be determined by stimulation frequency alone and may be related to phosphorylation of E-C coupling proteins by protein kinase A (PKA) or by Ca2+-dependent kinases. Whatever its mechanism, this switch in SR Ca2+ leak propensity by JCN-KO cardiomyocytes mirrors the biphasic response of junctin-devoid RyRs to luminal [Ca2+] in lipid bilayers and is likely to play a central role in the generation of the increased ventricular automaticity (arrhythmias) that is typical of this animal model (Yuan et al. 2007). Remarkably, the increased ventricular automaticity observed in vivo is triggered by β-adrenergic stimulation and appears related to the generation of DADs (Yuan et al. 2007). As part of the compensatory mechanisms elicited by genetic ablation of junctin, the JCN-KO mice exhibit increased Na+–Ca2+ exchange protein and current (Iti) (Yuan et al. 2007). Thus, by directly determining that SR Ca2+ leak and RyR Po are both increased in JCN-KO cardiomyocytes under high luminal [Ca2+], we provide a strong lead to support the notion that spontaneous Ca2+ release during β-adrenergic stimulation provides the substrate for triggered activity and increased ventricular automaticity. Indeed, the propensity for spontaneous Ca2+ release and aftercontractions was significantly higher in β-adrenergic stimulated JCN-KO cells than in WT cells (Fig. 5).
Dual interaction of junctin with RyR2
Our biochemical experiments with junctin and RyR2 constructs supported the notion that junctin interacts with at least two distinct domains of the RyR2 protein. RyR2 appears to have two intraluminal loops, I and II, with the second being bipartite (proximal and distal loop II) (Lee et al. 2004). Our GST–RyR2 constructs corresponded to these loops: GST–RyR2-A (4520–4553 aa) was RyR2 intraluminal loop I, while GST–RyR2-D (4789–4846 aa) was RyR2 intraluminal loop II. GST–RyR2-B (4789–4818 aa) and GST–RyR2-C (4819–4846 aa) were proximal and distal loop II, respectively. Our MBP–junctin constructs were MBP–junctin-A (47–77 aa) N-proximal intraluminal domain, and MBP–junctin-D (78–210 aa) KEKE-like intraluminal domain. Notice that MBP–junctin iso(1) (47–92 aa) was the isoform of junctin containing exon2A, while MBP–junctin-B (78–161 aa) and MBP–junctin-C (160–210 aa) were the proximal and distal KEKE-like domains. It has been reported that triadin's KEKE-motif interacts with intraluminal loop II of RyR1 (Lee et al. 2006; Goonasekera et al. 2007).
Our data (Fig. 7) suggest that junctin interacts with both intraluminal loops I and II but with different domains. The N-proximal intraluminal domain of junctin interacts with RyR2 intraluminal loop I, while the KEKE-like C-proximal domain of junctin interacts with RyR2 intraluminal loop II. It seems that an intact KEKE-motif and an intact intraluminal loop II are needed for junctin–RyR2 binding because their proximal and distal fractions did not interact (Fig. 7).
Integrated scheme of junctin regulation of RyR2 channels
Overall, because at low luminal [Ca2+] (<1 mmol l−1) junctin acts as an activator of RyR2 channels (in its absence, RyR2 activity is attenuated, Fig. 6), and at high luminal [Ca2+] (>1 mmol l−1) junctin inhibits RyR2 channel activity (in its absence, RyR2 channels are more active), junctin produces intricate effects that are difficult to understand with our limited knowledge of the dyadic architecture, RyR2–junctin–Csq2 stoichiometry, and other critical parameters. However, the luminal [Ca2+]-dependent dual role of junctin could be approximated with a mechanical model that proposes that junctin binds to two distinct sites in the RyR2 protein (Fig. 8). In this potential scenario, at low luminal [Ca2+], only one of the binding sites of junctin (labelled ‘1’ in Fig. 8) would be bound to RyR2 because the other site is likely to have stronger affinity for Csq2 (Zhang et al. 1997). At high luminal [Ca2+], when Csq2 dissociates from junctin due to polymerization (Wang et al. 1998), the second binding site of junctin (labelled ‘2’) would be exposed and available to interact with the RyR2. Thus, we propose that when the first binding site of junctin is bound to RyR2, junctin acts as a positive regulator of RyR2 channels, perhaps by increasing the affinity of their luminal Ca2+ binding site for Ca2+. When the second binding site is bound to RyR2, junctin acts as a negative regulator of RyR2s, perhaps again by decreasing the affinity of their luminal Ca2+ binding site for Ca2+. Because the crossover occurs at a luminal [Ca2+] that is close to that determined in resting cells (Shannon & Bers, 1997), it is possible that the activator–inhibitor role of junctin may be exerted under periods of prevalent parasympathetic and sympathetic activity, respectively. This hypothetical scheme may be used for further testing.
Figure 8. Proposed model for junctin effect on RyR2 channel activity.

Two binding sites for junctin (labelled ‘1’ and ‘2’ in the WT cartoons) on the luminal side of the cardiac RyR2 may mediate the dual role of junctin described in this study. Arrows on top of RyR2 represent Po of RyR2, Ca2+ spark frequency, or SR Ca2+ leak during diastole. Notice the variable thickness of the arrows. Left and right cartoon in each panel represent the SR Ca2+ loading during basal conditions and during β-adrenergic stimulation, respectively. See text for more details.
Summary and health relevance of our findings
In summary, we determined that the absence of junctin induces complex effects on single RyR2 activity, [Ca2+]i transients and SR Ca2+ leak that are dependent on the level of luminal (intra-SR) Ca2+. As supported by biochemical data with RyR2 constructs, most of these effects may be explained by a mechanical model in which junctin binds to two functionally different domains in the RyR2 protein, one that attenuates and the other that promotes Ca2+ release. In the JCN-KO mouse, junctin-devoid RyR2s increase their sensitivity to activation by intraluminal [Ca2+], thus enhancement of SR Ca2+ load by β-adrenergic stimulation leads to increased SR Ca2+ leak, enhanced cellular Ca2+ extrusion by Na+–Ca2+ exchange, and the development of spontaneous Ca2+ transients and aftercontractions. The pathophysiology of the JCN-KO mouse observed in response to high stress is reminiscent of the classical symptoms observed in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT), namely enhanced Iti, DADs, aftercontractions, and eventually, triggered cardiac arrhythmias in response to the increased propensity for spontaneous SR Ca2+ release. Interestingly, mutations in RyR2 and Csq2, two integral proteins of the Ca2+ release unit, are implicated in the development of CPVT (Priori & Chen, 2011). While junctin mutations have not been implicated in any human cases of CPVT, our results predict that junctin ablation, or mutations that alter its structural attributes, may mimic the etiology of CPVT by causing enhanced diastolic Ca2+ leak in the presence of β-adrenergic activation. Also, it is possible that in failing hearts, the down-regulation of junctin in the face of depressed SR Ca2+ content may constitute an important compensatory mechanism to increase contractility. However, such compensation may be costly, as we have shown that junctin down-regulation is likely to contribute to increased arrhythmias under β-adrenergic stimulation or stress conditions.
Acknowledgments
This investigation was supported by NIH grant NRSA-T32-HL07936 (University of Wisconsin Cardiovascular Research Training grant to B.A.A.); NIH grants RO1-HL055438 and PO1-HL094291 (to H.H.V.) and HL26057 and HL60418 (to E.G.K.); and the European Union 7th Framework Program EUTrigTreat No. HEALTH-F2–2009-241526 (to E.G.K.). We thank Ana M. Gómez for providing us with custom written software and for assisting with the analysis of Ca2+ sparks and transients data.
Glossary
Abbreviations
- CICR
Ca2+-induced Ca2+ release
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- Csq2
cardiac calsequestrin
- DAD
delayed after-depolarization
- DHPR
dihydropyridine receptor
- E-C
excitation–contraction
- FDHM
full duration at half-maximal amplitude
- FKBP12.6
FK506 binding protein 12.6 kDa
- FWHM
full width at half-maximal amplitude
- ICa
L-type Ca2+ current
- Iso
isoproterenol
- Iti
Na+–Ca2+ exchanger current
- JCN
junctin
- JCN-KO
junctin knock-out
- PKA
protein kinase A
- Po
open probability
- RyR
ryanodine receptor
- SR
sarcoplasmic reticulum
- TTP
time-to-peak
- WT
wild-type
Author contributions
B.A.A. and H.H.V.: conception, design, and performance of experiments, data interpretation and analysis, and manuscript writing; D.A.: performance of experiments and data analysis; O.F.: performance of experiments and data analysis; Q.Y.: providing the JCN-KO mouse model and data interpretation; E.G.K.: providing the JCN-KO mouse model, data interpretation, and manuscript writing. The majority of the work was done in the laboratory of Dr. Hector H. Valdivia at the University of Wisconsin-Madison. All authors approved the final version for publication.
References
- Altschafl BA, Beutner G, Sharma VK, Sheu SS, Valdivia HH. The mitochondrial ryanodine receptor in rat heart: A pharmaco-kinetic profile. Biochim Biophys Acta. 2007;1768:1784–1795. doi: 10.1016/j.bbamem.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, Powers PA, Valdivia HH. Intact β-adrenergic response and unaltered progression towards heart failure in mice with genetic ablation of a major PKA phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–829. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Capes EM, Loaiza R, Valdivia HH. Ryanodine Receptors. Skelet Muscle. 2011;1:18–31. doi: 10.1186/2044-5040-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- Cheng H, Song LS, Shirokova N, González A, Lakatta EG, Rios E, Stern MD. Amplitude distribution of calcium sparks in confocal images: theory and studies with an automatic detection method. Biophys J. 1999;76:606–617. doi: 10.1016/S0006-3495(99)77229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, Cattolica RA, Perez CF, Hlaing T, Knollmann-Ritschel BE, Jones LR, Pessah IN, Allen PD, Franzini-Armstrong C, Knollmann BC. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106:7636–7641. doi: 10.1073/pnas.0902919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulhunty A, Wei L, Beard N. Junctin–the quiet achiever. J Physiol. 2009;587:3135–3137. doi: 10.1113/jphysiol.2009.171959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell EF, Antaramian A, Rueda A, Gómez AM, Valdivia HH. Sorcin inhibits calcium release and modulates excitation-contraction coupling in the heart. J Biol Chem. 2003;278:34660–34666. doi: 10.1074/jbc.M305931200. [DOI] [PubMed] [Google Scholar]
- Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- Gergs U, Berndt T, Buskase J, Jones LR, Kirchhefer U, Müller FU, Schlüter KD, Schmitz W, Neumann J. On the role of junctin in cardiac Ca2+ handling, contractility, and heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H728–734. doi: 10.1152/ajpheart.01187.2006. [DOI] [PubMed] [Google Scholar]
- Goonasekera SA, Beard NA, Groom L, Kimura T, Lyfenko AD, Rosenfeld A, Marty I, Dulhunty AF, Dirksen RT. Triadin binding to the C-terminal loop of the ryanodine receptor is important for skeletal muscle excitation contraction coupling. J Gen Physiol. 2007;130:365–378. doi: 10.1085/jgp.200709790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györke I, Hester N, Jones LR, Györke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LR, Zhang L, Sanborn K, Jorgensen AO, Kelley J. Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J Biol Chem. 1995;270:30787–30796. doi: 10.1074/jbc.270.51.30787. [DOI] [PubMed] [Google Scholar]
- Kirchhefer U, Hanske G, Jones LR, Justus I, Kaestner L, Lipp P, Schmitz W, Neumann J. Overexpression of junctin causes adaptive changes in cardiac myocyte Ca2+ signaling. Cell Calcium. 2006;39:131–142. doi: 10.1016/j.ceca.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Kirchhefer U, Neumann J, Bers DM, Buchwalow IB, Fabritz L, Hanske G, Justus I, Riemann B, Schmitz W, Jones LR. Impaired relaxation in transgenic mice overexpressing junctin. Cardiovasc Res. 2003;59:369–379. doi: 10.1016/s0008-6363(03)00432-2. [DOI] [PubMed] [Google Scholar]
- Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010;2:a003996. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EH, Song DW, Lee JM, Meissner G, Allen PD, Kim do H. Occurrence of atypical Ca2+ transients in triadin-binding deficient-RyR1 mutants. Biochem Biophys Res Commun. 2006;351:909–914. doi: 10.1016/j.bbrc.2006.10.115. [DOI] [PubMed] [Google Scholar]
- Lee JM, Rho SH, Shin DW, Cho C, Park WJ, Eom SH, Ma J, Kim DH. Negatively charged amino acids within the intraluminal loop of ryanodine receptor are involved in the interaction with triadin. J Biol Chem. 2004;279:6994–7000. doi: 10.1074/jbc.M312446200. [DOI] [PubMed] [Google Scholar]
- Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–316. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- Overend CL, Eisner DA, O'Neill SC. The effect of tetracaine on spontaneous Ca2+ release and sarcoplasmic reticulum calcium content in rat ventricular myocytes. J Physiol. 1997;502:471–479. doi: 10.1111/j.1469-7793.1997.471bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–883. doi: 10.1161/CIRCRESAHA.110.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard TJ, Kranias EG. Junctin and the histidine-rich Ca2+ binding protein: potential roles in heart failure and arrhythmogenesis. J Physiol. 2009;587:3125–3133. doi: 10.1113/jphysiol.2009.172171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Bers DM. Assessment of intra-SR free [Ca] and buffering in rat heart. Biophys J. 1997;73:1524–1531. doi: 10.1016/S0006-3495(97)78184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Wang F, Bers DM. Regulation of cardiac sarcoplasmic reticulum Ca release by luminal [Ca] and altered gating assessed with a mathematical model. Biophys J. 2005;89:4096–4110. doi: 10.1529/biophysj.105.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijskens P, Jones LR, Franzini-Armstrong C. Junctin and calsequestrin overexpression in cardiac muscle: the role of junctin and the synthetic and delivery pathways for the two proteins. J Mol Cell Cardiol. 2003;35:961–974. doi: 10.1016/s0022-2828(03)00181-0. [DOI] [PubMed] [Google Scholar]
- Wang S, Trumble WR, Liao H, Wesson CR, Dunker AK, Kang CH. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat Struct Biol. 1998;5:476–483. doi: 10.1038/nsb0698-476. [DOI] [PubMed] [Google Scholar]
- Wei L, Hanna AD, Beard NA, Dulhunty AF. Unique isoform-specific properties of calsequestrin in the heart and skeletal muscle. Cell Calcium. 2009;45:474–484. doi: 10.1016/j.ceca.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Wolska BM, Solaro RJ. Method for isolation of mouse cardiac myocytes for studies of contraction and microfluorimetry. Am J Physiol Heart Circ Physiol. 1996;271:H1250–H1255. doi: 10.1152/ajpheart.1996.271.3.H1250. [DOI] [PubMed] [Google Scholar]
- Yuan Q, Fan GC, Dong M, Altschafl BA, Diwan A, Ren X, Hahn HH, Zhao W, Waggoner JR, Jones LR, Jones WK, Bers DM, Dorn IIGW, Wang HS, Valdivia HH, Chu G, Kranias EG. Sarcoplasmic reticulum calcium overloading in junctin deficiency enhances cardiac contractility but increases ventricular automaticity. Circulation. 2007;115:300–309. doi: 10.1161/CIRCULATIONAHA.106.654699. [DOI] [PubMed] [Google Scholar]
- Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272:23389–23397. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]
- Zhang L, Franzini-Armstrong C, Ramesh V, Jones LR. Structural alterations in cardiac calcium release units resulting from overexpression of junctin. J Mol Cell Cardiol. 2001;33:233–247. doi: 10.1006/jmcc.2000.1295. [DOI] [PubMed] [Google Scholar]