

Abstract
Monodisperse protein polymers engineered by biosynthetic techniques are well suited to serve as a basis for creating comb-like polymer architectures for biomaterial applications. We have developed a new class of linear, cationic, random-coil protein polymers designed to act as scaffolds for multivalent display. These polymers contain evenly spaced lysine residues that allow for chemical or enzymatic conjugation of pendant functional groups. Circular dichroism (CD) spectroscopy and turbidity experiments have confirmed that these proteins have a random coil structure and are soluble up to at least 65 °C. Cell viability assays suggest these constructs are non-toxic in solution up to a concentration of 100 μM. We have successfully attached a small bioactive peptide, a peptoid-peptide hybrid, a poly(ethylene glycol) (PEG) polymer, and a fluorophore to the protein polymers by chemical or enzymatic coupling, demonstrating their suitability to serve as multivalent scaffolds in solutions or as gels.
Keywords: Protein polymer, genetic engineering, multivalent scaffold, tissue transglutaminase
Introduction
Synthetic biomaterials that can display multiple bio-activating or bio-passivating elements are of increasing interest for the study of cell-material interactions and for drug delivery and tissue engineering applications. In biology, many types of recognition events require multiple interactions between ligands and receptors, where both avidity and specificity are affected by the valency.1–3 Increasing the number of bioactive agents often increases activity; for example, linear polymers with elements displayed in comb-like architectures have been shown to bind antibodies with increased antigenicity in comparison to the equivalent concentration of free epitopes.4 In drug delivery, the attachment of small biologically active compounds to a macromolecular scaffold can alter drug pharmacological properties, thereby increasing solubility and retention.5 These scaffolds can be functionalized with targeting moieties to enhance drug selectivity and receptor-mediated delivery.6 A variety of synthetic materials have been created to act as multivalent scaffolds for these applications, such as dendrimers,7–10 liposomes,11 and polymers2,12–15.
Scaffolds created by synthetic approaches can obtain high epitope valencies and allow versatile conjugation chemistries; however, traditional chemical synthesis results in materials that are inherently polydisperse. A broad distribution of molar mass can present a challenge for characterization and affects biological activity16 and in vivo toxicity17. New polymer synthesis techniques, such as ring-opening metathesis polymerization (ROMP) or atom transfer radical polymerization (ATRP), have been utilized to create linear multivalent scaffolds with a narrower molar mass distribution. These methods, however, allow little control over stereochemistry and still cannot achieve true monodispersity.
As a viable alternative to chemical synthesis, molecular biology and genetic engineering techniques can be employed to create homogeneous polymer-like protein (“protein polymer”) scaffolds. Genetic engineering utilizes the well-regulated machinery of cells to produce de novo proteins with remarkable control over protein structure and properties. The DNA template coding for the protein, can be dictated to precisely control sequence, stereochemistry, fold, and length; thus they are entirely monodisperse. When creating molecules for multivalent display, depending on the efficiency of the coupling reaction and the pendant moiety, the conjugate may no longer be monodisperse. However, the controlled backbone of the protein polymer limits one source of polydispersity, which is unable to be achieved by synthetic polymer backbones. Thus the precision of artificial protein polymer scaffolds allows for exact determination of valency when the protein polymers are decorated with moieties. Additionally, protein polymers may also exhibit less toxicity than synthetic polymers, as they are composed of natural amino acids and better mimic natural structures. Furthermore, the robustness of cloning methods allows for the inclusion of bioactive domains in the protein polymer backbone.
The modular biosynthetic strategy utilizing genetically engineered protein polymers as a backbone for chemical modification has gained attention as a source of new materials. This method has been used to create precisely defined macromolecules that self-assemble into liquid crystalline phases18,19 and as multivalent scaffolds. Recently, the Kiick group synthesized both alanine-rich α-helical20,21 and random coil22 protein polymer scaffolds containing evenly spaced glutamic acid residues for conjugation of saccharides to the carboxyl groups for toxin inhibition. The random coil proteins allowed the study of glycopolymer architecture and saccharide spacing on multivalent binding events and the α-helical polymers allowed for controlled three-dimensional placement of pendant groups. We are further exploring the use of random coil protein polymers for multivalent display, which allows the study of the protein polymer and pendant groups separately from protein structure.
Inspired by research with genetically engineered proteins containing single amino acid incorporation for increased functionality, we present a class of random-coil cationic protein polymers that contain evenly spaced reactive lysine groups. A random-coil protein polymer mimics the flexibility of synthetic polymers, unlike proteins with enhanced secondary structure, and still maintains a protein’s homogeneous properties. The effects of polymer flexibility on activity and recognition events could be better studied with an unordered multivalent protein scaffold. To date, the majority of research related to protein polymers is focused on molecules composed of amino acid sequences inspired by silk, collagen, and elastin that have folded secondary structures. Relatively little research has been conducted on random coil protein polymers, with the exception of their use as flexible spacer sequences23, and for toxin inhibition22.
Herein, we describe the synthesis and characterization of protein polymers that vary in chain length and lysine spacing. These proteins have been found to be water-soluble and random-coil in structure over a wide range of temperatures. Additionally, these proteins have low toxicity to mammalian cells at concentrations relevant to drug delivery applications. The protein polymers investigated have a chemically active group introduced by the primary amine of the lysine side chain at controlled intervals. The free amine allows for either chemical or enzymatic grafting of bioactive pendant groups. Chemical conjugation can be achieved by a wide range of amine-reactive chemistries, and has been used to covalently couple bioactive peptides, polymers, peptide-peptoid hybrids, and small organic molecules onto the cationic protein polymers.
In addition to providing a reactive group for chemical conjugation, the lysine in these protein polymers serves as a substrate for enzymatic grafting by tissue transglutaminase. Transglutaminase catalyzes the formation of bonds between glutamine and lysine residues under mild and biocompatible reaction conditions.24 This family of enzymes has been used to covalently link bioactive peptides to fibrin matrices25–27 and self-assembled fibrillar structures. 28 Enzymatic bond formation has recently been used with genetically engineered protein polymers to stabilize elastin-based hydrogels.29 We have created a protein polymer multivalent scaffold by enzymatic conjugation with a bioactive peptide.
Complete derivatization of the protein polymers allows for precise control over the spacing of pendant groups, not otherwise achieved with synthetic polymers. It is sometimes desirable to obtain partial coverage and although incomplete pendant group coupling does not result in precise control over placement, regardless, the randomly functionalized protein polymers nearly maintain their monodispersity and the valency is exactly determined. This class of de novo-designed protein polymers is a promising candidate for application as building blocks for multivalent display and tissue engineering hydrogels.
Materials and Methods
Standard molecular biology techniques were used for gene synthesis, protein expression, and protein purification, unless otherwise noted. DNA amplification was completed by polymerase chain reactions (PCR) with Taq polymerase (Promega, Madison, WI) according to manufacturer’s recommendations. All enzymes for cloning were supplied by New England Biolabs (Ipswich, MA).
DNA Gene Design and Synthesis
The DNA sequences encoding the monomers of the protein polymer amino acids were designed according to the codon preferences of Escherichia coli30 and to prevent DNA and RNA secondary structure formation. DNA oligonucleotides (Integrated DNA Technologies, Coralville, IA) encoding the three different sequences of protein polymers (Figure 1) were amplified by PCR, purified by electrophoresis on a 2% agarose gel, and obtained using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA). After EarI digestion and purification the monomers were self-ligated utilizing T4 ligase to form DNA multimers. The multimers were ligated into the cloning vector pUC18 (Life Technologies, USA) and transformed into NovaBlue cells (Novagen Inc., Madison, WI). Concatemer genes were created by the controlled cloning method31 with the restriction enzyme EarI serving as an analog of Eam1104 I. Briefly, the plasmids containing multimers were amplified by PCR primers designed to eliminate one of the flanking SapI sites. This was split into two fractions; one was digested by EarI and one with SapI. The EarI fraction was dephosphorylated with calf intestinal phosphatase and then ligated to the SapI-digested fraction. The 5′ terminal phosphate group was replaced on the resulting concatamer using T4 polynucleotide kinase and then the concatemer was digested with EarI. This gene was ligated into pUC18 and transformed into NovaBlue cloning cells. Single colonies were isolated and gene insertion was verified by an EarI digestion and visualized on a 2% agarose slab gel. This process was repeated to obtain genes of the desired lengths.
Figure 1.

DNA oligonucleotide sequences used to create repetitive genes, (A) K4 (B) K6 (C) K8. EarI restriction sites are in bold and underlined, and denotes the cleavage site.
The expression vector, pET-19b (Novagen Inc.) was modified to remove the internal Sap I site using QuickChange Site-Directed Mutagenesis (Stratagene, La Jolla, CA) to change a single base in the restriction site. The mutated vector was then amplified by PCR with primers containing EarI recognition sites as well as a stop codon. The amplified linear plasmid was digested with EarI to create complementary termini for the concatemer genes. These genes were ligated into the plasmid with T4 DNA ligase and transformed into NovaBlue cells. Correct insertion was verified by electrophoresis of genes that were doubly digested with BamHI and NdeI restriction enzymes and through DNA sequencing (SeqWright, Houston, TX). The ligation into the expression vector PET-19 (Novagen Inc.) results in the gene of interest expressed as a fusion protein with an N-terminal histidine tag (tag amino acid sequence: GH10SSGHIDDDDKHM).
Protein Expression and Purification
Plasmids containing the desired genes were transformed into E. coli expression strain BLR(DE3) cells (Novagen Inc.). A 5-ml starter culture in LB Broth (Fisher Scientific, Waltham, MA) was grown from a single colony for 8 hours. This was used to inoculate an overnight 100-ml LB broth culture, both supplemented with 200 μg/ml ampicillin (Sigma, St. Louis, MO) and 12.5 μg/ml tetracycline (Fisher Scientific). One-liter cultures of Difco Terrific Broth (Fisher Scientific) supplemented with 200 μg/ml ampicillin and 12.5 μg/ml tetracycline were inoculated with 15–20 ml from the overnight culture. The culture was induced with 0.5 mM isopropyl thiogalactoside (IPTG) (US Biologicals, Swampscott, MA) when the OD600 was between 0.6 to 0.8. The cells were harvested after 4 hours by centrifugation and the cell pellet was resuspended in 6M guanidine hydrochloride, 20 mM sodium phosphate, 500 mM NaCl, pH 7.8 buffer. The cells were lysed by three successive freeze (−80°C)-thaw (37°C) cycles followed by sonication. Cellular debris was removed by centrifugation for 40 minutes at 10,000 g at 4°C. The protein was purified with Chelating Sepharose Fast Flow nickel-charged resin (GE Healthcare, Piscataway, NJ) under denaturing conditions with imidazole-based competitive elution. The fractions containing purified protein, as determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), were dialyzed against deionized water and then lyophilized.
General Characterization
Protein molar mass was determined by matrix assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS) (Perseptive Biosystems Voyager Pro DE) using sinapinic acid matrix at Northwestern University’s Analytical Services Laboratory. Samples were dissolved in a 50% acetonitrile solution containing 0.1% trifluoroacetic acid. Amino acid analysis was conducted by the Yale Keck Facility (Yale University, New Haven, CT).
Turbidity
The phase behavior of protein polymers in solution was characterized as a function of temperature on a Varian Cary 500 UV-Vis-NIR spectrophotometer (Palo Alto, CA) with Peltier temperature control. Proteins were dissolved at 1 mg/ml in ddH 2O and placed in a 1 cm path length quartz cuvette. Temperature was ramped at 1°C/min from 5°C to 80°C and then back down to 5°C. The spectrophotometer recorded the optical density at 500 nm and automatically subtracted the blank optical density.
Circular Dichroism
Circular dichroism (CD) spectroscopy was conducted with a J-715 Jasco Inc. (Easton, MD) spectrometer with Peltier temperature control. Samples were placed in a quartz cuvette, with a path length of 1 mm. Scans ranging from 280 to 185 nm were collected with a bandwidth of 1.0 nm, a resolution of 0.2 nm, and a scan speed of 100 nm/min. An average of forty scans was reported for each condition. Samples were prepared from a stock solution of approximately 2 mg/ml in ddH2O. Protein dilutions were made between 1–10 μM for CD measurements. Stock solution and all dilutions were weighed in triplicate. Blank background scans were collected for every condition and manually subtracted from the sample scans. The results are given as mean residue molar ellipticity [Θ] in deg cm2 dmol −1 res−1 calculated by [Θ] = CD * 100,000/(L * C * N) where CD is the observed ellipticity in μdeg, L is the path length in cm, C is the protein concentration in μM and N is the number of amino acid residues in the protein.
Cell Culture and Viability
NIH 3T3 fibroblasts (ATCC, Manassas, VA) were cultured at 37 °C and 5% CO2 in DMEM (Gibco) supplemented with 1% sodium pyruvate, 1% penicillin-streptomycin, 1.5 g/L NaHCO3, and 10% fetal bovine serum (Gibco). Cells were grown to approximately 75% confluence and removed with 0.05% Trypsin-EDTA in Hanks’ Balanced Salt Solution with phenol red (Fisher Scientific). For cell viability experiments, cells were seeded at a density of 15,000 cells per well in 96-well plates (100 μl total volume) and cultured overnight. Cells were then washed with 100 μl of Hanks’ Balanced Salt Solution without phenol red (HBSS) (Fisher Scientific). Protein polymers in concentrations ranging from 100 μM to 5 mM were dissolved in HBSS and added to the wells containing cells. After 4 hours of incubation, the media was removed and the cells were washed with 100 μl HBSS. Cells were detached with 50 μl of 0.05% trypsin-EDTA (Invitrogen, Carlsbad, CA) solution. Approximately 150 μl of Guava reagent (Guava Technologies, Hayward, CA) was added per sample. Samples were added to a Guava EasyCyte Mini system and cell viability was determined by the Guava ViaCount™ assay according to manufacturers’ recommendations. At a minimum, each experimental condition was repeated in triplicate. Samples incubated without protein polymer served as positive treated controls.
Protease Degradation
Protein polymers in solution were incubated at 37°C with either trypsin or plasmin. For trypsin digestion, 400 μl of protein polymer at 1 mg/ml was added to 100 μl of trypsin solution (495 units/ml of trypsin) in 50 mM Tris-HCl pH 7.1. For plasmin digestion, 400 μl of protein polymer at 1.6 mg/ml was added to 100 μl of plasmin solution (0.5 units/ml) in 20 mM Tris 150 mM NaCl pH 7.6. Samples were collected at set time points and proteases were inactivated by incubation with 1% (w/v) SDS at 100 °C. Proteolytic degradation of protein polymers over time was monitored by electrophoretic analysis using a 12% SDS-PAGE gel.
Peptide and Peptoid-peptide Hybrid Synthesis and Purification
Peptide synthesis reagents were supplied by Aldrich (Milwaukee, WI) and Applied Biosystems (Foster City, CA). Resin and Fmoc-protected amino acids were purchased from NovaBiochem (San Diego, CA). The first peptide, (1) Ac-EGSGRGDSP-NH2 was synthesized using FastMoc™ (Applied Biosystems) chemistry on Rink amide resin on an ABI 433A automated peptide synthesizer (Applied Biosystems). The second peptide, (2) Ac-GQQQLGSEGRGDSP-NH2 was synthesized at the Northwestern University Institute for Bionanotechnology in the Medicine center. The peptoid-peptide hybrid, (3) Ac-(FKG)2-(NMEG)4-E-NH2, was also synthesized on the ABI 433A on Rink amide resin following a submonomer protocol.32 The peptides and peptoid-peptide hybrid were cleaved with a mixture of 2.5% triisopropylsilane 2.5% H2O, and 95% trifluoroacetic acid (TFA) for 1.5 hours at room temperature. The peptides and peptoid-peptide hybrid were purified on a Varian preparative RP-HPLC system with a Peeke C18 preparative column using a linear gradient of 0%–35% solvent B in solvent A over 70 minutes for peptides, and 10–50% solvent B over 60 minutes for the peptoid peptide hybrid, at 20 ml/min (solvent A is 0.1% TFA in water (v/v) and solvent B is 0.1% TFA in acetonitrile (v/v)). The peptides’ and peptoid-peptide hybrid’s molar masses and purities were confirmed using MALDI-TOF spectroscopy and electrospray ionization spectrometry at Northwestern University’s Analytical Services Laboratory.
Chemical Conjugation
The Ac-EGSGRGDSP-NH2 peptide (1) and the peptoid-peptide hybrid Ac-(FKG)2-(NMEG)4-E-NH2 (3) were grafted to the protein polymer scaffold using 1-ethyl-3-carbodiimide hydrochloride (EDC, Pierce, Rockford, IL) and N-hydroxysulfosuccinimide (Sulfo-NHS, Pierce). The protein polymer was dissolved at 1 mg/ml in aqueous 0.1 M 4-morpholineethanesulfonic acid (MES), 0.5 M NaCl, pH 6.0. A 2.5X molar excess of peptide 1 or peptoid-peptide hybrid 3 was added to the solution; reagent was added so that the final concentration resulted in 50 mM EDC and 65 mM Sulfo-NHS. The reactions proceeded for 24 hours before dialysis and analysis by MALDI-TOF spectroscopy. The number of molecules conjugated to the protein polymer then was determined by the shift in molar mass divided by the molar mass of the molecule.
An amine-reactive poly(ethylene glycol) (PEG) derivative, NHS-m-dPEG™ (molar mass 333 g/mol) was purchased from Quanta BioDesign Ltd.(Powell, OH). A solution was prepared of 97.6 mg NHS-m-dPEG™ diluted in 5 ml ddH2O. A second solution of protein polymer was dissolved at 10 mg/ml in 0.1 M Sodium Phosphate, 0.15 M NaCl, pH 7.2. The NHS-m-dPEG™ solution was added to 1 ml of protein polymer and reacted at room temperature for 1 hr. The degree of PEGylation was determined by MALDI-TOF spectroscopy as described above. The average number of conjugated PEGs was determined by the change in molar mass.
The Alexa-Fluor 488 dye 5-TFP (Invitrogen) was conjugated onto the protein polymer. Protein polymer was dissolved at 1.16 mg/ml in 0.1 M sodium bicarbonate, pH 9.0 and the AlexaFluor 488 5-TFP was dissolved at 1 mg/μl in DMF. 50 μl of the AlexaFluor solution was added to 5 ml of protein polymer solution. The reaction was incubated at room temperature with continuous stirring for 1 hour. Unreacted fluorophore and salt were removed using a Centrisep spin column from Princeton Separations (Adelphia, NJ). The average number of conjugated fluorophores then was estimated by MALDI-TOF spectroscopy.
The MALDI-TOF data were used to determine polydispersity as calculated by the following equations:
| Eq. 1 |
| Eq. 2 |
| Eq. 3 |
Mn = number average molecular weight, Mw = weight average molecular weight, N i = signal intensity at point i, Mi = mass at point i, D = polydispersity.
Enzymatic Conjugation
Peptide (2) Ac-GQQQLGSEGRGDSP-NH2 was conjugated to the protein polymer utilizing the enzyme tissue transglutaminase (tTG). Protein polymer was dissolved at 12 mg/ml in 200 mM 3-(N-Morpholino)propanesulfonic acid (MOPS), 20 mM CaCl2, pH 7.7 buffer and peptide (2) was dissolved at 50 mg/ml in 2 mM (Ethylenedinitrilo)tetraeacetic acid (EDTA) pH 7.3 buffer. Approximately 3.9 units of tTG were dissolved in 50 μl of 2 mM EDTA, 20 mM DL-Dithiothreitol (DTT), pH 7.7 buffer. Enzymatic grafting of peptide (2) was completed with 200 μl of protein polymer solution, 93 μl peptide (2) solution and 30 μl of tTG solution. The reaction was incubated at 37°C for 1 hour. MALDI-TOF spectroscopy was used to estimate the average number of peptides conjugated.
Gelation
Protein polymers were crosslinked at 10 wt% in 20 mM sodium phosphate 0.15 M NaCl by the addition of amine-reactive crosslinkers. Bis(sulfosuccinimidyl) suberate (BS3) was added to the protein polymer solution at approximately 80 mM. β-[Tris(hydroxymethyl)phosphino] propionic acid (THPP) was added to the protein polymer solution at a final concentration of 50 mM. For incorporation of the fluorophore, 1% of solution volume was the protein polymer-AlexaFluor conjugate dissolved in MOPS buffer. Solutions were mixed vigorously and incubated at 37°C for 1 hour. Gelation was tested by sample inversion. Image of fluorescent gel was captured on a Kodak Image Station (Rochester, New York) with UV transillumination, a 465 nm excitation filter, 535 nm emission filter, and a 5-second exposure.
Results and Discussion
A new class of protein polymers was designed to display chemically reactive functional groups with different spatial distributions. These reactive groups can be modified with short peptides, polymers, bioactive proteins, sugars, or organic complexes, allowing the protein polymers to serve as scaffolds for multivalent display for therapeutic applications. Our primary goal was to create flexible, random-coil proteins that have a controlled number and spacing of reactive functional groups. To accomplish this, we created a de novo-designed repetitive protein sequence expected to be soluble in aqueous media at the appropriate concentrations for the intended applications. A critical parameter in the de novo protein design is the choice of amino acid sequence, since sequence can determine the overall protein solubility and structure. The dependence of these parameters on amino acid sequence, however, can be challenging to predict accurately, and there can be unintended secondary and tertiary affects that influence the final protein solubility at physiological conditions.33 In addition, amino acid hydrophobicity and spacing may contribute to protein aggregation. This unpredictability may partially explain why previous research with genetically engineered protein polymers has been performed for the most part using well-characterized silk or elastin-like proteins (SLP or ELP).
The choice of the amino acid sequences employed in our protein polymers was based on previous research with random-coil proteins. Previously, we reported well-expressed, random-coil, water-soluble protein polymers with the repeat sequences GAGQGSA34, GAGQGEA34, and GKGSAQA31. Additional combinations of the amino acids (Gly, Ala, Gln, Ser, Thr, Lys, Glu, Val) were also investigated. These genes either did not express well, or formed insoluble products (results not shown). In the current investigation, the amino acid sequence was altered for better functionality, solubility, and stability. The glutamine residues were removed because they can undergo deamidation after exposure to acidic conditions.35 In addition, glutamine is a hydrogen bond-forming amino acid and may aid in protein polymer aggregation at elevated concentrations. The polar amino acids threonine and serine were added to promote water solubility. Evenly spaced lysine residues were included to introduce functional primary amine groups, as an alternative to carboxylate chemistry utilized by the Kiick group. Previous researchers have included sparsely spaced lysines in ELP proteins for chemical crosslinking and stabilization.36 Based on their results and on previous research with random-coil lysine-containing proteins,31 the lysine frequency was chosen to be every 4, 6, and 8 amino acids.
Gene Construction
Three synthetic oligonucleotides encoding repeats of K4, K6, and K8 (GKGASGKGA, GKGTGA, and GKAGTGSA respectively) (Figure 1) were designed and PCR-amplified. The genes flanked by EarI restriction sites were cleaved to create monomers. These monomers were isolated and ligated together to produce multimers for controlled cloning. Controlled cloning allows for the production of large genes without any particular amino acid sequence requirement. This technique enables the well-controlled creation of higher order DNA multimers (concatemers) by the reconcatemerization of pre-multimerized genes. In addition, this method prevents multimer intramolecular cyclization, which can otherwise occur during the self-ligation reaction and prevents gene insertion into the recipient vector.31 Genes encoding: 30 repeats of K4, 40 repeats of K6, and 30, 60, and 120 repeats of K8 were created by these techniques (Figure 2A).
Figure 2.

(A) 2% agarose gel of DNA genes digested from the pET19 plasmid with NdeI and BamHI. Lane 1: 100bp marker, lane 2: K640, lane 3: K430, lane 4: K830, lane 5: K860, lane 6: K8120 lane 7: 1 kbp marker (B) 12% SDS-PAGE gel of purified protein polymers. Lane 1: K640, lane 2: K430, lane 3: K830, lane 4: K860, lane 5: K8120 lane 6: protein standard.
Protein Expression and Purification
The proteins were successfully expressed in E. coli cells with the pET19 expression vector, despite previous concerns that expressing proteins with high numbers of lysine residues could cause increased toxicity to the bacterial host associated with the high concentration of the cationic amino acid.37 During expression, toxicity was not observed with any of the recombinant proteins (see supporting information). These proteins were easily purified by nickel affinity chromatography (Figure 2B). Although SDS-PAGE analysis shows the proteins migrating at higher than expected molecular weights, this is not uncommon for engineered proteins, and is most likely due to charge and solvent effects.38 Protein polymer molar mass and composition was compared with expectation and confirmed to match by MALDI-TOF MS analysis (Table 1) and amino acid analysis results (data not shown). The yields ranged from 42 to 75 milligrams of purified protein per liter of bacteria culture (Table 1).
Table 1.
Protein polymer yields and MALDI-TOF analysis results
| Yield (mg/L)a | Expected Mass (Da) | Observed Mass (Da) | |
|---|---|---|---|
| K430 | 54 | 22,110 | 22,086 |
| K640 | 42 | 21,702 | 21,786 |
| K830 | 75 | 21,732 | 21,789 |
| K860 | 65 | 40,622 | 40,671 |
| K8120 | 52 | 78,402 | 79,020 |
Yield given in mg of purified protein per liter of cell culture
Protein Characterization
The phase behavior of protein polymers in aqueous solution was investigated by temperature-dependent turbidimetry. The cationic protein polymers were designed based on amino acid sequence to be water-soluble over a wide range of temperatures. Protein solubility was monitored by UV absorbance as a function of temperature. Heating and cooling profiles for the protein polymer solutions at a concentration of 1 mg/ml (12.7 to 46 μM), relevant to study ligand-receptor interactions and drug delivery, were essentially flat (see supporting information). However, the protein polymers showed a slight increase in absorbance at 80°C. The increase in turbidity does not appear to be reversible as indicated by the small hysteresis observed when cooling, indicating some aggregation and precipitation. The protein polymer length affects the initial solution turbidity, where longer proteins with the same amino acid sequence had a higher initial absorbance, suggesting a decrease in protein polymer solubility as a function of increasing length. Protein amino acid sequence, in this set of constructs, had minimal influence on phase behavior. The three constructs, differing in lysine spacing, had essentially the same initial absorbance. However, K830 had a non-reversible and prominent shift in turbidity at elevated temperatures. The higher concentration of charged groups in the K4 and K6 constructs likely assisted in enhancing their water-solubility.
Circular dichroism (CD) was used to characterize the average secondary structures of the protein polymers in aqueous solution as a function of amino acid sequence, protein polymer length, and temperature. The CD spectra exhibit a minimum signal at 195 nm and a maximum at 212 nm, indicating that the proteins adopt a random coil conformation (Figure 3). As designed, these proteins do not adopt β-sheet or α-helical structure, which can lead to protein aggregation and decreased solubility. For all proteins, the per residue molar ellipticity was found to decrease as temperature was increased. As protein polymer length increased, the spectra maintained the spectral characteristics of a protein with a random coil conformation and the molar ellipticity per residue increased, suggesting a more disordered state for the longer molecules (Figure 3B). Protein polymer lysine spacing and amino acid sequence did not appreciably affect chain conformation (Figure 3C). However, the amino acid sequence appeared to influence the per residue molar ellipticity.
Figure 3.

Circular dichroic spectra of protein polymers in solution. (A) K830 over a range of temperatures. Spectra as a function of protein polymer (B) length and (C) lysine spacing and amino acid sequence.
Biocompatibility of Cationic Protein Polymers
For further studies, protein polymers with lysine residues spaced every 8 amino acids (K8 proteins) were chosen as the model system because we anticipated less toxicity for biological applications. The K8 protein polymers in solution at concentrations between 100 μM to 5 mM were incubated with mouse fibroblasts and cell viability was examined after 4 hours. The percent of viable cells was determined from a Guava ViaCount™ Assay that determines cell viability based on permeability of DNA-binding dyes and can distinguish viable, apoptotic, and dead cells. Cells incubated with K830 at a concentration as high as 5 mM had low toxicity (Figure 4). Proteins K860 and K8120 were unable to be tested at 5 mM concentration due to increased solution turbidity, which resulted in low cell counts with the Guava ViaCount™ system. Proteins with the same amino acid sequence displayed some toxicity at elevated concentrations with increased protein length. The increased toxicity is likely due to the concentration of cationic lysine groups. Poly(lysine) is known to be toxic to a wide range of mammalian cells.39,40 The K830 construct with 31 lysines per molecule showed the least toxicity. K8 60 with 61 lysines per molecule, showed some toxicity starting at 1 mM concentration. K8120, containing the most lysines at 121 per molecule showed an onset of toxicity at a concentration of 500μM.
Figure 4.
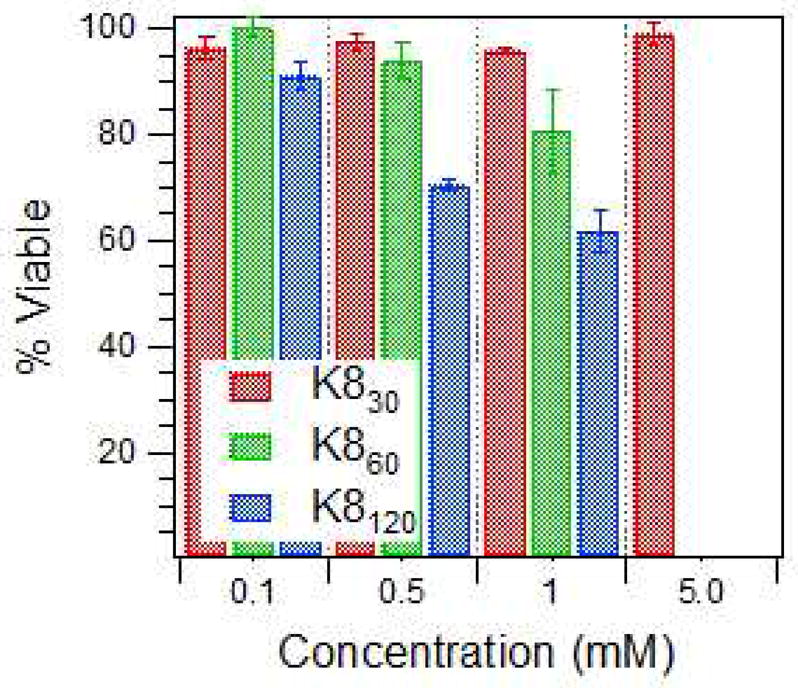
Relative cell viability after incubation with protein polymers. Viability determined by the Guava ViaCount™ Assay after a 4-hour incubation.
Protein Polymers as Protease Substrates
The protein polymers were determined to be degradable by two lysine-based proteases. Trypsin, a pancreatic endoprotease and member of the serine protease family, hydrolyzes peptide bonds on the carboxyl side of lysine and arginine residues. The protein polymer K830 was fully digested by trypsin in under 30 minutes as indicated by a Coomassie-stained SDS-PAGE gel of the protein construct and its degradation products (Figure 5A). The second protease, plasmin, is a lysine-based protease that cleaves on the carboxyl side of lysine residues and arginine residues with a higher specificity than trypsin. In vivo, plasmin converts fibrin into solubilized products for clearance. The lysine-based protein polymers were highly susceptible to plasmin cleavage; after a 24 hour incubation, virtually no full length K8 30 remained (Figure 5B). The multiple degradation sites in the protein allow for rapid degradation and potentially rapid polymer clearance in vivo. This would facilitate the release of the pendant molecules conjugated to the protein backbone.
Figure 5.

Protease degradation of K830 in solution with (A) trypsin and (B) plasmin. Control samples incubated without proteases are displayed in odd lane numbers for each time point. Full-length protein is indicated by the arrows.
Chemical Conjugation
These cationic protein polymers were designed to have reactive lysines that are susceptible to a variety of linking chemistries; the primary amines from the lysines can react with chemical groups such as N-hydroxysuccinimide ester (NHS) and with imidoester reactions. In this example, coupling was completed using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), which links primary amines with carboxyl groups. The water-soluble EDC was used to form active ester functional groups with carboxyl groups on the pendant molecules using N-hydroxysulfosuccinimide (sulfo-NHS). A stable amide linkage was created in the presence of the protein polymer amine group, which attacks the carbonyl group of the ester and eliminates the NHS group. EDC in the presence of sulfo-NHS was efficient and as is known to increase the yield of conjugation versus EDC alone by mitigating the competitive reaction with water.41
Protein polymers were conjugated with a short bioactive peptide or peptoid-peptide hybrid using the EDC/Sulfo-NHS coupling method. The peptide (1), Ac-EGSGRGDSP-NH2, was employed as prototypical bioactive ligand for conjugation to the protein polymer backbone. This peptide contained the GRGDSP sequence, which is derived from the cell attachment site of fibronectin and recognized by a number of integrin receptors.42 This particular version of the bioactive RGD peptide was designed to have a water-soluble glycine-serine spacer, positioned after the glutamic acid residue, which afforded an additional carboxyl group for chemical conjugation. The bioactive peptide was coupled to K830; Figure 6A shows the incorporation of an average of 8 RGD-containing peptides (1) onto the 31 amine sites in K830 for 26% grafting. Peptide (1) is capable of being crosslinked at the aspartic acid residue, and this may impact the ability for bioactive recognition and disturb receptor binding. To overcome this, orthogonal protecting groups could be used during peptide synthesis which would allow for shielding of the RGD sequence from the coupling chemistry and prevent crosslinking at both the D and E sites.
Figure 6.

MALDI-TOF spectra of chemically conjugated protein polymer scaffolds. The protein K830 was conjugated with (A) a RGD peptide (1) (black), PEG (blue), RGD and PEG (red), (B) peptoid peptide hybrid (3) (C) Alexa-Fluor 488. The unconjugated MALDI spectrum of K830 is shown in green in all graphs.
In the second example, a peptoid-peptide hybrid (3) was chemically crosslinked to the protein polymer. Peptoids, or poly-N-substituted glycines, have their amino acid side chains appended to the nitrogen rather than the α-carbon in the peptide backbone.43,44 Certain peptoids have non-fouling properties45 and all peptoids are extremely stable even to large temperature changes, proteases, and other denaturing conditions.46,47 The peptoid-peptide hybrid potentially introduces non-fouling properties into the scaffold to possibly prevent protein and cell adsorption. The peptoid-peptide hybrid (3), Ac-(FKG)2-(NMEG)4-E-NH2 was synthesized on resin to include a free carboxylate group on the C′-terminus, by the inclusion of the glutamic acid residue. The amine terminus, contained an FKG repeat that could act as either an additional amine site for chemical grafting or as an enzymatic recognition site for transglutaminase.48 Through EDC/sulfo-NHS coupling, an average of six peptoid-peptide hybrid molecules (3) were conjugated onto the K830 scaffold, resulting in 20% coverage (Figure 6B). The low efficiency of EDC coupling may be due to the side reaction with water, or the crosslinking of (3) to itself. To increase the extent of grafting, a greater excess of both NHS and the coupling peptide could be used. In addition, the use of an orthogonal protecting group on the K residue could prevent cross reactions between K and E. In principle, any peptide or peptoid-peptide hybrid (not containing glutamic or aspartic acid in the bioactive region) could be coupled using this technique, introducing a range of functionality to the protein polymer scaffolds. Recently, using the same conjugation chemistry, our lab has reported the coupling of gadolinium chelators to these protein polymer scaffolds for use as MRI contrast agents. 49
The third example, a synthetic polymer, poly(ethylene glycol) (PEG) was grafted onto the protein polymer. PEG has been shown to be biocompatible and to increase the in vivo circulation time of drug carriers due to its slower clearance from the body.50 A multivalent scaffold that is PEGylated may have some advantages in increasing scaffold solubility and retention, reducing aggregation and providing non-immunogenicity. PEG was incorporated onto K830 by the reaction of a low-molar mass, monodisperse PEG functionalized with an NHS group to the lysines in the protein polymer, forming stable amide bonds. We incorporated an average of 12 PEG chains onto K830 resulting in 39% grafting (Figure 6A).
In the fourth example, a low-molar mass fluorophore was added to the protein polymer scaffold to allow in vivo and in vitro visualization. The Alexa Fluor 488 5-TFP has been used in applications such as photolithographic patterning of PEG-based hydrogels51 and for labeling proteins for electrophoresis on microchips.52 The Alexa Fluor 488 5-TFP contained a tetrafluorophenyl ester for amine labeling. The fluorophore was successfully conjugated onto an average of 4 sites on the K830 protein polymer, for 13% conjugation. Since the fluorophore is very sensitive, the particularly low level of derivatization is desired since it leaves more lysine sites available for conjugation with other moieties or for aqueous solubility.
In these examples, the active ester conjugation techniques did not react to completion, allowing for multivalent coupling. Multivalent conjugation can be performed in a step-by-step or batch-wise coupling. For illustration, K830 was first conjugated with the bioactive RGD peptide (1) by the methods described above, and then in a second step was coupled with NHS-m-PEG. The resulting multivalent scaffold had an average of 8 out of 31 sites occupied by the bioactive peptide (1) and 7 sites with PEG, resulting in 48% total coverage (Figure 6A). The remaining free amines in the scaffold aided in aqueous solubility.
As an alternative to chemical conjugation, enzymatic coupling by tissue transglutaminase (tTG) was investigated. Transglutaminase catalyzes the formation of an isopeptide bond between the γ-carboxamide group on a glutamine residue and the ε-amino group on a lysine residue under biocompatible reaction conditions.24 The covalent bonds created by tTG are highly protease-resistant.53 The protein polymers were tested as an acyl acceptor (lysine substrate) for tTG by coupling with an acyl donor (glutamine substrate) short peptide. Figure 7 shows tTG grafting of 15 peptides (2) onto the protein polymer K830 resulting in 48% coverage. Successful enzymatic grafting is favored since the tTG sequence specificity is less stringent for amines than for glutamine residues. 54 The glutamine substrate on the short peptide sequence (2) was chosen based on research by Hu and Messersmith,48 where the sequence GQQQL was found to have high specificity towards tTG. In addition, the glutamine substrate peptide (2) contained the RGD cell adhesion sequence for presentation of an integrin-binding domain.
Figure 7.

MALDI-TOF spectra of unconjugated (green) and conjugated (red) K830 protein polymers. The protein K830 was conjugated with peptide 2 by tTG.
The derivatization of the protein polymers by these methods may alter solution behavior depending on number and type of pendant groups. For instance, grafting at the K sites would reduce the cationic charge and potentially decrease toxicity. Also, protein polymer solubility may be altered by the specific pendant groups present. For example, PEGylation of the scaffold resulted in higher solubility than unconjugated protein polymer (results not shown). We hypothesize that given the flexibility of the backbone and the initially random coil structure of the protein polymer, the conformation is unlikely to change significantly with the addition of a few pendant groups, depending on the structure of the conjugated moieties (preliminary data with Gd conjugated49 protein polymers). Furthermore, we hypothesize that the low efficiency of grafting in the conjugates described here should lead to only minor changes in solution properties.
One advantage of protein polymer scaffolds is their inherent monodispersity. The derivatization of these non-natural proteins, however, does introduce some distribution of molecular weight. For this reason, the polydispersity index (PDI) was calculated for the constructs using equations (1–3). Table 2 shows the calculated PDI based on the MALDI-TOF MS spectra for each conjugate. In the case of polymers with low PDI (<1.1), MALDI-TOF analysis gives a good estimate of molecular weight distribution.55 The unconjugated protein had a PDI of 1.00 which is the predicted value for genetically engineered protein polymers. The PEG conjugation introduced very little polydispersity due the monodispersity of the conjugate moiety. Similarly, the inclusion of the fluorophore did not appreciably affect dispersity. The RGD peptides grafted by both chemical and enzymatic conjugation engendered the highest conjugate PDI. As expected, the step-wise conjugation of two different types of molecules increased polydispersity, as compared to a single-molecule conjugation. Although PDI does increase with conjugation, it still remains extremely close to 1.00. The very narrow range of molecular weight can be attributed to the initially totally monodisperse protein that was the basis for the multivalent scaffold. The ability to conjugate a wide variety of materials to a scaffold and still maintain a nearly monodisperse product is advantageous for many biological applications.
Table 2.
Estimated PDI for Protein Polymer Scaffolds Based on MALDI-TOF Spectra
| PDI | |
|---|---|
| K830 | 1.000 |
| K830-RGD peptide | 1.005 |
| K830-PEG | 1.001 |
| K830-RGD peptide-PEG | 1.008 |
| K830-peptoid-peptide hybrid | 1.000 |
| K830-Alexa Fluor 488 | 1.001 |
| K830-Q-RGD | 1.011 |
Protein polymer backbones can serve as well-defined multivalent scaffolds, whereas synthetic polymer backbones have a heterogeneous molecular weight, stereochemistry, ligand spacing and ligand density. Genetically engineered polypeptides allow the fine-tuning of molecular length and weight, ligand valency and backbone conformation. Backbone conformation appears to play an important role in some multivalent events. For example, Lui and Kiick created a glycopolymer with an α-helical core that provided a relatively rigid backbone and controlled spacing of pendant groups for toxin inhibition. The glycopolymer had up to a 340-fold improvement in inhibition of cholera toxin binding than galactose monomers,20,21 which was a 2-fold improvement over similarly spaced pendant groups on a flexible random coil protein backbone. Although the α-helical conformation was favorable in this case, it is important to note that this conclusion cannot necessarily be generalized. Additionally, the glycopolymer conformation was controlled by the α-helical backbone, and this technique is not applicable under all conditions, since α-helical stability is dependent on temperature and amino acid sequence. The random coil proteins presented herein create a flexible multivalent scaffold that may be advantageous for the display of some binding moieties.
Gelation
The protein polymers were able to form self-supporting hydrogels upon the addition of amine-reactive crosslinkers. The first crosslinker investigated was bis(sulfosuccinimidyl) suberate (BS3), a water-soluble homobifunctional NHS ester that reacts with primary amine groups at pH 7–9 to form stable amide bonds. The unconjugated protein K830 was reacted with BS3, and a hydrogel formed within minutes (Figure 8a). The second chemical crosslinker tested was β-[Tris(hydroxymethyl)phosphino] propionic acid (THPP). THPP is a water-soluble trifunctional crosslinker that reacts with both primary and secondary amines to produce covalent linkages by a Mannich-type reaction. In addition, the THPP crosslinker has been shown to be biocompatible with fibroblast cells,56 suggesting that THPP crosslinking may be used for safe in situ (in vivo) hydrogel formation. The unconjugated protein K830 was dissolved in aqueous media and crosslinked with THPP; gelation was observed to occur in less than 10 minutes which is ideal for in situ therapies (Figure 8b). The ability to chemically crosslink multivalent scaffolds with our protein polymers was recently shown with Gd(III) chelators conjugated to K830. Gels crosslinked with glutaraldehyde showed increased MRI contrast over controls.49 In the present study, we incorporated K830-AF488 into the protein polymer hydrogel by chemical crosslinking with BS 3. As expected, the fluorescence of AF488 was not affected by the incorporation of the protein polymer scaffold into a chemically crosslinked hydrogel (Figure 9C).
Figure 8.

Photographs of K830 precursor solution before and after gelation with (A) THPP (B) BS3 chemical crosslinking.
Figure 9.

Photographs of K830-AF488 precursor solution (A) before and (B) after gelation with BS3 chemical crosslinking. (C) Fluorescent image after gelation.
Conclusions
This study has demonstrated the synthesis and characterization of a new class of cationic protein polymers with precisely defined molecular weight and reactive group spacing. We have cloned and expressed protein polymers with varying lengths and reactive lysine group spacings. The protein polymer length and amino acid sequence were shown to affect their bulk properties in solution, where longer polymers had slightly decreased solubility and greater random coil per-mole-residue ellipticity. As designed, the proteins were confirmed to be water-soluble and to adopt random chain configurations, i.e., to be natively unfolded at physiological conditions. The protein polymers were biocompatible to mammalian cells at concentrations necessary for delivery applications. The cationic nature of the proteins resulted in some observable toxicity at elevated protein polymer (lysine) concentrations, but this toxicity could be modulated by derivitization of the lysines. We have shown the ability to chemically and enzymatically couple a range of molecules to the protein polymers. The versatility of these engineered protein scaffolds for multivalent display was shown by attaching a bioactive peptide, a non-fouling peptoid-peptide hybrid, a non-immunogenic polymer (PEG), and a small organic molecule (fluorophore). We also demonstrated the ability to create a polyvalent scaffold by the step-wise conjugation of a bioactive peptide and a non-immunogenic polymer onto the same protein polymer backbone. Even after this two-step conjugation, these protein polymers offered remaining free reactive sites that could undergo chemical crosslinking to form a self-sustaining hydrogel. These results suggest the ability to create a multitude of materials with a variety and significant number of pendant groups for biological applications. These materials currently are being investigated as multivalent display surfaces and as the basis for a novel class of highly modular hydrogels with controlled swelling, degradation, and mechanical properties, that we hope will be useful for biomedical research and development projects.
Supplementary Material
Acknowledgments
We thankfully acknowledge the use of instruments in the Keck Biophysics Facility at Northwestern University [http://www.biochem.northwestern.edu/Keck/keckmain.html], the Northwestern University Analytical Services Laboratory, and Northwestern University Institute for Bionanotechnology in Medicine. We thank Jennifer Rea for the NIH 3T3 cells, and Russell Haynes and Nate Brown for assistance with peptide and peptoid synthesis. This work was funded by NIH/NIBIB grant 1 R01EB003806.
Footnotes
Supporting Information Available. Experimental details of protein polymer growth curves, turbidity graphs, MALDI-TOF spectra and chemical structures. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bertozzi CR, Kiessling LL. Science. 2001;291:2357–2364. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 2.Mammen M, Dahmann G, Whitesides GM. Journal of Medicinal Chemistry. 1995;38:4179–4190. doi: 10.1021/jm00021a007. [DOI] [PubMed] [Google Scholar]
- 3.Mourez M, Kane RS, Mogridge J, Metallo S, Deschatelets P, Sellman BR, Whitesides GM, Collier RJ. Nature Biotechnology. 2001;19:958–961. doi: 10.1038/nbt1001-958. [DOI] [PubMed] [Google Scholar]
- 4.Duncan R. Nature Reviews Drug Discovery. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 5.Maeda H, Seymour LW, Miyamoto Y. Bioconjugate Chemistry. 1992;3:351–362. doi: 10.1021/bc00017a001. [DOI] [PubMed] [Google Scholar]
- 6.Allen TM. Nature Reviews Cancer. 2002;2:750–763. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- 7.Cloninger MJ. Current Opinion in Chemical Biology. 2002;6:742–748. doi: 10.1016/s1367-5931(02)00400-3. [DOI] [PubMed] [Google Scholar]
- 8.Crespo L, Sanclimens G, Pons M, Giralt E, Royo M, Albericio F. Chemical Reviews. 2005;105:1663–1681. doi: 10.1021/cr030449l. [DOI] [PubMed] [Google Scholar]
- 9.Lee CC, MacKay JA, Frechet JMJ, Szoka FC. Nature Biotechnology. 2005;23:1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- 10.van Baal I, Malda H, Synowsky SA, van Dongen JLJ, Hackeng TM, Merkx M, Meijer EW. Angewandte Chemie-International Edition. 2005;44:5052–5057. doi: 10.1002/anie.200500635. [DOI] [PubMed] [Google Scholar]
- 11.Crommelin DJA, Storm G. Journal of Liposome Research. 2003;13:33–36. doi: 10.1081/lpr-120017488. [DOI] [PubMed] [Google Scholar]
- 12.Kiessling LL, Gestwicki JE, Strong LE. Current Opinion in Chemical Biology. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 13.Haag R, Kratz F. Angewandte Chemie-International Edition. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- 14.Hersel U, Dahmen C, Kessler H. Biomaterials. 2003;24:4385–4415. doi: 10.1016/s0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- 15.Kanai M, Mortell KH, Kiessling LL. Journal of the American Chemical Society. 1997;119:9931–9932. [Google Scholar]
- 16.Seymour LW, Duncan R, Strohalm J, Kopecek J. Journal of Biomedical Materials Research. 1987;21:1341–1358. doi: 10.1002/jbm.820211106. [DOI] [PubMed] [Google Scholar]
- 17.Godwin A, Hartenstein M, Muller AHE, Brocchini S. Angewandte Chemie-International Edition. 2001;40:594–597. doi: 10.1002/1521-3773(20010202)40:3<594::AID-ANIE594>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 18.Zhuravel MA, Davis NE, Nguyen ST, Koltover I. Journal of the American Chemical Society. 2004;126:9882–9883. doi: 10.1021/ja046965q. [DOI] [PubMed] [Google Scholar]
- 19.Yu SJM, Conticello VP, Zhang GH, Kayser C, Fournier MJ, Mason TL, Tirrell DA. Nature. 1997;389:167–170. doi: 10.1038/38254. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Kiick KL. Macromolecules. 2008;41:764–772. doi: 10.1021/ma702128a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Kiick KL. Journal of the American Chemical Society. 2005;127:16392–16393. doi: 10.1021/ja055102+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polizzotti BD, Maheshwari R, Vinkenborg J, Kiick KL. Macromolecules. 2007;40:7103–7110. doi: 10.1021/ma070725o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Science. 1998;281:389–392. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 24.Lorand L, Conrad SM. Molecular and Cellular Biochemistry. 1984;58:9–35. doi: 10.1007/BF00240602. [DOI] [PubMed] [Google Scholar]
- 25.Sakiyama-Elbert SE, Hubbell JA. Journal of Controlled Release. 2000;65:389–402. doi: 10.1016/s0168-3659(99)00221-7. [DOI] [PubMed] [Google Scholar]
- 26.Schense JC, Hubbell JA. Bioconjugate Chemistry. 1999;10:75–81. doi: 10.1021/bc9800769. [DOI] [PubMed] [Google Scholar]
- 27.Schense JC, Hubbell JA. Journal of Biological Chemistry. 2000;275:6813–6818. doi: 10.1074/jbc.275.10.6813. [DOI] [PubMed] [Google Scholar]
- 28.Collier JH, Messersmith PB. Bioconjugate Chemistry. 2003;14:748–755. doi: 10.1021/bc034017t. [DOI] [PubMed] [Google Scholar]
- 29.McHale MK, Setton LA, Chilkoti A. Tissue Engineering. 2005;11:1768–1779. doi: 10.1089/ten.2005.11.1768. [DOI] [PubMed] [Google Scholar]
- 30.Henaut A, Danchin A, NF, editors. ASM press. Vol. 2. Washington, D.C: 1996. pp. 2047–2066. [Google Scholar]
- 31.Won JI, Barron AE. Macromolecules. 2002;35:8281–8287. [Google Scholar]
- 32.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. Journal of the American Chemical Society. 1992;114:10646–10647. [Google Scholar]
- 33.Rizzi SC, Hubbell JA. Biomacromolecules. 2005;6:1226–1238. doi: 10.1021/bm049614c. [DOI] [PubMed] [Google Scholar]
- 34.Won JI, Meagher RJ, Barron AE. Electrophoresis. 2005;26:2138–2148. doi: 10.1002/elps.200410042. [DOI] [PubMed] [Google Scholar]
- 35.Won JI, Meagher RJ, Barron AE. Biomacromolecules. 2004;5:618–627. doi: 10.1021/bm034442p. [DOI] [PubMed] [Google Scholar]
- 36.Trabbic-Carlson K, Setton LA, Chilkoti A. Biomacromolecules. 2003;4:572–580. doi: 10.1021/bm025671z. [DOI] [PubMed] [Google Scholar]
- 37.Farmer RS, Argust LM, Sharp JD, Kiick KL. Macromolecules. 2006;39:162–170. doi: 10.1021/ma051534t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Creel HS, Fournier MJ, Mason TL, Tirrell DA. Macromolecules. 1991;24:1213–1214. [Google Scholar]
- 39.Putnam D, Gentry CA, Pack DW, Langer R. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:1200. doi: 10.1073/pnas.031577698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolfert MA, Dash PR, Nazarova O, Oupicky D, Seymour LW, Smart S, Strohalm J, Ulbrich K. Bioconjugate Chemistry. 1999;10:993–1004. doi: 10.1021/bc990025r. [DOI] [PubMed] [Google Scholar]
- 41.Staros JV, Wright RW, Swingle DM. Analytical Biochemistry. 1986;156:220–222. doi: 10.1016/0003-2697(86)90176-4. [DOI] [PubMed] [Google Scholar]
- 42.Ruoslahti E. Annual Review of Cell and Developmental Biology. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 43.Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, SR, et al. Proc Natl Acad Sci U S A. 1992;89:9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]
- 45.Statz AR, Meagher RJ, Barron AE, Messersmith PB. Journal of the American Chemical Society. 2005;127:7972–7973. doi: 10.1021/ja0522534. [DOI] [PubMed] [Google Scholar]
- 46.Borman S. Chem Eng News. 1998;76:56–57. [Google Scholar]
- 47.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Drug Development Research. 1995;35:20–32. [Google Scholar]
- 48.Hu BH, Messersmith PB. Journal of the American Chemical Society. 2003;125:14298–14299. doi: 10.1021/ja038593b. [DOI] [PubMed] [Google Scholar]
- 49.Karfeld LS, Bull SR, Davis NE, Meade TJ, Barron AE. Bioconjug Chem. 2007;18:1697–700. doi: 10.1021/bc700149u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris JM, Chess RB. Nature Reviews Drug Discovery. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 51.Hahn MS, Taite LJ, Moon JJ, Rowland MC, Ruffino KA, West JL. Biomaterials. 2006;27:2519–2524. doi: 10.1016/j.biomaterials.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 52.Roman GT, Carroll S, McDaniel K, Culbertson CT. Electrophoresis. 2006;27:2933–2939. doi: 10.1002/elps.200500795. [DOI] [PubMed] [Google Scholar]
- 53.Griffin M, Casadio R, Bergamini CM. Biochemical Journal. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greenberg CS, Birckbichler PJ, Rice RH. Faseb Journal. 1991;5:3071–3077. doi: 10.1096/fasebj.5.15.1683845. [DOI] [PubMed] [Google Scholar]
- 55.Montaudo G, Montaudo MS, Puglisi C, Samperi F. Rapid Communications in Mass Spectrometry. 1995;9:453–460. [Google Scholar]
- 56.Lim DW, Nettles DL, Setton LA, Chilkoti A. Biomacromolecules. 2007;8:1463–1470. doi: 10.1021/bm061059m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.