

Abstract
Background
We previously demonstrated that vascular smooth muscle cells (VSMC) proliferation and development of neointimal hyperplasia as well as the ability of nitric oxide (NO) to inhibit these processes is dependent on sex and hormone status. The aim of this study was to evaluate the role of estrogen receptor (ER) in mediating proliferation in male and female VSMC.
Materials and Methods
Proliferation was assessed in primary rat aortic male and female VSMC using 3H-thymidine incorporation in the presence or absence of ER alpha (α) inhibitor methyl-piperidino-pyrazole, the ER beta (β) inhibitor (R,R)-5,11-Diethyl-5,6,11,12-tetrahydro-2,8-chrysenediol, the combined ERαβ inhibitor ICI 182,780, and/or the NO donor DETA/NO. Proliferation was also assessed in primary aortic mouse VSMC harvested from wildtype (WT), ERα knockout (ERα KO), and ERβ knockout (ERβ KO) mice in the presence or absence of DETA/NO and the ERα, ERβ, and ERαβ inhibitors. Protein levels were assessed using Western blot analysis.
Results
Protein expression of ERα and ERβ was present and equal in male and female VSMC, and did not change after exposure to NO. Inhibition of either ERα or ERβ had no effect on VSMC proliferation in the presence or absence of NO in either sex. However, inhibition of ERαβ in rat VSMC mitigated NO-mediated inhibition in female but not male VSMC (p<0.05). Evaluation of proliferation in the knockout mice revealed distinct patterns. Male ERαKO and ERβKO VSMC proliferated faster than male WT VSMC (p<0.05). Female ERβKO proliferated faster than female WT VSMC (p<0.05), but female ERαKO VSMC proliferated slower than female WT VSMC (p<0.05). Last, we evaluated the effect of combined inhibition of ERα and ERβ in these knockout strains. Combined ERαβ inhibition abrogated NO-mediated inhibition of VSMC proliferation in female WT and knockout VSMC (p<0.05), but not in male VSMC.
Conclusions
These data clearly demonstrate a role for the ER in mediating VSMC proliferation in both sexes. However, these data suggest that the antiproliferative effects of NO may be regulated by the ER in females but not males.
Keywords: neointimal hyperplasia, vascular smooth muscle, nitric oxide, hormones, estrogen receptors
Introduction
Atherosclerosis is the number one health care concern in the United States.[1] Cardiovascular disease (CVD) causes a tremendous amount of morbidity and mortality and costs over $475 billion dollars to treat per year.[1] In addition to medical therapy to delay the progression, there are a number of surgical interventions that can be performed to improve blood flow including: angioplasty, stenting, endarterectomy, and bypass grafting. Unfortunately, these procedures can fail due to restenosis caused by neointimal hyperplasia.[2-4] Nitric oxide (NO) is a small gaseous molecule normally produced by endothelial cells that is known to inhibit the development of neointimal hyperplasia in animals following arterial balloon injury and vein bypass grafting in small and large animal models.[5-16] Thus, NO provides a promising translational therapeutic intervention for the treatment and prevention of restenosis following vascular intervention in our expanding cardiovascular patient population.
In our previous experiments, we examined the effects of NO on vascular smooth muscle cell (VSMC) proliferation, migration, and cell cycle regulation in vitro and neointimal hyperplasia in vivo based on sex and hormone status of the animal.[17] The results of this study show that NO is effective at inhibiting these vascular processes in both sexes, however we found that NO is more effective in males versus females and more effective in a hormonally intact versus deficient environment. At baseline, in a hormone replete environment, male VSMC proliferated and migrated more and had less cell cycle arrest compared to female VSMC. After exposure to NO, male VSMC experienced greater inhibition of proliferation, migration, and cell cycle arrest than female VSMC. In a hormone deplete environment, while females proliferated more than males, NO was still more effective at inhibiting VSMC proliferation in males compared to females. In our in vivo model, NO therapy resulted in a greater reduction in neointimal hyperplasia in males than females, and in hormone intact versus castrated rats. In summary, our previous work indicates that VSMC growth and the efficacy of NO at inhibiting neointimal hyperplasia is sex- and hormone-dependent.
To address the differences in proliferation and neointimal hyperplasia between the sexes and following exposure to NO, we directed our attention to the sex hormone receptors. It remains unknown whether the sex hormone receptors are responsible for these observed differences. Since sex and hormone differences in the cardiovascular system exist in both humans and animals, it stands to reason that the sex hormone receptors could explain these differences. Thus, the goal of our study is to determine if estrogen receptor (ER) signaling regulates VSMC proliferation at baseline and following exposure to NO. Because we observed less proliferation and neointimal hyperplasia at baseline and following NO exposure in females, we hypothesized that the ER modulates proliferation as well as the antiproliferative effects of NO in females, but not males.
Methods
Nitric oxide donor
The diazeniumdiolate utilized in this study, 1-[N-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (DETA/NO), was provided by Larry K. Keefer. DETA/NO was chosen based on its half-life, efficacy in our prior publications, and to maintain consistency among our experiments.[17] DETA/NO was stored in a desiccator in the -20°C freezer. For the studies in this manuscript, a 10 mM stock solution was made fresh for each experiment and diluted accordingly. Of note, DETA/NO releases 2 moles of NO per mole of compound.
Cell culture
VSMC were isolated and cultured from the aortas of 8-week-oldmale and female Sprague-Dawley rats (Harlan, Indianapolis, IN) using the collagenase method[18] and maintained as previously described.[19;20] VSMC were also isolated and cultured from the aortas of 8-week-old male and female mice. VSMC from three different mouse strains were cultured: wild-type C57BL6/J (Taconic, Hudson, NY), ER alpha knockout mice (ERaKO), and ER beta knockout mice (ERβKO). The ERαKO mice were generously supplied by Larry Jameson, MD (Northwestern University) and Pierre Chambon, MD (Institut de Génétique et de Biologie Moléculaire et Cellulaire) and the ERβKO mice were generously supplied by Douglas Losordo, MD (Northwestern University). Please refer to the details of the creation of these KO mice as described by Dupont et al.[21] Purity of each lot of VSMC was confirmed using anti-smooth muscle α-actin monoclonal antibodies (Sigma; St. Louis, MO) and by specific morphology for VSMC. Cells were maintained in media containing equal volumes of DMEM-low glucose (SAFC Biosciences; Lenexa, KS) and Ham's F12 (JRH; Lenexa, KS) supplemented with 10% fetal bovine serum (FBS, Invitrogen; Carlsbad, CA), 100 U/mL penicillin (Invitrogen), 100 ug/mL streptomycin (Invitrogen) and 4 mM L-glutamine (VWR; West Chester, PA) and incubated at 37°0, 95% air and 5% CO 2. Rat VSMC were used between passages 3-8 for all experiments and mouse VSMC were used between passages 5-8. Every experiment was performed in male VSMC and female VSMC lines simultaneously under identical conditions.
Proliferation assay
Tritiated (3H) thymidine incorporation was assessed as a surrogate for cellular proliferation. Rat (4 × 104 cells/well) and mouse (1 × 104 cells/well) VSMC were plated in 12-well plates and were growth-arrested for 24 hours with starvation media containing no FBS. These densities were chosen in order to achieve 30-40% subconfluence on the treatment date and 60-70% subconfluence by the end of the experiment. Mouse VSMC were smaller than rat VSMC, however, proliferated faster. Thus, mouse VSMC were plated at lower densities. Mouse VSMC were plated in 12-well plates (1 × 104 cells/well), and were growth-arrested for 24 hours with starvation media (containing no FBS). Cells were then exposed to media ± DETA/NO (31-1000 μM) ± sex hormone receptor inhibitor in the presence of 3H-thymidine (5 μCi/mL, PerkinElmer, Wellesley, MA) for an additional 24 hours. Inhibitors to the following sex hormone receptors were used: estrogen receptor alpha (ERα), estrogen receptor beta (ERβ), combined ERα and ERB (ERαβ), androgen receptor (AR), and aromatase. The ERα inhibitor, Methyl-piperidino-pyrazole (MPP, Tocris Bioscience; Ellisville, MO), was dissolved in ethanol and used between 10-1000 nM. The ERβ inhibitor, (R,R)-5,11-Diethyl-5,6,11,12-tetrahydro-2,8-chrysenediol (THC, Tocris Bioscience), was dissolved in ethanol and used between 100-2500 nM. The ERαβ inhibitor, ICI 182,780 (ICI, Sigma, St. Louis, MO), was dissolved in ethanol and used between 10-1000 nM. The AR inhibitor, flutamide (Sigma), was dissolved in ethanol and used between 1-100 nM. The aromatase inhibitor, formestane (Sigma), was dissolved in methanol and used between 1-100 nM. The concentrations of the inhibitors were initially determined by evaluating a range of concentrations around the known Ki for each compound and ensuring no cell death using trypan blue exclusion. The cells were pre-treated with the inhibitors 1-2 hours prior to treatment with DETA/NO. After 24 hours of exposure to the treatment groups, 3H-thymidine incorporation into trichloroacetic acid–precipitated DNA was quantified by scintillation counting. Of note, cell counting was also performed and the results of cell counting correlated with 3H-thymidine incorporation.
Western blot analysis
VSMC were collected after 24 hours of exposure to DETA/NO by scraping and were resuspended in 20 mM Tris with 100 μM phenylmethylsulfonylflouride (Sigma), 1 μM leupeptin (Sigma), and 1 μM sodium orthovanadate (Sigma). Protein was quantified with the bicinchoninic acid protein assay according to manufacturer's instructions (Pierce, Rockford, IL). Whole cell samples (20 μg) were subjected to sodium dodecylsulfate-polyacrylamide gel electrophoresis on 8-13% gels and transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH). Membranes were hybridized using antibodies to ERα (rabbit polyclonal, 1:500, Santa Cruz Biotechnology, lnc.; Santa Cruz, CA), ERβ (mouse monoclonal, 1:1000, Abcam; Cambridge, MA), AR (rabbit monoclonal, 1:2000, Abcam), and aromatase (rabbit polyclonal, 1:750, Abcam). Primary antibodies were followed by horseradish peroxidase-linked goat anti-rabbit immunoglobulin (1:10,000; Pierce). Proteins were visualized using chemiluminescent reagents according to the manufacturer's instructions (Supersignal Substrate; Pierce), and the membranes were exposed to film and developed. Western blot films were scanned to JPEG images. Densitometry was performed on representative images using ImageJ (National Institutes of Health (NIH), Bethesda, MD). Protein levels were adjusted to beta actin levels, and normalized to the male starvation group to allow for comparisons between different experiments.
Statistical analysis
Results are expressed as mean ± standard error of the mean (SEM). For baseline properties between male and female VSMC, the male control was compared to the female control. Each NO group was compared to the same-sex control group. In order to compare the effects of NO between the male and female groups, the male group was calculated as a percent of its control and the female group was calculated as a percent of its control. The percent reduction of each male NO group was compared to the female counterpart. Studies with inhibitors were compared to DETA/NO 250 μM treatment group within the same sex. Differences between two groups were analyzed using t-tests. Differences between multiple groups were analyzed using one-way analysis of variance (ANOVA) with the Student-Newman-Keuls post hoc test for all pairwise comparisons (SigmaStat; SPSS, Chicago, IL). Statistical significance was assumed when P < 0.05.
Results
No difference in hormone receptors was found between male and female rat VSMC
Given the differences in proliferation that were observed between the sexes and based on hormone status, hormone receptor levels were compared based on sex and after NO therapy using Western blot analysis (Figure 1). Aromatase converts testosterone to estrogen, thus is responsible for local hormone levels. Therefore, we assessed expression of aromatase as well. The expression of ERα, ERβ, AR, and aromatase was found in both male and female rat VSMC. However, no differences were found based on sex or after NO therapy in ERα, ERβ, AR, and aromatase levels. Thus, levels of these proteins do not appear to be responsible for observed differences in proliferation based on sex.
Figure 1.
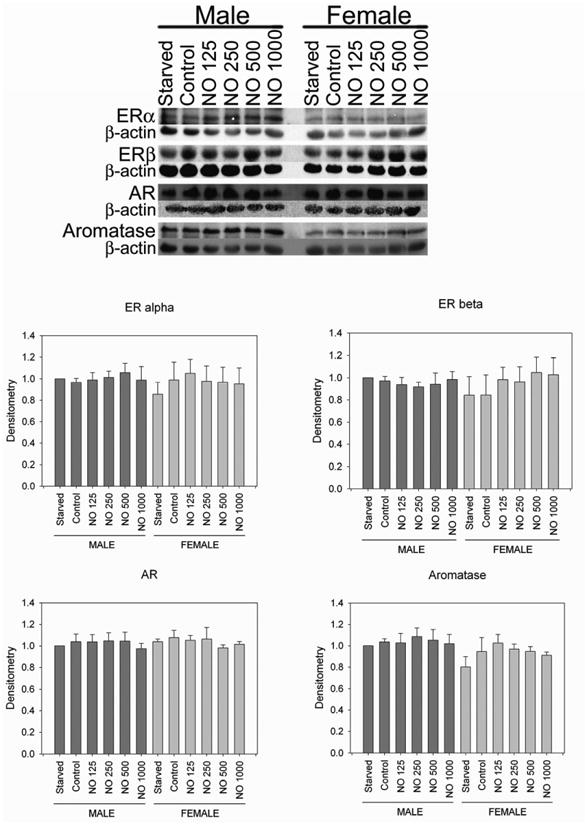
Western blots showing the presence of estrogen receptor alpha (ERα), estrogen receptor beta (ERβ), androgen receptor (AR), and aromatase present in male and female vascular smooth muscle cells. No differences in any of these proteins levels were noted between the sexes or following treatment with NO. NO = DETA/NO (μM). Western blot image is representative of 3-4 separate experiments. Graphs represent the densitometry data from 3-4 separate experiments adjusted for beta actin levels and normalized to the male starvation group to allow for comparisons between experiments and groups.
Estrogen receptors mediate the antiproliferative effects of NO in females
Since hormone receptors were present and expressed equally in males and females, we wanted to determine if their activity differed between sexes or after NO therapy. Therefore, proliferation assays were conducted in the presence of inhibitors to ERα, ERβ, ERαβ, AR, and aromatase (Figures 2A-E, respectively). None of the inhibitors (MPP, THC, ICI, Flutamide, and Formestane) had any effect on proliferation compared to control, and neither solvent (ethanol nor methanol) had any effect on proliferation compared to control. Treatment with MPP (Figure 2A) at 10 nM, 100 nM, and 1000 nM did not alter NO-mediated inhibition of VSMC proliferation in males (74%, 73%, and 72% respectively vs. 81%) or females (67%, 74%, and 73% respectively vs. 69%) compared to NO alone. Treatment with THC (Figure 2B) at 100 nM, 1000 nM, and 2500 nM did not alter NO-mediated inhibition of VSMC proliferation in males (71%, 76%, and 75% respectively vs. 81%) or females (68%, 77%, and 70% respectively vs. 69%) compared to NO alone. Treatment with flutamide (Figure 2D) at 1 nM, 10 nM, and 100 nM did not alter NO-mediated inhibition of VSMC proliferation in males (93%, 93%, and 81% respectively vs. 91%) or females (88%, 81%, and 83% respectively vs. 79%) compared to NO alone. Treatment with formestane (Figure 2E) at 1 nM, 10 nM, and 100 nM did not alter NO-mediated inhibition of VSMC proliferation in males (73%, 77%, and 77% respectively vs. 81%) or females (70%, 70%, and 71% respectively vs. 69%) compared to NO alone. However, using ICI (Figure 2C), males had less reduction in proliferation at the highest concentration (1000 nM) with DETA/NO compared to DETA/NO alone (61% vs. 89% respectively, P < 0.05). In females, all concentrations of ICI (10 nM, 100 nm, 1000 nM), the combined ERαβ inhibitor, resulted in a decrease in the antiproliferative effects of NO on VSMC proliferation compared to DETA/NO alone (21%, 24%, and 30% respectively vs. 40%, P < 0.05). Thus, blocking all ERs in females abrogates the antiproliferative effects of NO on VSMC.
Figure 2.
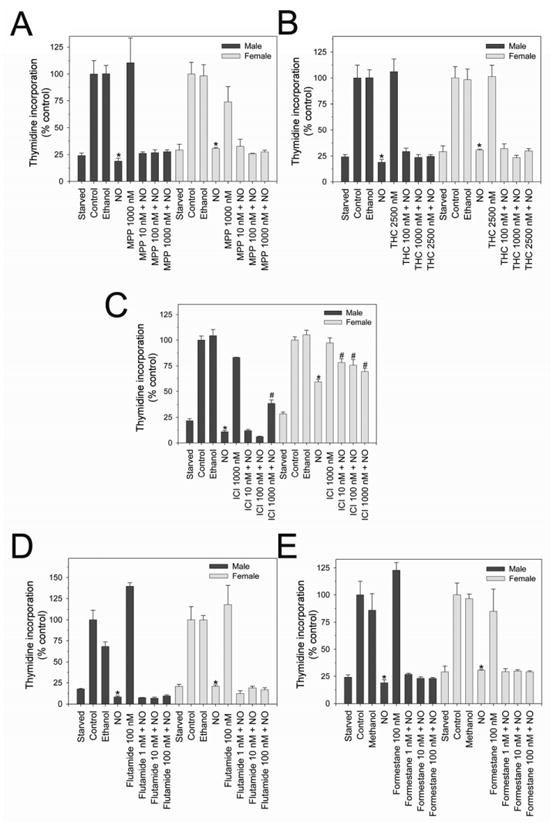
Proliferation of male and female rat vascular smooth muscle cells (VSMC) exposed to inhibitors to ERα, ERβ, or ERαβ. No reversal of NO-mediated inhibition of VSMC proliferation was observed after treatment with the A) ERα inhibitor, MPP or B) the ERβ inhibitor, THC. C) The ERαβ inhibitor ICI resulted in partial reversal of NO-mediated inhibition of proliferation in female VSMC at all concentrations but only male VSMC at the highest concentration (*P < 0.05 compared to same sex NO). No reversal of NO-mediated inhibition of VSMC proliferation was observed after use of the D) androgen receptor inhibitor, flutamide, or E) the aromatase inhibitor, formestane. NO = DETA/NO 500 μM. Data are representative of at least 3 separate experiments. n=3/treatment group.
ERα and ERβ have different roles in mediating proliferation between the sexes
Given that combined inhibition of ERα and ERβ using ICI caused partial reversal of NO-mediated inhibition of VSMC, but not ERα or ERβ alone, we wanted to examine this further. Since MPP is not a complete ERα inhibitor and THC is not a complete ERβ inhibitor and these inhibitors have not been used extensively in cultured VSMC, we harvested VSMC from ERαKO and ERβKO mice which have complete knockdown of their respective receptors.[21] Proliferation assays were conducted in the presence of DETA/NO in male and female VSMC from three strains: WT, ERαKO, and ERβKO (Figures 3A-C). In males, ERαKO (41.0 × 103 ± 3,465 cpm) and ERβKO (41.0 x 103 ± 3,465 cpm) VSMC proliferated at greater rates than WT VSMC (4.4 × 103 ± 774 cpm, *P < 0.05, Figure 3A), indicating that ERα and ERβ regulate basal proliferation in males. In females, ERβKO (32.1 × 103 ± 3,013 cpm) VSMC proliferated at greater rates than WT VSMC (12.6 × 103 ± 451 cpm, *P < 0.05, Figure 3B). However, ERαKO (1.2 × 103 ± 341 cpm) VSMC proliferated at lower rates than WT VSMC (*P < 0.05, Figure 3B). These data suggest that ERβ may function to repress proliferation or ERα may function to stimulate proliferation in females.
Figure 3.
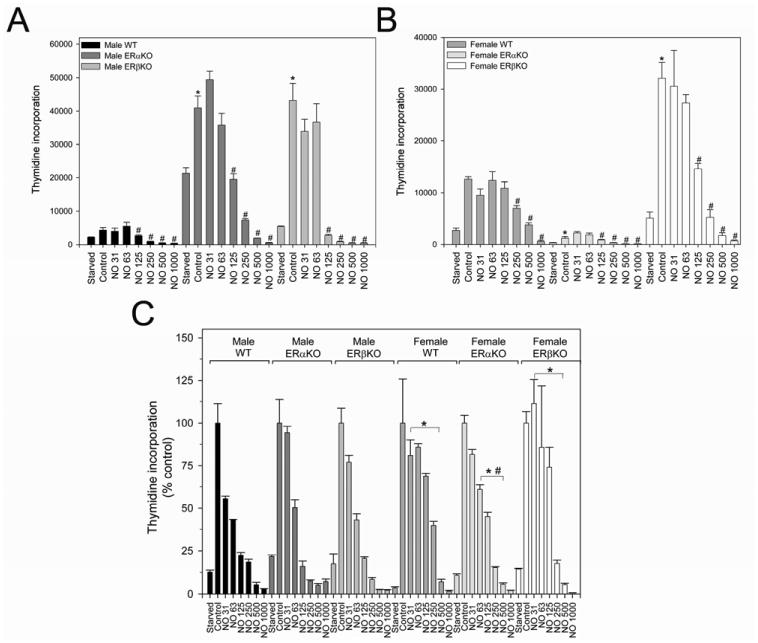
Proliferation of male and female mouse vascular smooth muscle cells (VSMC) harvested from ERα and ERβ knockout (KO) mice. A) Male ERaKO and ERpKO VSMC proliferate at greater rates than male wild-type (WT) VSMC (*P < 0.05 vs. male WT VSMC). NO caused a dose-dependent inhibition of proliferation in all mice groups (#P < 0.05 vs. same group control). B) Female ERβKO VSMC proliferate at greater rates than female WT VSMC, and female ERαKO VSMC. ERαKO VSMC proliferated at lower rates than WT cells (*P < 0.05 vs. female WT VSMC). NO caused a dose-dependent inhibition of proliferation in all mice groups (#P < 0.05 vs. same group control). C) NO had a general trend toward greater VSMC inhibition in male cells than their female counterpart (*P < 0.05 vs. corresponding male NO and mouse group). In the male groups, NO was equally effective in all male mouse cells. In the female groups, NO was most effective in the ERαKO mice (#P < 0.05 vs. corresponding female WT and ERβKO groups). NO = DETA/NO. Data are representative of at least 3 separate experiments. n=3/treatment group.
Similar to previous experiments, exposure to NO (DETA/NO 31-1000 uM) resulted in a dose-dependent inhibition of VSMC proliferation in all six cell types (#P < 0.05; Figure 3). In males, there was no difference in the antiproliferative effects of NO in the ERα, ERβ, and WT VSMC (78%, 84%, 80% reduction, respectively, DETA/NO 125 μM, P=NS), indicating that NO does not appear to signal through ERα or ERβ. Also, similar to our prior work,[17] at equivalent NO dosing the antiproliferative effect of NO is greater in male compared to female VSMC (Figure 3C, *P < 0.05). However in females, NO is most effective in ERαKO compared to WT and ERβKO (65% vs. 31% and 26% reduction, respectively, DETA/NO 125 μM, #P < 0.05). These data confirm a dependency of NO on the ERs and suggest that NO may mediate its antiproliferative effects on VSMC proliferation through ERβ in females.
Knockout mice experiments confirm that NO may signal through ERs in female VSMC
In an effort to confirm the above findings with the KO VSMC and with the inhibitors in rat VSMC, we performed additional proliferation assays using a combination of ER inhibitors and KO mice [Figure 4]. All mouse VSMC groups were treated to achieve combined inhibition of ERα and ERβ. WT VSMC were treated with ICI. ERaKO VSMC were treated with THC. ERβKO VSMC were treated with MPP. Inhibitors and solvents alone had no effect on proliferation compared to controls (data not shown). Similar to the data presented above using rat VSMC, partial reversal of the antiproliferative effects of NO was observed in only one male group, ERαKO VSMC exposed to the ERβ inhibitor, THC (10 nM, *P < 0.05). However, combined inhibition of ERα and ERβ abrogated the antiproliferative effects of NO in all female cell types (Figure 4, *P < 0.05 compared to the corresponding NO alone group). These data demonstrate that the ERs play some role in mediating the antiproliferative effects of NO in female VSMC.
Figure 4.
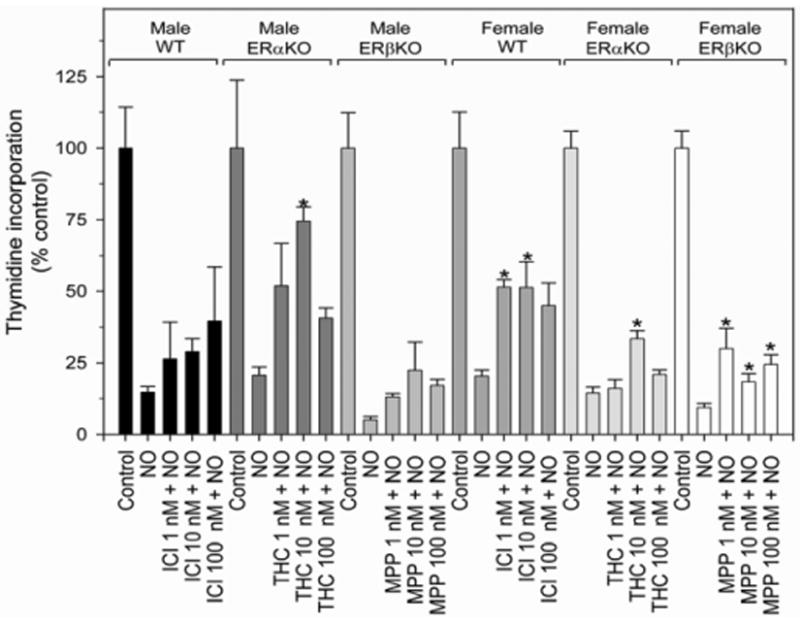
Proliferation of male and female mouse vascular smooth muscle cells (VSMC) harvested from ERα and ERβ knockout (KO) mice exposed to chemical inhibitors to ERα and ERβ. All mice groups were treated to achieve combined inhibition of ERα and ERβ. Wild-type (WT) VSMC were treated with combined estrogen receptor inhibitor, ICI. ERαKO were treated with the ERβ inhibitor, THC. ERβKO VSMC were treated with ERα inhibitor, MPP. This combined inhibition caused partial reversal of NO-mediated inhibition of VSMC proliferation in female WT and KO VSMC (*P < 0.05 vs. same group NO alone). NO = DETA/NO 250 μM. Data are representative of at least 3 separate experiments. n=3/treatment group.
Discussion
In this study, we examined the presence of hormone receptor proteins in VSMC and sought to determine whether these proteins regulate proliferation. We found that there were no differences in the protein levels of ERα, ERβ, AR, and aromatase at baseline or following exposure to NO in male and female VSMC. However, through investigations using chemical inhibitors and/or KO cells, we determined that ERα and ERβ may serve to repress proliferation in males, whereas only ERβ may do so in females. Furthermore, ERα appears to mediate the antiproliferative effects of NO in VSMC more than ERβ, yet, combined inhibition of ERα and ERβ consistently decreased the efficacy of NO in female but not male VSMC in two different assays.
Estrogen receptors exist in male and female VSMC and have been proven to be responsible for estrogen-induced vasoprotection in models of vascular injury. Estrogen also shares many vasoprotective properties in common with NO.[22-24] Thus, we sought to determine whether NO mediates its antiproliferative effects on VSMC through ER-mediated signaling.[25-33] Krom et al evaluated the role for the ER in the arterial injury response by using ERα and ERβ agonists and an ERα antagonist in a mouse model of arterial injury.[32] In this model, the investigators induced arterial injury and delivered drugs locally via an external cuff applied to the periadventitial surface of the femoral artery in male mice.[32] An ERα agonist decreased neointimal formation at low concentrations but an ERβ agonist decreased neointimal formation in a dose-dependent response for all concentrations studied.[32] Adding an ERα antagonist (MPP) produced no effect, mitigated the effect of the ERα agonist, and did not change the effects of the ERβ agonist alone.[32] These data suggest that both ERα and ERβ activation are involved in the inhibition of neointimal hyperplasia. However, ERβ may play a more prominent role in the process than ERα.
To explore the direct role of the ER in estrogen-mediated inhibition of neointimal hyperplasia, studies have been conducted in KO mice. The first ERαKO that was created was done so by the Korach laboratory.[34] Initial experiments performed in this incomplete ERαKO did not show any difference with respect to estrogen-mediated inhibition of intimal thickness or VSMC proliferation compared to WT mice.[29] ERβKO also did not show any difference with respect to intimal thickness or VSMC proliferation compared to WT mice.[30] This was a surprising finding given the results of the Krom study described above, and that Lindner et al showed that ERβ mRNA expression markedly increased after vascular injury in males.[25;32] Karas et al then created an ERα and ERβ double knockout mouse (ERαβKO).[31] These investigators reported that estrogen did not inhibit neointimal hyperplasia to the same degree in the ERαβKO compared to WT controls, yet VSMC proliferation was inhibited. These data demonstrated that ERs do play an important role in estrogen-mediated inhibition of neointimal hyperplasia, but combined inhibition may be necessary due to redundant signaling.[31] This ERαKO mouse created by Korach's group was actually an incomplete knockout. Subsequently, Chambon's laboratory created a complete ERαKO.[28] In vivo experiments performed in the complete ERαKO showed no detectable effect on any measure of vascular injury, including medial area and VSMC proliferation after estrogen therapy.[28] Therefore, a clear role for the ER was established for estrogen-mediated inhibition of neointimal hyperplasia.
Before embarking on costly animal experiments using large numbers of KO mice, our lab wanted to determine if any effect could be ascertained in cell culture that may indicate that NO signals through the ER in mediating its antiproliferative effects on VSMC. Our results consistently showed that combined inhibition of ERαβ brought partial reversal of NO-mediated inhibition of VSMC proliferation in females. These results are consistent with data indicating vasoprotective properties of the ER. Expression of these receptors was not different based on sex, but activity may be. However, NO may be less effective at inhibiting neointimal hyperplasia in female rats because NO signals through ERs. Since it has already been proven that estrogen mediates vasoprotection through the ER; higher local estrogen concentrations found in females could be competitively inhibiting NO signaling, thus making the therapy less effective in our female model.
What our prior in vivo data show is the greater the serum concentration of testosterone, the more effective NO is at inhibiting neointimal hyperplasia.[17] This could potentially establish a role of testosterone in NO-induced vasoprotection. However, it could also mean that testosterone stimulates VSMC proliferation and that NO is more effective in cells that proliferate at greater rates. Alternatively, since testosterone is converted to estrogen locally via aromatase, serum testosterone levels may ultimately affect local estrogen levels. Aromatase is an enzyme whose function is to aromatize androgens thereby producing estrogen. Most recently, VSMC and endothelial cells have also been found to be involved in this process.[35] Furthermore, aromatase expression was found to be present in both male and female VSMC and endothelial cells.[36-39] Since aromatase is an estrogen-producing enzyme in the vasculature, local hormone concentrations may be more important than plasma hormone concentrations in dictating final outcome. For example, exogenous administration of testosterone to VSMC in vitro elicits significant production of estrogen due to conversion by aromatase.[35] This may explain the conflicting results observed in the studies above administering systemic testosterone to inhibit neointimal hyperplasia, as local concentrations may be more significant.[40-42] We showed that aromatase was expressed in rat VSMC, but inhibiting this protein did not alter NO's effect on VSMC proliferation. Aromatase can still play a role in this process, but differences in VSMC proliferation based on sex and hormone status cannot be explained by aromatase alone.
Ultimately, VSMC proliferation is a result of multiple cellular and extracellular processes. It has been well established that NO, both endogenous and exogenous, clearly plays a role. Additionally, we have already shown that sex and hormones, estrogen and testosterone, clearly influence this process.[17] These experiments show that ERs play an additional role in this process and may interact with NO. The exact mechanism still needs to be delineated. The NO and ER signaling pathways are complex and redundant. For example, the ER is known to stimulate the MAPK and the PI3K/Akt signaling pathways, the latter of which stimulates NO production from eNOS. NO is known to regulate many intracellular signaling pathways, including the cGMP, cAMP, MAPK, PI3K/Akt, and protein phosphatase pathways, to name but a few. The purpose of our current study was to determine if NO signals through ERα and ERβ as one pathway to inhibit VSMC proliferation, given the downstream similarities in the effect of both estrogen and NO in the vasculature. Our data suggest that it is possible, but may depend on the specific ER subunit, sex, and local hormone environment. Additional studies are planned including: measuring hormone concentrations in tandem with these experiments, analyzing downstream signaling including MAPK and Akt pathways, and evaluating the role of reactive oxygen species including superoxide dismutase.
In conclusion, ER signaling is likely involved in regulating sex-based differences in VSMC proliferation as well as the response of NO. Ultimately our goal is to develop a therapy that will prevent restenosis following vascular interventions, and to be able to translate this therapy to all patient populations. Thus exploring specific challenges to all patient populations on a cellular level is important to product development. Overall, NO-based therapies have promising clinical potential for an ever-growing population of patients with vascular disease, but more specific evaluation into mechanisms and efficacy of the therapy based on sex, hormone, and ER status are warranted.
Acknowledgments
This work was supported in part by funding from the Institute for Women's Health Research at the Feinberg School of Medicine, Northwestern University, Chicago, Illinois through the Pioneer Award competitive funding mechanism. This work was also supported in part by funding from the National Institutes of Health (K08HL084203 - MRK); Department of Veterans Affairs Merit Review award (MRK); the Society for Vascular Surgery Foundation (MRK), and an American Medical Association Seed Grant (MEH). Lastly, this work was supported in part by funding from the National Cancer Institute, NIH, under contract NO1-CO-12400 with SAIC-Frederick Inc. and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The authors would also like to express their thanks to the Institute for BioNanotechnology in Medicine and Lynnette Dangerfield for her “tireless” hours.
Footnotes
Presented at the 5th Annual Academic Surgical Congress, San Antonio, TX
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lloyd-Jones D, Adams R, Carnethon M, De SG, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O'Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y. Circulation. 2008. Heart Disease and Stroke Statistics--2009 Update. A Report From the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. [DOI] [PubMed] [Google Scholar]
- 2.Allaire E, Clowes AW. Endothelial cell injury in cardiovascular surgery: The intimal hyperplastic response. Annals of Thoracic Surgery. 1997;63:582–591. doi: 10.1016/s0003-4975(96)01045-4. [DOI] [PubMed] [Google Scholar]
- 3.Clowes AW, Reidy MA, Clowes MM. Mechanisms of stenosis after arterial injury. Lab Invest. 1983;49:208–215. [PubMed] [Google Scholar]
- 4.Clowes AW. Intimal Hyperplasia and Graft Failure. Cardiovasc Pathol Cardiovasc Pathol. 1993;2:S179–S186. [Google Scholar]
- 5.Kibbe MR, Tzeng E, Gleixner SL, Watkins SC, Kovesdi I, Lizonova A, Makaroun MS, Billiar TR, Rhee RY. Adenovirus-mediated gene transfer of human inducible nitric oxide synthase in porcine vein grafts inhibits intimal hyperplasia. J Vasc Surg. 2001;34:156–165. doi: 10.1067/mva.2001.113983. [DOI] [PubMed] [Google Scholar]
- 6.Shears LL, Kibbe MR, Murdock AD, Billiar TR, Lizonova A, Kovesdi I, Watkins SC, Tzeng E. Efficient inhibition of intimal hyperplasia by adenovirus-mediated inducible nitric oxide synthase gene transfer to rats and pigs in vivo. J Am Coll Surg. 1998;187:295–306. doi: 10.1016/s1072-7515(98)00163-x. [DOI] [PubMed] [Google Scholar]
- 7.von der Leyen HE, Gibbons GH, Morishita R, Lewis NP, Zhang L, Nakajima M, Kaneda Y, Cooke JP, Dzau VJ. Gene-Therapy Inhibiting Neointimal Vascular Lesion - In-Vivo Transfer of Endothelial-Cell Nitric-Oxide Synthase Gene. P Natl Acad Sci USA P Natl Acad Sci USA. 1995;92:1137–1141. doi: 10.1073/pnas.92.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies MG, Kim JH, Dalen H, Makhoul RG, Svendsen E, Hagen PO. Reduction of experimental vein graft intimal hyperplasia and preservation of nitric oxide-mediated relaxation by the nitric oxide precursor L-arginine. Surgery. 1994;116:557–568. [PubMed] [Google Scholar]
- 9.Marks DS, Vita JA, Folts JD, Keaney JFJ, Welch GN, Loscalzo J. Inhibition of neointimal proliferation in rabbits after vascular injury by a single treatment with a protein adduct of nitric oxide. J Clin Invest. 1995;96:2630–2638. doi: 10.1172/JCI118328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaul S, Cercek B, Rengstrom J, Xu XP, Molloy MD, Dimayuga P, Parikh AK, Fishbein MC, Nilsson J, Rajavashisth TB, Shah PK. Polymeric-based perivascular delivery of a nitric oxide donor inhibits intimal thickening after balloon denudation arterial injury: role of nuclear factor-kappaB. J Am Coll Cardiol. 2000;35:493–501. doi: 10.1016/s0735-1097(99)00543-4. [DOI] [PubMed] [Google Scholar]
- 11.Chaux A, Ruan XM, Fishbein MC, Ouyang Y, Kaul S, Pass JA, Matloff JM. Perivascular delivery of a nitric oxide donor inhibits neointimal hyperplasia in vein grafts implanted in the arterial circulation. J Thorac Cardiovasc Surg. 1998;115:604–612. doi: 10.1016/S0022-5223(98)70325-3. [DOI] [PubMed] [Google Scholar]
- 12.Kown MH, Yamaguchi A, Jahncke CL, Miniati D, Murata S, Grunenfelder J, Koransky ML, Rothbard JB, Robbins RC. L-arginine polymers inhibit the development of vein graft neointimal hyperplasia. J Thorac Cardiovasc Surg. 2001;121:971–980. doi: 10.1067/mtc.2001.112532. [DOI] [PubMed] [Google Scholar]
- 13.Fleser PS, Nuthakki VK, Malinzak LE, Callahan RE, Seymour ML, Reynolds MM, Merz SI, Meyerhoff ME, Bendick PJ, Zelenock GB, Shanley CJ. Nitric oxide-releasing biopolymers inhibit thrombus formation in a sheep model of arteriovenous bridge grafts. J Vasc Surg. 2004;40:803–811. doi: 10.1016/j.jvs.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Masters KS, Lipke EA, Rice EE, Liel MS, Myler HA, Zygourakis C, Tulis DA, West JL. Nitric oxide-generating hydrogels inhibit neointima formation. J Biomater Sci Polym Ed. 2005;16:659–672. doi: 10.1163/1568562053783722. [DOI] [PubMed] [Google Scholar]
- 15.Lee JS, Adrie C, Jacob HJ, Roberts JDJ, Zapol WM, Bloch KD. Chronic inhalation of nitric oxide inhibits neointimal formation after balloon-induced arterial injury. Circ Res. 1996;78:337–342. doi: 10.1161/01.res.78.2.337. [DOI] [PubMed] [Google Scholar]
- 16.Davies MG, Dalen H, Kim JH, Barber L, Svendsen E, Hagen PO. Control of accelerated vein graft atheroma with the nitric oxide precursor: L-arginine. J Surg Res. 1995;59:35–42. doi: 10.1006/jsre.1995.1129. [DOI] [PubMed] [Google Scholar]
- 17.Hogg ME, Varu VN, Vavra AK, Popowich DA, Banerjee MN, Martinez J, Jiang Q, Saavedra JE, Keefer LK, Kibbe MR. Effect of nitric oxide on neointimal hyperplasia based on sex and hormone status. Free Radic Biol Med. 2011;50:1065–1074. doi: 10.1016/j.freeradbiomed.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu SM, Hung LM, Lin CC. cGMP-elevating agents suppress proliferation of vascular smooth muscle cells by inhibiting the activation of epidermal growth factor signaling pathway [see comments] Circulation. 1997;95:1269–1277. doi: 10.1161/01.cir.95.5.1269. [DOI] [PubMed] [Google Scholar]
- 19.Kibbe MR, Li J, Nie S, Watkins SC, Lizonova A, Kovesdi I, Simmons RL, Billiar TR, Tzeng E. Inducible nitric oxide synthase (iNOS) expression upregulates p21 and inhibits vascular smooth muscle cell proliferation through p42/44 mitogen-activated protein kinase activation and independent of p53 and cyclic guanosine monophosphate [In Process Citation] J Vasc Surg. 2000;31:1214–1228. doi: 10.1067/mva.2000.105006. [DOI] [PubMed] [Google Scholar]
- 20.Gunther S, Alexander RW, Atkinson WJ, Gimbrone MA. Functional angiotensin-Ii receptors in cultured vascular smooth-muscle cells. J Cell Biol. 1982;92:289–298. doi: 10.1083/jcb.92.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors alpha (ER alpha) and beta (ER beta) on mouse reproductive phenotypes. Development. 2000;127:4277–4291. doi: 10.1242/dev.127.19.4277. [DOI] [PubMed] [Google Scholar]
- 22.Mendelsohn ME. Genomic and nongenomic effects of estrogen in the vasculature. Am J Cardiol. 2002;90:3F–6F. doi: 10.1016/s0002-9149(02)02418-9. [DOI] [PubMed] [Google Scholar]
- 23.Mendelsohn ME. Protective effects of estrogen on the cardiovascular system. Am J Cardiol. 2002;%20(89):12E–17E. doi: 10.1016/s0002-9149(02)02405-0. [DOI] [PubMed] [Google Scholar]
- 24.Ahanchi SS, Tsihlis ND, Kibbe MR. The role of nitric oxide in the pathophysiology of intimal hyperplasia. J Vasc Surg. 2007;45 A:A64–73. A64–A73. doi: 10.1016/j.jvs.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 25.Lindner V, Kim SK, Karas RH, Kuiper GGJM, Gustafsson JA, Mendelsohn ME. Increased expression of estrogen receptor-beta mRNA in male blood vessels after vascular injury. Circulation Research. 1998;83:224–229. doi: 10.1161/01.res.83.2.224. [DOI] [PubMed] [Google Scholar]
- 26.Karas RH, Patterson BL, Mendelsohn ME. Human Vascular Smooth-Muscle Cells Contain Functional Estrogen-Receptor. Circulation. 1994;89:1943–1950. doi: 10.1161/01.cir.89.5.1943. [DOI] [PubMed] [Google Scholar]
- 27.Karas RH, Baur WE, Mendelsohn ME. Human Vascular Smooth-Muscle Cells Express A Novel Estrogen-Receptor Isoform. Circulation. 1994;90:462. [Google Scholar]
- 28.Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, Mendelsohn ME. Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circulation Research. 2002;90:1087–1092. doi: 10.1161/01.res.0000021114.92282.fa. [DOI] [PubMed] [Google Scholar]
- 29.Iafrati MD, Karas RH, Aronovitz M, Kim S, Sullivan TR, Lubahn DB, Odonnell TF, Korach KS, Mendelsohn ME. Estrogen inhibits the vascular injury response in estrogen receptor alpha-deficient mice. Nature Medicine. 1997;3:545–548. doi: 10.1038/nm0597-545. [DOI] [PubMed] [Google Scholar]
- 30.Karas RH, Hodgins J, Kwoun M, Krege J, Aronovitz M, Mackey W, Gustafsson JA, Korach K, Smithies O. Estrogen inhibits the vascular injury response in estrogen receptor beta-deficient mice. Circulation. 1999;100:703. doi: 10.1073/pnas.96.26.15133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karas RH, Schulten H, Pare G, Aronovitz MJ, Ohlsson C, Gustafsson JA, Mendelsohn ME. Effects of estrogen on the vascular injury response in estrogen receptor alpha,beta (double) knockout mice. Circulation Research. 2001;89:534–539. doi: 10.1161/hh1801.097239. [DOI] [PubMed] [Google Scholar]
- 32.Krom YD, Pires NMM, Jukema JW, de Vries MR, Frants RR, Havekes LM, van Dijk KW, Quax PHA. Inhibition of neointima formation by local delivery of estrogen receptor alpha and beta specific agonists. Cardiovascular Research. 2007;73:217–226. doi: 10.1016/j.cardiores.2006.10.024. [DOI] [PubMed] [Google Scholar]
- 33.Bakir S, Mori T, Durand J, Chen YF, Thompson JA, Oparil S. Estrogen-induced vasoprotection is estrogen receptor dependent - Evidence from the balloon-injured rat carotid artery model. Circulation. 2000;101:2342–2344. doi: 10.1161/01.cir.101.20.2342. [DOI] [PubMed] [Google Scholar]
- 34.Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of Reproductive Function But Not Prenatal Sexual Development After Insertional Disruption of the Mouse Estrogen-Receptor Gene. P Natl Acad Sci USA P Natl Acad Sci USA. 1993;90:11162–11166. doi: 10.1073/pnas.90.23.11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun D, Yan CD, Jacobson A, Jiang H, Carroll MA, Huang A. Contribution of epoxyeicosatrienoic acids to flow-induced dilation in arteries of male ER alpha knockout mice: role of aromatase. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2007;293:R1239–R1246. doi: 10.1152/ajpregu.00185.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sasano H, Murakami H, Shizawa S, Satomi S, Nagura H, Harada N. Aromatase and sex steroid receptors in human vena cava. Endocrine Journal. 1999;46:233–242. doi: 10.1507/endocrj.46.233. [DOI] [PubMed] [Google Scholar]
- 37.Nathan L, Shi WB, Dinh H, Mukherjee TK, Wang XP, Lusis AJ, Chaudhuri G. Testosterone inhibits early atherogenesis by conversion to estradiol: Critical role of aromatase. P Natl Acad Sci USA P Natl Acad Sci USA. 2001;98:3589–3593. doi: 10.1073/pnas.051003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diano S, Horvath TL, Mor G, Register T, Adams M, Harada N, Naftolin F. Aromatase and estrogen receptor immunoreactivity in the coronary arteries of monkeys and human subjects. Menopause-the Journal of the North American Menopause Society. 1999;6:21–28. [PubMed] [Google Scholar]
- 39.Bayard F, Clamens S, Meggetto F, Blaes N, Delsol G, Faye JC. Estrogen Synthesis, Estrogen Metabolism, and Functional Estrogen-Receptors in Rat Arterial Smooth-Muscle Cells in Culture. Endocrinology. 1995;136:1523–1529. doi: 10.1210/endo.136.4.7895662. [DOI] [PubMed] [Google Scholar]
- 40.Chen SJ, Li HB, Durand J, Oparil S, Chen YF. Estrogen reduces myointimal proliferation after balloon injury of rat carotid artery. Circulation. 1996;93:577–584. doi: 10.1161/01.cir.93.3.577. [DOI] [PubMed] [Google Scholar]
- 41.Bowles DK, Ivey J, Casati J, Turk JR. Testosternone attenuates neointimal formation after coronary balloon angioplasty in male swine. The FASEB Journal. 2007;21:A18. [Google Scholar]
- 42.Dorsett-Martin WA, Hester RL. Sex hormones and aortic wall remodeling in an arteriovenous fistula. Gend Med. 2007;4:157–169. doi: 10.1016/s1550-8579(07)80029-5. [DOI] [PubMed] [Google Scholar]