

Abstract
Glioblastoma multiforme (GBM) is a commonly occurring brain tumor with a poor prognosis. GBM can develop both “de novo” or evolve from a previous astrocytoma and is characterized by high proliferation and infiltration into the surrounding tissue. Following treatment (surgery, radiotherapy, and chemotherapy), tumors often reappear. Glioma-initiating cells (GICs) have been identified in GBM and are thought to be responsible for tumors initiation, their continued growth, and recurrence. β-catenin, a component of the cell-cell adhesion complex and of the canonical Wnt pathway, regulates proliferation, adhesion, and migration in different cell types. β-catenin and components of the Wnt canonical pathway are commonly overexpressed in GBM. Here, we review previous work on the role of Wnt/β-catenin signalling in glioma initiation, proliferation, and invasion. Understanding the molecular mechanisms regulating GIC biology and glioma progression may help in identifying novel therapeutic targets for GBM treatment.
1. Introduction
Gliomas are the most common primary malignancies in the central nervous system, comprising a heterogeneous group of tumors that display some histological similarities to glia (mainly, astrocytes in astrocytomas and oligodendrocytes in oligodendrogliomas). Astrocytomas account for the majority of gliomas, which can be classified into four different grades according to the World Health Organization (WHO) classification system [1, 2]: grade I and grade II astrocytomas are slow-growing less aggressive tumors, whereas grade III and IV gliomas are malignant tumors characterized by high proliferation rate (grade III) and the presence of necrotic tissue and/or angiogenic activity (grade IV). The most malignant form, glioblastoma multiforme (GBM, grade IV), is one of the most aggressive and lethal forms of cancer with an average survival time of 15 months after diagnosis [3, 4]. Standard treatment consists of surgical removal of the tumor, followed by chemotherapy and radiotherapy. Temozolomide, an oral alkylating agent, is the most commonly used chemotherapy treatment [5]. Importantly, a high infiltration capacity of individual cells over long distances, already present in grade II gliomas, hinders complete tumor resection and most likely contributes to recurrence [6].
GBMs can present as primary or secondary. Primary or “de novo” GBMs, representing the majority of GBM cases, arise without any prior evidence of a lower-grade precursor lesion and more commonly affect older patients (mean age of 62 years). Secondary GBMs progress from a lower-grade glioma and typically develop in younger patients (median age of 45 years). Gliomas exhibit a vast array of genetic changes that contribute to the malignant phenotype [6–10]. These include loss of function mutations in the p53 tumor suppressor and hyperactivation of receptor tyrosine kinase (RTK) signalling, such as epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), and the receptor for hepatocyte growth factor c-Met. The signalling cascades downstream of activated RTK often result in activation of Ras and AKT. Mutations in tumor suppressors such as phosphatase and tensin homolog (PTEN) and neurofibromatosis 1 (NF1) that normally control these pathways further contribute to oncogenesis [2, 3, 6–10]. Based on its gene expression profile, GBM can be further classified into proneural, neural, mesenchymal, and classical types [8, 9]. This classification should allow a molecular stratification of GBM cases with important therapeutic implications.
Here we focus our attention on Wnt/β-catenin signalling, a pathway primarily involved in embryogenesis and displaying important functions in adulthood when aberrant Wnt signalling has been linked to disease and cancer [11–14]. Understanding how β-catenin signalling regulates gliomagenesis and tumor progression may lead to novel therapeutic interventions in GBM. Firstly, therefore, we discuss the role of glioma-initiating cells (GICs), a type of “cancer stem cells,” in glioma development and evidence suggesting that Wnt/β-catenin signalling regulates GICs biology. We then review progress in the understanding of the involvement of Wnt/β-catenin signalling in the proliferation and invasion of glioma tumor cells.
2. Glioma-Initiating Cells (GICs) and Signalling Pathways in GICs
The existence of brain tumor stem cells was proposed about a decade ago, following advances in the stem cell field and the discovery that neurogenesis persists in the adult brain [15–18]. Glioma-initiating cells (GICs) share the features of neural stem cells that have been identified in GBM, including the expression of CD133 (prominin), the ability to form neurospheres, and the reproduction of tumors [19, 20]. However, the role of CD133+ and CD133− GIC subpopulations in tumor initiation is not clear. CD133− cells from the C6 glioma cell line showed clonogenic, self-renewal, and tumorigenic capacities [21]. Nonetheless, CD133− GICs isolated from primary GBM were as capable of producing tumors as CD133+ cells [22]. Individual GBM may contain CD133+ and CD133− GICs that represent different stages of differentiation [23]. Furthermore, multipotent CD133+ GICs contain a CD144+ (vascular endothelial cadherin positive) subpopulation that can give rise to tumor endothelial cells [24].
Signalling by several morphogens and cytokines (including leukemia inhibitory factor (LIF), fibroblast growth factor (FGF) and members of the Wnt, transforming growth factor-β (TGF-β)/bone morphogenetic proteins (BMP) families) maintains the self-renewal capacity of embryonic stem cells and supports cancer stem cell growth [25]. Thus, TGF-β signalling through the induction of LIF and the JAK-STAT pathway promotes the self-renewal of patient-derived GICs [26]. Seoane and coworkers recently identified a population of CD44 high/Id1 high GICs in GBM that locates in the perivascular niche. Depletion of this cell population by TGF-β inhibitors prevents tumor initiation and recurrence. This work identifies CD44 and Id1 levels as prognosis markers in GBM and shows that TGF-β signalling is key to maintain this GIC population [27]. Aberrant activation of sonic hedgehog signaling (another morphogen involved in embryogenesis and brain development) in committed cerebellar granule neuron precursors is responsible for aggressive medulloblastoma, a pediatric cerebellar tumor [28–30]. Hedgehogs signal through Gli transcription factors, with Gli1 and Gli2 acting as activators and Gli3 as a repressor factor. Consistent with the isolation of Gli1 from glioma cells, activation of the Hedgehog-Gli1 pathway is reported in GBM, which is required for the clonogenicity and formation of secondary neurospheres of CD133+ GICs [31].
Wnt factors are a family of secreted glycoproteins (19 members exist in humans) that regulate embryonic patterning and play different roles throughout development of the nervous system [32, 33]. Wnts signal through at least three different pathways [11, 33], the best known being the Wnt/β-catenin canonical pathway (Figure 1). In the absence of Wnt, the Ser/Thr kinase glycogen-synthase kinase (Gsk-3β) in the so-called destruction complex (comprising of Gsk-3β, adenomatosis polyposis coli (APC), Axin and β-catenin) phosphorylates β-catenin, which is then targeted for proteasomal degradation. Upon Wnt binding to Frizzled (Fz) (of which there are 11 family members in humans) and low-density-lipoprotein-related protein (LRP)5/6 receptors, the scaffolding protein Dishevelled (Dvl) and LRP5/6 become phosphorylated by Gsk-3 and Casein-Kinase Iγ. Consequently, the destruction complex components are recruited instead to the receptor complex, leading to β-catenin stabilization [34]. The protooncogene Frat/GBP further prevents the phosphorylation and degradation of β-catenin because it competes with Axin to bind Gsk-3 and removes it from the destruction complex. Stabilized β-catenin translocates to the nucleus, where it binds to lymphoid enhancer factor-1 Lef-1/T-cell factor (Tcf) transcription factors and regulates expression of Wnt target genes. In the absence of nuclear β-catenin, Tcf/Lef factors suppress the expression of target genes through their binding to members of the Groucho/transducin-like enhancer of split (TLE) family of transcriptional corepressors. β-catenin does not have a DNA binding domain but it has a potent transcription activation domain. Conversely, Lef/Tcf transcription factors do not have a strong transcription activation domain, but they do have a good DNA binding/bending domain [35]. Thus, when β-catenin binds to a Lef/Tcf protein, a potent transcription regulatory complex is formed. Nuclear translocation of β-catenin converts Tcf proteins into potent transcriptional activators by displacing Groucho/TLE and recruiting an array of coactivator proteins, including CBP, TBP, BRG1, BCL9/PYGO, Legless, Mediator, and Hyrax [36] (Figure 1).
Figure 1.
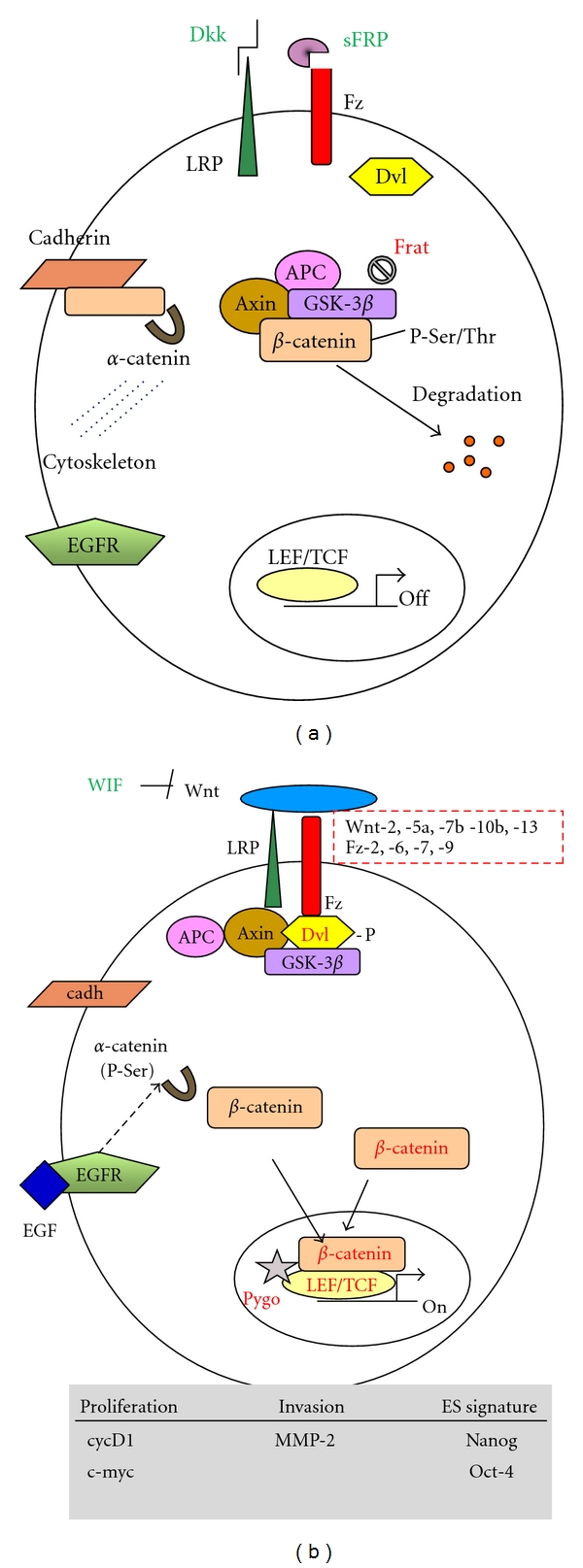
Wnt/β-catenin signalling and β-catenin role in adhesion in glioma cells. A) In the absence of Wnt ligands or in the presence of Wnt antagonists Dkk and sFRP that bind to the respective Wnt receptors Fz and LRP5/6, β-catenin is in a complex together with Axin, APC, and Gsk-3β. Here, β-catenin is phosphorylated by Gsk-3β in key Ser and Thr residues and is thus targeted for proteasomal degradation. Frat prevents the phosphorylation of β-catenin. Transcription by Lef/Tcf is off due to the binding of repressors. In the absence of growth factor signaling, a pool of β-catenin is engaged in the cadherin/β-catenin/α-catenin complex that is linked to the cytoskeleton. B) Following Wnt binding, the Fz-LRP5/6 complex is formed upon Dvl phosphorylation that recruits Gsk-3β, Axin, and APC to the membrane. This results in free β-catenin that accumulates in the cytosol and translocates to the nucleus, where it binds to Tcf and recruits transcriptional activators (including Pygo). Lef/Tcf transcriptional activation results in the regulation of Wnt target genes. The box shows Wnt target genes implicated in proliferation and invasion of glioma cells or conferring ES cell signature to GICs that might be related to aggressive growth and recurrence [37–39] EGF signalling through EGFR, ERK1/2, and CK2 results in the phosphorylation of α-catenin and promotes β-catenin transactivation [40]. Whether the Wnt-induced and growth factor-induced β-catenin nuclear pools collaborate in glioma cells remains to be studied. Text in red indicates Wnt pathway components that are overexpressed and green indicates Wnt antagonists repressed in high-grade astrocytomas and GBM. Wnt factors and Fz that have been reported to be upregulated in high-grade astrocytomas and GBM are shown (see references in the text).
Canonical Wnt/β-catenin signalling is crucially involved in embryonic development and controls stem cell biology, thus inducing self-renewal properties in embryonic stem (ES) cells and regulating adult stem cells [11, 14, 33, 41–45]. Nanog and Oct-4, two of the four transcription factors required to generate the pluripotency and self-renewal of ES cells, are Tcf3 targets [46, 47]. Interestingly, the ES signature characterized by the expression of Nanog, Oct-4, Sox-2, and c-Myc also associates with aggressive tumors, including poorly differentiated GBM [37]. As aberrant activation of the Wnt/β-catenin signalling pathway is a hallmark of many tumors [12, 14], these findings suggest that Wnt-regulated genes may contribute to the stem cell-like phenotypes displayed by brain tumors. Furthermore, the novel protooncogene PLAG2 is amplified in GBM and promotes GICs proliferation and gliomagenesis. PLAG2 increases the expression of Wnt-6, Fz-9, and Fz-2, inhibits differentiation, and increases proliferation of neural progenitors [48]. It is worth noting that PLAG2 amplification correlates with increased β-catenin levels in GBM samples. These results indicate that PLAG2 imparts stem-cell properties to glioma cells by regulating Wnt signalling. Another gene regulating Wnt signalling in glioma is PEG3 (paternally expressed gene 3), an imprinted gene with a tumor suppressor activity. Hypermethylation of PEG3 promoter in glioma decreases PEG3 mRNA expression and correlates with high-grade gliomas [49]. In turn, low PEG3 expression increases β-catenin that promotes the proliferation of CD133+ GICs [49]. Finally, the interaction between the transcription factor Forkhead box M1 and β-catenin that promotes β-catenin nuclear localization in tumor cells and maintains GIC self-renewal has been recently described [50]. Novel therapeutic interventions for GBM could inhibit Wnt/β-catenin signalling in GICs to decrease GIC proliferation and stop glioma growth, while increasing GICs differentiation.
3. Targeting GIC Chemoresistance and Radioresistance Mechanisms as an Approach to Treat GBM Recurrence
Following surgery, chemotherapy, and radiotherapy GBM recurrence is common. Therefore, a particularly relevant feature of the cancer stem cells is their ability to export drugs and develop resistance mechanisms to cytotoxics and irradiation [17]. Current knowledge suggests that resistance to temozolomide is promoted by enhanced O-6-methylguanine-DNA-methyltransferase- (MGMT-) mediated DNA repair of mismatches [51]. Thus, MGMT promoter methylation status improves the benefits of chemotherapy. According to Liu eta l., CD133 expression in tumor tissue is higher in recurrent GBM than in newly diagnosed tumors and CD133+ GICs are chemoresistant to temozolomide [52]. However, these findings are inconsistent with a report indicating that CD133+ GICs are depleted after temozolomide treatment [53]. Consequently, at present it is not clear which glioma cells are responsible for the resistance to temozolomide.
Another mechanism of drug resistance is the expression of the ATP-binding cassette (ABC) transporters by cancer stem cells. BCRP1 and MDR1 ABC transporters allow the exclusion of Hoechst 33342 dye, a feature that defines the pluripotential side population (SP) originally reported in haematopoietic stem cells and now used to identify cancer stem cells. Expression of these ABC transporters accounts for the chemoresistance of some cancers and high drug efflux capacity. CD133+ GICs express higher levels of BCRP1 compared to CD133− cells [52]. Consistent with this, a cancer stem-cells cell line (WJ2) derived from GBM showed increased expression of BCRP1, CD133 and the neural precursor marker Nestin and at the same time maintained Wnt-1 expression [54]. Interestingly, overexpression of MDR1 downstream of Wnt-1/Fz-1 signalling mediates chemoresistance in neuroblastoma cells [55]. This suggests that a similar mechanism could be operating in the chemoresistance mechanisms of gliomas.
As regards radioresistance, the CD133+ stem cell fraction is enriched after glioma radiation [56]. Furthermore, the CD133+ subpopulation is able to repair radiation-induced DNA damage more efficiently than CD133− tumor cells [57]. These results indicate that the CD133+ tumor cell population confers radioresistance to GBM and most likely accounts for glioma recurrence. Wnt-1 ectopic expression triggers DNA damage response in epithelial mammary cells [58], while activation of Wnt/β-catenin signalling confers radioresistance to mammary progenitors cells through survivin upregulation [59]. In addition, Gsk-3β inhibition enhances DNA repair of double-strand breaks following radiation of hippocampal neurons [60]. Taken together, these findings suggest that Wnt signalling may be involved in the chemo- and radioresistance mechanisms developed by GICs. Expanding our understanding of the molecular mechanisms supporting GICs resistance to conventional glioma treatment will allow the design of novel therapeutic tools to decrease tumor recurrence and improve patient survival.
4. Wnt/β-Catenin Signalling in the Proliferation of Glioma Cells
GICs represent a small percentage of the brain tumor mass, which is thought to contain a heterogeneous mixture of tumor cells with limited proliferation capacity. Molecular analysis on whole tumor samples is expected to mainly represent non-GIC cells. Wnt/β-catenin signalling plays a role in the proliferation of glioma tumor cells and tumor progression. β-catenin has been proposed as a prognostic marker in GBM, as both mRNA and protein levels increase in high-grade astrocytomas and GBM, thus correlating with malignancy [38, 61, 62]. In addition, the expression of other positive regulators of the Wnt pathway (including Dvl-3, FRAT-1, Pygo-2, Tcf4, and Lef-1) [38, 63, 64] and of Wnt target genes (namely the regulators of cell proliferation Cyclin D1 and c-myc) [38, 62] also increases in high-grade astrocytomas and GBM (see Figure 1). Using immunohistological techniques, a nuclear fraction of β-catenin was observed that associates with high-grade astrocytoma and GBM [62]. This result suggests increased cytoplasmic stabilization of β-catenin that escapes proteasomal degradation, in addition to the elevated β-catenin mRNA levels reported in GBM. Silencing β-catenin, Wnt-2, and Pygo-2 expression demonstrated the involvement of Wnt/β-catenin signalling in the proliferation of U251 glioma cell line [63, 65]. Together, these findings point to the activation of nuclear β-catenin signalling as a mediator of Wnt-induced proliferation of glioma cells. Moreover, expression of noncanonical Wnt-5a is also upregulated in high grade gliomas, in which Wnt-5a stimulates cell proliferation [66]. Wnt-5a signalling can inhibit canonical Wnt signalling during development [67, 68]. How Wnt canonical and noncanonical pathways interact in glioma cells remains to be studied.
In contrast to other cancers, no mutations have been found in β-catenin exon 3, a hot spot affecting the GSK-3 phosphorylation sites and β-catenin degradation that renders β-catenin active in glioma samples and cell lines [69, 70]. Truncation of APC, a mechanism causing polyposis and predisposing for Wnt/β-catenin-driven colorectal carcinoma, has not been associated with gliomagenesis (with the exception of Turcot syndrome patients) [71]. These observations suggest the deregulation of the pathway by unbalanced ligand/antagonist expression during tumor initiation and progression. Indeed, in addition to regulation of the expression of Wnt family members, Wnt antagonists often appear repressed in GBM (Figure 1). Expression of the Wnt antagonist and tumor suppressor Wnt inhibitory factor (WIF) decreases with malignancy in astrocytomas, which has been linked to aberrant promoter hypermethylation [72]. Also, hypermethylation of the secreted-Frizzled-related protein (sFRP) promoters is a significant event in primary “de novo” GBM, whereas hypermethylation of the promoter of the LRP antagonist Dickkopf (Dkk) associates with secondary GBM [70]. Similar epigenetic modifications are common to other Wnt-driven cancers [73, 74]. In addition, a novel mechanism for β-catenin nuclear localization and transcriptional activation (both constitutive and Wnt-induced) that controls Wnt target gene expression and glioma tumorigenesis has been described, which involves the interaction of β-catenin with FoxM1 [50].
5. β-Catenin and Wnt Signalling in Glioma Invasion
As a component of the cell adhesion complex, β-catenin binds to cadherin, thus regulating cell-cell adhesion. Altering the binding of β-catenin to cadherin or to α-catenin downregulates cell adhesion, while promoting cell migration and epithelial-mesenchymal transition [75]. However, β-catenin nuclear signalling is not only achieved by Wnt factors in tumor development [76]. Growth factor signalling can induce the phosphorylation of specific tyrosine residues of β-catenin, resulting in increased migration [75, 77–79]. EGFR expression is upregulated in primary GBM correlating with malignancy [15]. EGF/EGFR signalling through extracellular signal-regulated kinases 1/2 (ERK1/2) and casein kinase-2 (CK2) in glioma cells results in the phosphorylation of α-catenin at serine 641, which correlates with glioma malignancy [40]. Interestingly, α-catenin phosphorylation promotes β-catenin transactivation and glioma cell invasion [40]. These results highlight the involvement of β-catenin signalling not only as a mediator of Wnt but also downstream of growth factor signalling in glioma invasion. On the other hand, enhanced expression of the Fz antagonist sFRP2 reduced glioma invasion by decreasing β-catenin tyrosine phosphorylation and downregulating matrix metalloprotease-2 (MMP-2) [39]. However, sFRP2 did not affect β-catenin levels, its cytoplasmic/nuclear distribution, or its serine phosphorylation status [39]. How sFRP2 signalling modulates β-catenin tyrosine phosphorylation requires further investigation.
Noncanonical Wnt-5a, which signals through β-catenin independent pathways (including the planar cell polarity and the calcium pathways [33]), enhances the migration of glioma cells by regulating the expression of MMP-2 [80]. Moreover, silencing the expression of Wnt-2, Wnt-5a, and Fz-2 in the U251 glioma cell line decreases invasion [65, 80]. These findings are consistent with Wnt-5a function in invasion and metastasis in other cancers [11, 81, 82]. Together, these results point to metalloprotease regulation as important downstream targets of β-catenin and Wnt signalling pathways in glioma invasion [39, 80, 83].
We are accumulating knowledge on the signalling pathways responsible for the maintenance of GICs, sustaining the proliferation of bulk tumor cells and dictating the invasive properties of glioma cells. Current and future research should offer novel opportunities for anticancer drug discovery. Undoubtedly, the cancer stem cell hypothesis has provided a promising framework for investigations into an incurable disease. Future combined therapies including cytotoxics, tumor-targeted drugs, and agents that target GICs should be expected to reduce glioma growth and recurrence, raising hopes for glioma patients.
Acknowledgments
Work in the authors laboratory is supported by funding from Instituto de Salud Carlos III (ISCIII) to L. Medina, J. Herreros and Grups de Recerca Consolidats (Agaur-Generalitat de Catalunya) to L. Medina, M. Nager is funded by ISCIII. D. Bhardawaj is a predoctoral fellow of the Generalitat de Catalunya. The authors thank John Eastham and Theresa Higgins for proofreading the paper.
References
- 1.Kleihues P, Louis DN, Scheithauer BW, et al. The WHO classification of tumors of the nervous system. Journal of Neuropathology and Experimental Neurology. 2002;61(3):215–225. doi: 10.1093/jnen/61.3.215. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN. Molecular pathology of malignant gliomas. Annual Review of Pathology. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 3.Zhu Y, Parada LF. The molecular and genetic basis of neurological tumours. Nature Reviews Cancer. 2002;2(8):616–626. doi: 10.1038/nrc866. [DOI] [PubMed] [Google Scholar]
- 4.Maher EA, Furnari FB, Bachoo RM, et al. Malignant glioma: genetics and biology of a grave matter. Genes and Development. 2001;15(11):1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 5.Yung WKA. Future directions for temozolomide therapy. Seminars in Oncology. 2001;28(4):43–46. doi: 10.1016/s0093-7754(01)90070-3. [DOI] [PubMed] [Google Scholar]
- 6.Holland EC. Glioblastoma multiforme: the terminator. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(12):6242–6244. doi: 10.1073/pnas.97.12.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim SK, Llaguno SRA, McKay RM, Parada LF. Glioblastoma multiforme: a perspective on recent findings in human cancer and mouse models. Biochemistry and Molecular Biology Reports. 2011;44(3):158–164. doi: 10.5483/BMBRep.2011.44.3.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brennan C, Momota H, Hambardzumyan D, et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE. 2009;4(11) doi: 10.1371/journal.pone.0007752. Article ID e7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.TCGA Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moon RT, Kohn AD, de Ferrari GV, Kaykas A. WNT and β-catenin signalling: diseases and therapies. Nature Reviews Genetics. 2004;5(9):691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 12.Polakis P. Wnt signaling and cancer. Genes and Development. 2000;14(15):1837–1851. [PubMed] [Google Scholar]
- 13.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nature Reviews Cancer. 2008;8(5):387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 14.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 15.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nature Reviews Cancer. 2006;6(6):425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 16.Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. GLIA. 2002;39(3):193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 17.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nature Reviews Cancer. 2008;8(10):755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 18.Lie DC, Song H, Colamarino SA, Ming GL, Gage FH. Neurogenesis in the adult brain: new strategies for central nervous system diseases. Annual Review of Pharmacology and Toxicology. 2004;44:399–421. doi: 10.1146/annurev.pharmtox.44.101802.121631. [DOI] [PubMed] [Google Scholar]
- 19.Singh SK, Clarke ID, Hide T, Dirks PB. Cancer stem cells in nervous system tumors. Oncogene. 2004;23(43):7267–7273. doi: 10.1038/sj.onc.1207946. [DOI] [PubMed] [Google Scholar]
- 20.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Research. 2003;63(18):5821–5828. [PubMed] [Google Scholar]
- 21.Beier D, Hau P, Proescholdt M, et al. CD133+ and CD133- glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Research. 2007;67(9):4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 22.Sakariassen PO, Prestegarden L, Wang J, et al. Angiogenesis-independent tumor growth mediated by stem-like cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(44):16466–16471. doi: 10.1073/pnas.0607668103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen R, Nishimura MC, Bumbaca SM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17(4):362–375. doi: 10.1016/j.ccr.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 24.Wang R, Chadalavada K, Wilshire J, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468(7325):829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- 25.Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nature Reviews Clinical Oncology. 2011;8(2):97–106. doi: 10.1038/nrclinonc.2010.196. [DOI] [PubMed] [Google Scholar]
- 26.Bruna A, Darken RS, Rojo F, et al. High TGFβ-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11(2):147–160. doi: 10.1016/j.ccr.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 27.Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, et al. TGF-β receptor inhibitors target the CD44high/Id1high glioma-initiating cell population in human glioblastoma. Cancer Cell. 2010;18(6):655–668. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 28.Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277(5329):1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 29.Schüller U, Heine VM, Mao J, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form shh-induced medulloblastoma. Cancer Cell. 2008;14(2):123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang ZJ, Ellis T, Markant SL, et al. Medulloblastoma can be initiated by deletion of patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14(2):135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Current Biology. 2007;17(2):165–172. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annual Review of Cell and Developmental Biology. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 33.Ciani L, Salinas PC. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nature Reviews Neuroscience. 2005;6(5):351–362. doi: 10.1038/nrn1665. [DOI] [PubMed] [Google Scholar]
- 34.Cadigan KM, Liu YI. Wnt signaling: complexity at the surface. Journal of Cell Science. 2006;119(3):395–402. doi: 10.1242/jcs.02826. [DOI] [PubMed] [Google Scholar]
- 35.Waterman ML. Lymphoid enhancer factor/T cell factor expression in colorectal cancer. Cancer and Metastasis Reviews. 2004;23(1-2):41–52. doi: 10.1023/a:1025858928620. [DOI] [PubMed] [Google Scholar]
- 36.Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nature Reviews Drug Discovery. 2006;5(12):997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 37.Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nature Genetics. 2008;40(5):499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sareddy GR, Panigrahi M, Challa S, Mahadevan A, Babu PP. Activation of Wnt/β-catenin/Tcf signaling pathway in human astrocytomas. Neurochemistry International. 2009;55(5):307–317. doi: 10.1016/j.neuint.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 39.Roth W, Wild-Bode C, Platten M, et al. Secreted Frizzled-related proteins inhibit motility and promote growth of human malignant glioma cells. Oncogene. 2000;19(37):4210–4220. doi: 10.1038/sj.onc.1203783. [DOI] [PubMed] [Google Scholar]
- 40.Ji H, Wang J, Nika H, et al. EGF-induced ERK activation promotes CK2-mediated disassociation of α-catenin from β-catenin and transactivation of β-catenin. Molecular Cell. 2009;36(4):547–559. doi: 10.1016/j.molcel.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wend P, Holland JD, Ziebold U, Birchmeier W. Wnt signaling in stem and cancer stem cells. Seminars in Cell and Developmental Biology. 2010;21(8):855–863. doi: 10.1016/j.semcdb.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 42.Toledo EM, Colombres M, Inestrosa NC. Wnt signaling in neuroprotection and stem cell differentiation. Progress in Neurobiology. 2008;86(3):281–296. doi: 10.1016/j.pneurobio.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 43.David MD, Canti C, Herreros J. Wnt-3a and Wnt-3 differently stimulate proliferation and neurogenesis of spinal neural precursors and promote neurite outgrowth by canonical signaling. Journal of Neuroscience Research. 2010;88(14):3011–3023. doi: 10.1002/jnr.22464. [DOI] [PubMed] [Google Scholar]
- 44.Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nature Medicine. 2004;10(1):55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- 45.Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nature Reviews Neuroscience. 2010;11(2):77–86. doi: 10.1038/nrn2755. [DOI] [PubMed] [Google Scholar]
- 46.Cole MF, Johnstone SE, Newman JJ, Kagey MH, Young RA. Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes and Development. 2008;22(6):746–755. doi: 10.1101/gad.1642408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pereira L, Yi F, Merrill BJ. Repression of Nanog gene transcription by Tcf3 limits embryonic stem cell self-renewal. Molecular and Cellular Biology. 2006;26(20):7479–7491. doi: 10.1128/MCB.00368-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng H, Ying H, Wiedemeyer R, et al. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell. 2010;17(5):497–509. doi: 10.1016/j.ccr.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang X, Yu Y, Yang HW, Agar NYR, Frado L, Johnson MD. The imprinted gene PEG3 inhibits Wnt signaling and regulates glioma growth. Journal of Biological Chemistry. 2010;285(11):8472–8480. doi: 10.1074/jbc.M109.069450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang N, Wei P, Gong A, et al. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20(4):427–442. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bredel M, Zentner J. Brain-tumour drug resistance: the bare essentials. Lancet Oncology. 2002;3(7):397–406. doi: 10.1016/s1470-2045(02)00786-6. [DOI] [PubMed] [Google Scholar]
- 52.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Molecular Cancer. 2006;5(67) doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beier D, Röhrl S, Pillai DR, et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Research. 2008;68(14):5706–5715. doi: 10.1158/0008-5472.CAN-07-6878. [DOI] [PubMed] [Google Scholar]
- 54.Wang J, Wang X, Jiang S, et al. Partial biological characterization of cancer stem-like cell line (WJ 2) of human glioblastoma multiforme. Cellular and Molecular Neurobiology. 2008;28(7):991–1003. doi: 10.1007/s10571-008-9273-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flahaut M, Meier R, Coulon A, et al. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wnt/β-catenin pathway. Oncogene. 2009;28(23):2245–2256. doi: 10.1038/onc.2009.80. [DOI] [PubMed] [Google Scholar]
- 56.Tamura K, Aoyagi M, Wakimoto H, et al. Accumulation of CD133-positive glioma cells after high-dose irradiation by γ knife surgery plus external beam radiation. Journal of Neurosurgery. 2010;113(2):310–318. doi: 10.3171/2010.2.JNS091607. [DOI] [PubMed] [Google Scholar]
- 57.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 58.Ayyanan A, Civenni G, Ciarloni L, et al. Increased Wnt signaling triggers oncogenic conversion of human breast epithelial cells by a Notch-dependent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(10):3799–3804. doi: 10.1073/pnas.0600065103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/β-catenin mediates radiation resistance of mouse mammary progenitor cells. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(2):618–623. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang ES, Nowsheen S, Wang T, Thotala DK, Xia F. Glycogen synthase kinase 3β inhibition enhances repair of DNA double-strand breaks in irradiated hippocampal neurons. Neuro-Oncology. 2011;13(5):459–470. doi: 10.1093/neuonc/nor016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu X, Wang L, Zhao S, Ji X, Luo Y, Ling F. β-Catenin overexpression in malignant glioma and its role in proliferation and apoptosis in glioblastma cells. Medical Oncology. 2011;28(2):608–614. doi: 10.1007/s12032-010-9476-5. [DOI] [PubMed] [Google Scholar]
- 62.Liu C, Tu Y, Sun X, et al. Wnt/β-Catenin pathway in human glioma: expression pattern and clinical/prognostic correlations. Clinical and Experimental Medicine. 2010:1–8. doi: 10.1007/s10238-010-0110-9. [DOI] [PubMed] [Google Scholar]
- 63.Wang ZX, Chen YY, Li BA, et al. Decreased pygopus 2 expression suppresses glioblastoma U251 cell growth. Journal of Neuro-Oncology. 2010;100(1):31–41. doi: 10.1007/s11060-010-0144-6. [DOI] [PubMed] [Google Scholar]
- 64.Guo G, Mao X, Wang P, et al. The expression profile of FRAT1 in human gliomas. Brain Research. 2010;1320:152–158. doi: 10.1016/j.brainres.2010.01.037. [DOI] [PubMed] [Google Scholar]
- 65.Pu P, Zhang Z, Kang C, et al. Downregulation of Wnt2 and β-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Therapy. 2009;16(4):351–361. doi: 10.1038/cgt.2008.78. [DOI] [PubMed] [Google Scholar]
- 66.Yu JM, Jun ES, Jung JS, et al. Role of Wnt5a in the proliferation of human glioblastoma cells. Cancer Letters. 2007;257(2):172–181. doi: 10.1016/j.canlet.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 67.Mikels A, Minami Y, Nusse R. Ror2 receptor requires tyrosine kinase activity to mediate Wnt5A signaling. Journal of Biological Chemistry. 2009;284(44):30167–30176. doi: 10.1074/jbc.M109.041715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mikels AJ, Nusse R. Purified Wnt5a protein activates or inhibits β-catenin-TCF signaling depending on receptor context. PLoS biology. 2006;4(4) doi: 10.1371/journal.pbio.0040115. Article ID e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Polakis P. The oncogenic activation of β-catenin. Current Opinion in Genetics and Development. 1999;9(1):15–21. doi: 10.1016/s0959-437x(99)80003-3. [DOI] [PubMed] [Google Scholar]
- 70.Gotze S, Wolter M, Reifenberger G, Muller O, Sievers S. Frequent promoter hypermethylation of Wnt pathway inhibitor genes in malignant astrocytic gliomas. International Journal of Cancer. 2010;126(11):2584–2593. doi: 10.1002/ijc.24981. [DOI] [PubMed] [Google Scholar]
- 71.Sarin S, Bernath A. Turcot syndrome (glioma polyposis): a case report. Southern Medical Journal. 2008;101(12):1273–1274. doi: 10.1097/SMJ.0b013e3181883853. [DOI] [PubMed] [Google Scholar]
- 72.Yang Z, Wang Y, Fang J, et al. Expression and aberrant promoter methylation of Wnt inhibitory factor-1 in human astrocytomas. Journal of Experimental & Clinical Cancer Research. 2010;29:p. 26. doi: 10.1186/1756-9966-29-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suzuki H, Watkins DN, Jair KW, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nature Genetics. 2004;36(4):417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 74.Roman-Gomez J, Jimenez-Velasco A, Agirre X, et al. Transcriptional silencing of the Dickkopfs-3 (Dkk-3) gene by CpG hypermethylation in acute lymphoblastic leukaemia. British Journal of Cancer. 2004;91(4):707–713. doi: 10.1038/sj.bjc.6602008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nelson WJ, Nusse R. Convergence of Wnt, β-catenin, and cadherin pathways. Science. 2004;303(5663):1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu Z, Hunter T. Wnt-independent β-catenin transactivation in tumor development. Cell Cycle. 2004;3(5):571–573. [PubMed] [Google Scholar]
- 77.Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harbor Perspectives in Biology. 2010;2(2) doi: 10.1101/cshperspect.a002915. Article ID a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of β-catenin. Current Opinion in Cell Biology. 2005;17(5):459–465. doi: 10.1016/j.ceb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 79.David MD, Yeramian A, Dunach M, et al. Signalling by neurotrophins and hepatocyte growth factor regulates axon morphogenesis by differential β-catenin phosphorylation. Journal of Cell Science. 2008;121(16):2718–2730. doi: 10.1242/jcs.029660. [DOI] [PubMed] [Google Scholar]
- 80.Kamino M, Kishida M, Kibe T, et al. Wnt-5a signaling is correlated with infiltrative activity in human glioma by inducing cellular migration and MMP-2. Cancer Science. 2011;102(3):540–548. doi: 10.1111/j.1349-7006.2010.01815.x. [DOI] [PubMed] [Google Scholar]
- 81.Pukrop T, Klemm F, Hagemann T, et al. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(14):5454–5459. doi: 10.1073/pnas.0509703103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weeraratna AT, Jiang Y, Hostetter G, et al. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell. 2002;1(3):279–288. doi: 10.1016/s1535-6108(02)00045-4. [DOI] [PubMed] [Google Scholar]
- 83.Crawford HC, Fingleton BM, Rudolph-Owen LA, et al. The metalloproteinase matrilysin is a target of β-catenin transactivation in intestinal tumors. Oncogene. 1999;18(18):2883–2891. doi: 10.1038/sj.onc.1202627. [DOI] [PubMed] [Google Scholar]