

Abstract
The TRiC/CCT chaperonin is a 1-MDa hetero-oligomer of 16 subunits that assists the folding of proteins in eukaryotes. Low-resolution structural studies confirmed the TRiC particle to be composed of two stacked octameric rings enclosing a folding cavity. The exact arrangement of the different proteins in the rings underlies the functionality of TRiC and is likely to be conserved across all eukaryotes. Yet despite its importance it has not been determined conclusively, mainly because the different subunits appear nearly identical under low resolution. This work successfully addresses the arrangement problem by the emerging technique of cross-linking, mass spectrometry, and modeling. We cross-linked TRiC under native conditions with a cross-linker that is primarily reactive toward exposed lysine side chains that are spatially close in the context of the particle. Following digestion and mass spectrometry we were able to identify over 60 lysine pairs that underwent cross-linking, thus providing distance restraints between specific residues in the complex. Independently of the cross-link set, we constructed 40,320 (= 8 factorial) computational models of the TRiC particle, which exhaustively enumerate all the possible arrangements of the different subunits. When we assessed the compatibility of each model with the cross-link set, we discovered that one specific model is significantly more compatible than any other model. Furthermore, bootstrapping analysis confirmed that this model is 10 times more likely to result from this cross-link set than the next best-fitting model. Our subunit arrangement is very different than any of the previously reported models and changes the context of existing and future findings on TRiC.
Keywords: group II chaperonins, protein folding, violation distances
In eukaryotes and archaea the folding of nascent and mis-folded polypeptide chains is assisted by a group II chaperonin system known as the thermosome in archea and TRiC (or CCT) in eukaryotes (1). These large protein complexes consist of two stacked octameric rings (2) with flexible integral lids that can open and close through a cycle of ATP binding and hydrolysis (3). The open state of the complex binds the polypeptide substrate (4, 5), whereupon closing the substrate is sequestered into a large interior cavity where folding can occur. TRiC has been implicated in the folding pathways of many cytosolic proteins (6), most notably actin and tubulin (7, 8).
Many archaeal species have just one thermosome gene and a simple homo-oligomeric architecture (9). Other archaeal species have at most three different types of subunits, and there is evidence that the multiple genes are redundant (10). In stark contrast, the eukaryotic TRiC consists of eight different subunit types (CCT1 to CCT8), all of which are essential. The subunit specialization occurred very early in eukaryote evolution (11) and is conserved to such an extent that the sequence identity between mammalian and yeast subunits of the same type is nearly 60%, whereas the sequence identity between the different subunit types in the same organism is only 30%. The diversity of eight distinct subunits allows for specific substrate binding modes (4, 12, 13) as well as other cycle-related activities such as differential ATP hydrolysis (14, 15).
Underlying the TRiC functionality is a defined particle arrangement where each ring is composed of all eight subunits in a precise order (16), and the upper and lower rings are stacked in specific registration. This defined architecture puts the substrate binding in a precise spatial context and confers fidelity to substrate folding. Despite its great functional importance, the subunit arrangement in TRiC has not been demonstrated conclusively. TRiC is very refractory to crystallization and a high-resolution crystal structure of the complex is not available. Due to their high sequence similarity, the different subunits look nearly identical in lower-resolution studies (3, 17, 18) and make reliable identification especially difficult. An alternative biochemical approach that analyzed the content of spontaneously occurring TRiC fragments (19) suggested a possible ring arrangement that has not been reaffirmed so far. Furthermore, all previous suggestions for the particle arrangement are completely incompatible with our recent inter-subunit interface analysis that is based on evolutionary conservation (20).
An emerging structure determination technique that has the potential to conclusively determine the subunit order in TRiC is cross-linking coupled with mass spectrometry (MS) (21, 22). In this technique, the complex is incubated under native conditions with a cross-linker that is capable of forming specific covalent bonds with side chains on the surface of the complex. Next, the complex is digested and submitted to MS analysis, with a fraction of the peptides still linked in pairs by cross-linkers. These linked peptides are identified by detailed analysis of the MS data, and the specific residues that are cross-linked are determined to be in close spatial proximity in the intact complex. It is this structural information that guides molecular modeling.
Recent advances in MS instrumentation and the availability of analysis software have contributed to a growing number of studies where this method is being applied. At the same time, the complexity and size of the investigated protein complexes increased (23, 24), with recent studies probing the 600 kDa PolIIF transcription system (25) and the intact proteasome (26). The combination of the technique with particle reconstruction by cryo-EM appears to be especially powerful because the cross-link data is helpful in assigning specific subunits to unassigned electron density.
Previous cross-linking studies took two different approaches in transforming the cross-links into a model. For simpler systems involving cross-linking of a pair of proteins, multiple docking models were derived and then filtered by the cross-link sets (23, 27). However, studies of more complex systems with multiple proteins only reported a single model in very coarse terms (24, 25). The latter approach is understandable given the great computational complexity of modeling such large systems but clearly has the disadvantage of not allowing one to gauge the confidence in the reported model compared to alternative models. Here we propose a third approach, termed combinatorial homology modeling, to resolve this issue and allow the best model to be determined with appropriate confidence limits. In this approach we model exhaustively all the 40,320 (= 8 factorial or 8!) possible arrangements of TRiC and compare each one against the cross-linking data. Such comparison objectively points toward the model or models that best fit the data while at the same time giving a confidence measure of how better they are over the alternatives.
This work reports on the successful application of cross-linking, MS, and combinatorial homology modeling to the subunit arrangement problem in TRiC. We were able to identify over 60 cross-links in our MS data, and through combinatorial homology modeling we prove that only a single subunit arrangement is compatible with them. To our surprise this arrangement is very different from what has been previously suggested. We show it is compatible with many aspects of the evidence that is available so far on the TRiC architecture. Finally, we thoroughly discuss the prospects of using cross-linking and combinatorial modeling in other systems beside TRiC.
Results
Cross-Linking and MS Analysis.
We incubated a sample of close-state TRiC with nonlabeled BS3 cross-linker under native conditions. The cross-linked complex was then denatured, digested with trypsin, and submitted to MS analysis. Both ends of BS3 reacts predominantly with the primary amines in lysine side-chains and the N termini of polypeptide chains. The significantly weaker reactivity toward other residue types (28) was not considered in this study. The specificities of the cross-linking and digestion reactions allowed us to compile a list of all possible pairs of cross-linked peptides expected from the TRiC complex. We then compared this list to the MS data in search of entries that fit the data well in both their expected MS1 and MS/MS fragmentation spectra (Fig. 1A). With more than 100 million possibilities for cross-linked peptide pairs, we run the serious risk that a good fit with the MS data might occur frequently by chance. A powerful advantage of the MS analysis pipeline is its ability to provide a reliable estimate of the false positive rate given the set of sequences and the raw MS data. To that end, the analysis pipeline is repeated with erroneous cross-linker masses or with the correct mass but with the sequences reversed (29, 30). Fig. 1B shows that a change of just a few Daltons to the cross-linker mass leads to a sharp drop in the number of matches passing our selection criteria (range 0–4, median 2). Thus, at an MS/MS score (SMS2) threshold of 0.5 our analysis is estimated to have approximately two false positives (3% rate). Similar estimates of false positive rates for thresholds of 0.4 and 0.35 were 9% and 12.5%, respectively. We opted to work at the regime of fewer false positives and used the 0.5 threshold on the MS/MS score.
Fig. 1.

Mass spectrometry (MS) data analysis. (A) Assignment of a cross-link between peptides from CCT2 (red) and CCT4 (green). The precursor ion (box) matches the expected mass of the cross-linked peptides to 1.9 ppm, and their fragmentation pattern is highly consistent with the MS/MS spectrum shown. (B) The data analysis identified 63 cross-links above our confidence criteria of SMS2≥0.5 (central “0”-bar). When the analysis is repeated with small, deliberate errors in the cross-linker mass or with the reversed sequences (REV), very few cross-links are identified above this confidence threshold. The false positive rate (dashed line) for these specific dataset, sequences and threshold can be estimated to be 3% or about two cross-links. Lower SMS2 thresholds will lead to more true positives (Fig. 2) but also to a rapid increase in the false positive rate.
Our analysis found 63 peptide pairs (Table S1) that passed the fit criteria to the MS data. This set consists of 27 pairs where the two peptides are of different subunits and 36 pairs where both peptides are of the same subunit. However, throughout this work we allow for the possibility that some of the latter pairs originate from two subunits with the same sequence on different rings. Peptides from each of the eight TRiC genes are represented in the cross-link set, although with different frequencies. The most represented subunit is CCT6(ζ) with 40 appearances, while CCT2(β) and CCT5(ϵ) only appear six times each. The variability in the representation of specific lysines is also quite large with K530 in CCT6(ζ) and K24 in CCT4(δ) appearing seven times, while many other lysine residues appear just once.
Combinatorial Modeling of TRiC.
The eight genes of TRiC are highly homologous (typical sequence identity of 38%) to those of simpler archaeal chaperonins whose close-state structures were solved by either high-resolution crystallography (2) or cryo-EM (9). Moreover, a recent cryo-EM study of TRiC itself has shown the close-state of TRiC to be highly similar to that of the archaeal chaperonins (17). Thus, we can expect homology modeling to faithfully represent the closed-state TRiC particle. We used straightforward homology modeling to get the backbone model of any TRiC subunit at any position in the particle. Hence, we can model a full particle with any consistency of the 16 subunits. Our models cover most of the TRiC sequences except for up to 20 residues at the N and C termini of each subunit, which are unstructured in the template. Any cross-link with either of its cross-linked lysine residues occurring in an unstructured part of the model was discarded. A list of the remaining cross-links between different subunits is shown in Fig. 2.
Fig. 2.

Twenty-one identified cross-links between subunits that have associated distances. The specific cross-linked lysine residues are listed together with their MS parameters. Cross-links that involved Lys residues not seen in the X-ray structure of the 1Q3R template are omitted. The first 18 entries are used for the combinatorial analysis (Fig. 3). Mox is oxidized methionine. aThe column headers are r for the rank in this list, S1 for the name of the first subunit in the cross-link, R1 for the lysine residue in S1, S2 and R2 for the name and position of the second subunit, SMS2 is the MS/MS fit score (see text), ppm is the mass error of the precursor ion in parts per million, Q is the ion charge, M is the molecular mass of the ion rounded to the nearest dalton, d12 is the distance between the Cα atoms of the cross-linked lysine residues R1 and R2 in the OMS model of TRiC (Fig. 4). bThe most likely cross-linked lysine is underlined in Peptide 1 and Peptide 2. cThis one cross-link was included in spite of its low MS/MS fit score (0.32) as there are at least four identified MS/MS fragmentation sites on each peptide. dThe last cross-link marked in pink highlight is not used in any analysis. The inset on the right shows that the six most reliable unique cross-links between subunit pairs: BD, AG, BE, AD, GZ and QZ assemble to ring arrangement EBDAGZQH, the correct arrangement (after rotation), without need for any modeling. Modeling we do here is essential to show this arrangement is unique and statistically significant.
In TRiC each ring contains all the eight genes in an unknown but precise order, and both rings have identical gene order (16). Under these premises the total number of ways to arrange the 16 subunits of TRiC is 40,320 (7! different ring arrangements and eight different registrations of the ring stacking). This number, although high, is within the scope of current computing, and we were able to build a backbone model for every possible particle arrangement. On a specific full-particle model (arrangement), any cross-link can be assigned a distance between the Cα atoms of the cross-linked lysine residues. Previous studies of cross-linked proteins with known crystal structures have established that this distance is less than 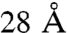 for most cross-links with few instances being as high as
for most cross-links with few instances being as high as 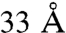 because of local protein flexibility (21, 25, 31). These numbers are in complete accord with the cross-linker length plus twice the length of the lysine side chain. Here, we define an arrangement to be in violation of a cross-link if the corresponding Cα atoms are more than
because of local protein flexibility (21, 25, 31). These numbers are in complete accord with the cross-linker length plus twice the length of the lysine side chain. Here, we define an arrangement to be in violation of a cross-link if the corresponding Cα atoms are more than 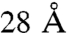 apart and the violation distance as the Cα–Cα distance minus
apart and the violation distance as the Cα–Cα distance minus 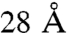 . Accordingly, the extent to which the entire cross-link set is consistent with an arrangement is defined by two measures: the tally of individual violations and the sum of their violation distances. Fig. 3 shows the values of these two measures on every one of the 40,320 possible TRiC arrangements. The distributions of these values reveal that a single arrangement stands out above the rest as having the highest consistency with the cross-link set. We will refer to this arrangement as the “optimal mass-spectrometry arrangement” (OMS) from here on, and its subunit order is shown in Fig. 4C. We also note that the center panel in Fig. 3 is “funneled” toward the OMS arrangement in the sense that its lower-left part is enriched with arrangements that are highly similar to it. For example, the arrangements with the second and third lowest sums of their violation distances have the same ring order as the OMS arrangement and a one subunit shift (in either direction) in the inter-ring registration (termed M1 and P1).
. Accordingly, the extent to which the entire cross-link set is consistent with an arrangement is defined by two measures: the tally of individual violations and the sum of their violation distances. Fig. 3 shows the values of these two measures on every one of the 40,320 possible TRiC arrangements. The distributions of these values reveal that a single arrangement stands out above the rest as having the highest consistency with the cross-link set. We will refer to this arrangement as the “optimal mass-spectrometry arrangement” (OMS) from here on, and its subunit order is shown in Fig. 4C. We also note that the center panel in Fig. 3 is “funneled” toward the OMS arrangement in the sense that its lower-left part is enriched with arrangements that are highly similar to it. For example, the arrangements with the second and third lowest sums of their violation distances have the same ring order as the OMS arrangement and a one subunit shift (in either direction) in the inter-ring registration (termed M1 and P1).
Fig. 3.

Assessment of fit between model and the inter-subunit cross-link dataset in A, for each of the 40,320 full-particle models representing all the possible arrangements of TRiC. Two measures of fit are used for each model: (y axis) the Number of Violations (cross-links in the set spanning more than 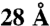 on the model) and (x axis) the Sum of Violation Distances above
on the model) and (x axis) the Sum of Violation Distances above 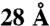 for these violating cross-links. Top and right panels plot the histograms of these measures across arrangements (in gray), with histograms of the log-counts (orange) to show the small number of high-fitting arrangements. A single arrangement stands above the rest in quality of fit (pink star) and is referred to in this work as the optimal mass-spectrometry arrangement (OMS). The second and third best-fitting arrangements (pink circles) are the OMS arrangement with an inter-ring registration shift of one subunit in either directions (P1 and M1). In B we show the arrangements previously found using electron microscopy: LFC (17) and X-ray crystallography: PW (18). It is easier to visualize the subunit arrangements by treating the TRiC sphere as a cylinder and then unwrapping the subunit names to form an 2 × 8 array. We rotate TRiC so that subunit A is on the upper left. In keeping with our previous work (20), we use Latin letters for the Greek letters used to name the TRiC subunits as follows: Alpha α = A, Beta β = B, Gamma γ = G, Delta δ = D, Epsilon ϵ = E, Zeta ζ = Z, Eta η = H, and Theta θ = Q.
for these violating cross-links. Top and right panels plot the histograms of these measures across arrangements (in gray), with histograms of the log-counts (orange) to show the small number of high-fitting arrangements. A single arrangement stands above the rest in quality of fit (pink star) and is referred to in this work as the optimal mass-spectrometry arrangement (OMS). The second and third best-fitting arrangements (pink circles) are the OMS arrangement with an inter-ring registration shift of one subunit in either directions (P1 and M1). In B we show the arrangements previously found using electron microscopy: LFC (17) and X-ray crystallography: PW (18). It is easier to visualize the subunit arrangements by treating the TRiC sphere as a cylinder and then unwrapping the subunit names to form an 2 × 8 array. We rotate TRiC so that subunit A is on the upper left. In keeping with our previous work (20), we use Latin letters for the Greek letters used to name the TRiC subunits as follows: Alpha α = A, Beta β = B, Gamma γ = G, Delta δ = D, Epsilon ϵ = E, Zeta ζ = Z, Eta η = H, and Theta θ = Q.
Fig. 4.

The full-particle model of TRiC in the OMS arrangement is shown in top (A) and side (B) views, with a different color for each subunit type. The inter-subunit cross-links are drawn to scale with their atoms as black Van der Waals spheres. Two solid slices through both the upper and lower rings (C) are annotated with the subunit order. The CCT2(β) subunits (green) in the upper and lower rings are in contact as are the CCT6(ζ) subunits (brown). For clarity, the back half of the particle in each view is not shown and the Van der Waals radii of the atoms in the cross-links are increased by 30%. The order of subunits in the top ring is αγζθηϵβδ or CCT1, CCT3, CCT6, CCT8, CCT7, CCT5, CCT2, CCT4 (13687524) that we prefer to write as AGZQHEBD.
We employed bootstrapping to quantify the significance of the OMS arrangement over other arrangements with good fit to the 63 cross-link dataset. To that aim, we randomly redrew with repeats from the original dataset a new dataset with identical size, ran the combinatorial modeling analysis, and recorded the arrangement that had the best fit. We repeated this step 1,000 times and found that the OMS arrangement had the best fit in 743 (P1 = 74%) of the steps. The arrangement ranking second in the number of best fits came on top in only 74 of the steps (P2 = 7.4%). Thus, we can estimate that the OMS arrangement is at least 10 times (= P1/P2) more likely to be consistent with the dataset than the any other arrangement and 2.9 times (= P1/(1-P1)) more likely to be consistent than all the other arrangements combined.
The OMS Arrangement.
In the cross-link set, 52 of the cross-links are between lysine residues corresponding to structured parts in the template and can therefore be assigned with a distance on the OMS arrangement model (Table S1). Six (11%) of these are defined as violations with distance values of: 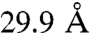 ,
, 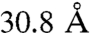 ,
, 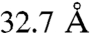 ,
, 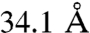 ,
, 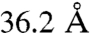 , and
, and 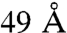 . The distance histogram of the entire set (Fig. 5) suggests that the last violation is very likely to be a mis-assignment of the MS data, a number consistent with our false positive analysis. The other five violations are less likely to be misassignments and more likely report on flexibilities in the TRiC complex that are not accounted for by our model. Indeed, three of these five violations involve residues from the flexible N termini of three different subunits. The remaining two violations both involve K317 in CCT6(ζ) and can both be resolved if the apical domain of CCT6(ζ) is rotated slightly.
. The distance histogram of the entire set (Fig. 5) suggests that the last violation is very likely to be a mis-assignment of the MS data, a number consistent with our false positive analysis. The other five violations are less likely to be misassignments and more likely report on flexibilities in the TRiC complex that are not accounted for by our model. Indeed, three of these five violations involve residues from the flexible N termini of three different subunits. The remaining two violations both involve K317 in CCT6(ζ) and can both be resolved if the apical domain of CCT6(ζ) is rotated slightly.
Fig. 5.

The histograms of the distributions of Cα–Cα distances within the OMS model for both the 63 observed cross-linked lysine pairs (red, left y axis scale) or nonspecific lysine pairing (blue, right y axis scale).
Discussion
TRiC Architecture.
In this work we cross-linked TRiC under native conditions and made efforts to use low cross-linker concentration (Fig. S1). We therefore have no reason to suspect that the cross-linking had seriously deformed the particle or otherwise altered its subunit order. TRiC presents a special circumstance in which the overall shape of the particle as well as the structures of each of its subunits can be modeled reliably to high accuracy, yet the subunit order is unknown. Under these premises we were able to model all the 40,320 possible subunit arrangements and compare the consistency of each of them with the cross-link dataset without any bias. This comparison revealed that one arrangement is significantly more consistent with the dataset than the rest, and bootstrapping analysis had quantified the chance of such a “best fit” to occur by random as low. We are therefore confident that the OMS arrangement (Fig. 4C) is in fact the native subunit arrangement of TRiC.
We were surprised to find the native subunit arrangement to be so different from previously suggested models (16–19). Yet some of the previously published data certainly support our new model. We have previously reported (20), based on evolutionary conservation of the subunit interfaces, that subunits CCT3(γ) and CCT6(ζ) are very likely to appear in that order within the ring as indeed they do in the top arrangement. Two other recent structural studies, one in cryo-EM (17) and the other in crystallography (18), independently showed that the twofold symmetry axis of the particle put two subunits within each ring on top of their homotypic counterparts in the opposite ring. In the OMS model, we see such abutting for subunits CCT2(β) and CCT6(ζ). Finally, our inter-ring registration is consistent with three-dimensional particle reconstructions of TRiC obtained by cryoelectron microscopy with attached monoclonal antibodies (16).
Combinatorial Homology Modeling.
Cross-linking and MS currently give only a small set of low-resolution structural clues on any complex of interest. Thus, unlike NMR, de novo structure determination is not possible and external structural information is required for a meaningful model. Previous cross-linking studies inherently suffered from lack of high-resolution knowledge on the subunit structures (25, 32), their interfacing modes (23–25), overall shape of the complex (24, 25, 33) or combinations of these factors. In the TRiC system all these factors are known to a much higher resolution than the maximum cross-linking length (approximately 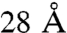 ). Consequently, this study exhaustively evaluated all the subunit arrangements, whereas previous studies were limited to a small sampling of the conformation space (23, 27) or even just to a single model (25). The merits of combinatorial modeling are evident: The top ranking model is chosen without bias, and its significance over the alternatives is clearly quantified. In contrast, with previous studies it was very hard to assess how much the cross-link set validated the model as opposed to guide its formation. It was also not ascertained that alternative models do not exist.
). Consequently, this study exhaustively evaluated all the subunit arrangements, whereas previous studies were limited to a small sampling of the conformation space (23, 27) or even just to a single model (25). The merits of combinatorial modeling are evident: The top ranking model is chosen without bias, and its significance over the alternatives is clearly quantified. In contrast, with previous studies it was very hard to assess how much the cross-link set validated the model as opposed to guide its formation. It was also not ascertained that alternative models do not exist.
The ability to perform combinatorial modeling is enabling smaller cross-link sets to produce more informative models. The cross-link set we report here is larger than most of the datasets recently published. Yet it is dwarfed in comparison with the number of all possible cross-links that span less than 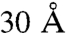 in the OMS arrangement model, which is about 2,000 (Fig. 5 and Fig. S2). Remarkably, combinatorial modeling is able to reveal the complex TRiC architecture with this tiny sample of the cross-link space.
in the OMS arrangement model, which is about 2,000 (Fig. 5 and Fig. S2). Remarkably, combinatorial modeling is able to reveal the complex TRiC architecture with this tiny sample of the cross-link space.
Combinatorial modeling of the TRiC system offers a unique opportunity to investigate the interplay between the false positive rate (a function of the MS/MS score threshold) and the quality of the resulting model. To that end, we plot the P1/P2 value of the bootstrapping analysis as a function of the number of cross-links in the dataset (Fig. 6). The ability to discriminate the OMS arrangement from the next candidate peaks close to the dataset size of 63 that we used. The highest P1/P2 values of 12.8 and 12.4 occur with 58 and 65 cross-links, respectively; this indicates that our choice for false positive rate was close to optimal. Interestingly, for just slightly larger datasets, the P1/P2 ratio deteriorates rapidly. We conclude that false positive rates of 5% or higher are undesirable.
Fig. 6.

Showing the bootstrap P1/P2 score as a function of the cutoff threshold in the list of identified non-self cross-links. One thousand bootstrap samples were used for each data point. The arrangement most probable for a particular number of cross-links is illustrated; when P1/P2 = 1, several arrangements are equally probable.
The favorable circumstances for combinatorial modeling that occur in TRiC are by no means unique. For example, the AAA-ATP ring module of the S19 regulatory fraction of the proteasome (26) shows similar circumstances with the availability of an overall cryo-EM density map and reliable homology models for each subunit in the ring. Yet ambiguity exists in the ring arrangement and its registration to the S20 subunit. In an even broader context, we foresee that combinatorial modeling will be applicable to a growing number of assemblies with less regular architectures. The drive for this growth is the improvement in the resolution of cryo-EM density maps of large complexes. Such maps show clearly defined borders for many of their constituent subunits. Yet the resolution is still too low to identify the subunits with confidence based on secondary structures or other clues from their folds. Because the folds of most subunits can usually be modeled reliably by homology, an enumeration of all the possibilities to dock each into the density map may be tractable.
Methods
Cross-Linking.
Bovine TRiC was purified as described (34). One hundred eighty μg of the purified complex were transferred into 60 μL of HEPES buffer (20 mM HEPES-KOH pH = 7.5, 50 mM KCl, 5 mM MgCl, 10% glycerol). The complex was arrested in the closed state by incubation with 1 mM ATP, 2.5 mM Al(NO3)3, and 15 mM NaF at 37 °C for 10 min (3). Bis (sulfosuccinimidyl) suberate (BS3, 2 mg, no-weigh format, Thermo Fisher Scientific) was freshly dissolved into HEPES buffer without glycerol and immediately added to the TRiC sample at 2 mM final concentration. The cross-linking reaction proceeded on ice for 2 h and was quenched by ammonium bicarbonate at 25 mM final concentration. The solution was dialyzed (molecular weight cutoff of 20,000) overnight against HEPES buffer, and submitted for digestion and MS.
MS Analysis.
Full methods of the MS preparation, measurement, and analysis appear in SI Text. Briefly, the mass of each precursor ion was compared to the list of possible cross-linked species. Matching candidates within a mass tolerance of 6 ppm were further analyzed for MS/MS fit. The expected b and y series were compared to the measured MS/MS spectrum with the same 6-ppm tolerance. The MS/MS score is
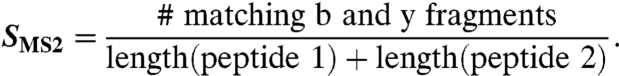 |
A candidate match is assigned to a specific cross-link entry if (i) the SMS2≥0.5 or at least four matching b or y fragments occur on each of the two peptides, and (ii) no other match is available with the same quality of fit.
Combinatorial Modeling.
The distance in a structural model assigned to a cross-link is taken as the Cα–Cα distance between the two cross-linked lysine residues. Because every subunit occurs twice in the TRiC particle, the distance between a particular pair of lysine residues is calculated between subunits within a ring as well as between the same subunits on different rings. The shorter of the two distances is assigned as the cross-link distance in the particular model. Two measures are used to score the fit of a model to the cross-link set: (i) the total number of violations, which occur when the Cα–Cα distance between the two cross-linked lysine residues exceeds 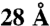 ; (ii) the sum of violation distances, defined as the cross-link distance minus
; (ii) the sum of violation distances, defined as the cross-link distance minus 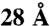 for violations and zero otherwise. The dependency of the number of violations on the distance threshold defining a violation is shown in Fig. S3.
for violations and zero otherwise. The dependency of the number of violations on the distance threshold defining a violation is shown in Fig. S3.
Combinatorial modeling follows ref. 20. The eight subunits in any ring can be arranged in 7! ways. By convention we start with the A or α subunit (CCT1) and then can place the second subunit in seven ways, the third in six ways, etc. The lower ring is assumed to have the same subunit ordering as the upper ring. In forming the arrangement of the 16 subunit TRiC particle, each of the eight subunits in the lower ring can be placed below the A subunit of the upper ring so that the total number of arrangements is 8 × 7 != 8 != 40,320. Subunit structures and position in the particle followed straightforward homology modeling from the template structure, PDB ID code 1Q3R (35), using manually curated alignments between the TRiC and thermosome sequences (Dataset S1). Very minor changes to the deposited TRiC sequences (SI Text and Table S2) were made following MS analysis of post translational modifications on native TRiC..
By scoring all 40,320 models against the cross-links, we can obtain the arrangement or arrangements that have the best score. Our confidence in this particular best arrangement will depend on how much better it fits than the second best arrangement. We evaluate this using the bootstrapping method (36) to generate an artificial set of cross-links selected randomly and with duplications from the original measured set. On average, this is equivalent to randomly deleting 37% (1/e) of the cross-link dataset and replacing the deleted entries with duplications of the remaining cross-links chosen by random. The new dataset is then the basis for a complete scoring of all the possible models. The procedure is repeated (typically 1,000 times) and the number of times that each arrangement scores best is counted. The probability or frequency of this best model is termed P1, while the probability of the second best model is termed P2.
Supplementary Material
Acknowledgments.
We thank Dr. Judith Frydman and Ramya Kumar for the TRiC and mm-CPN samples. We also thank Dr. Kenji Murkami and other members of the Kornberg lab for technical assistance and helpful discussions. This work was supported by grants from the National Institutes of Health through the Nanomedicine Development Center (PN1EY016525) and the BioX Interdisciplinary Initiative Program. M.L. is the Robert W. and Vivian K. Cahill Professor of Cancer Research.
Note.
The subunit arrangement in OMS is definitively confirmed by our ongoing Sentinel Correlation Analysis and refinement of the X-ray data from ref. 18.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1119472109/-/DCSupplemental.
References
- 1.Spiess C, Meyer AS, Reissmann S, Frydman J. Mechanism of the eukaryotic chaperonin: Protein folding in the chamber of secrets. Trends Cell Biol. 2004;14:598–604. doi: 10.1016/j.tcb.2004.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ditzel L, et al. Crystal structure of the thermosome, the archaeal chaperonin and homolog of CCT. Cell. 1998;93:125–138. doi: 10.1016/s0092-8674(00)81152-6. [DOI] [PubMed] [Google Scholar]
- 3.Booth CR, et al. Mechanism of lid closure in the eukaryotic chaperonin TRiC/CCT. Nat Struct Mol Biol. 2008;15:746–753. doi: 10.1038/nsmb.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llorca O, et al. Analysis of the interaction between the eukaryotic chaperonin CCT and its substrates actin and tubulin. J Struct Biol. 2001;135:205–218. doi: 10.1006/jsbi.2001.4359. [DOI] [PubMed] [Google Scholar]
- 5.Reissmann S, Parnot C, Booth CR, Chiu W, Frydman J. Essential function of the built-in lid in the allosteric regulation of eukaryotic and archaeal chaperonins. Nat Struct Mol Biol. 2007;14:432–440. doi: 10.1038/nsmb1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yam AY, et al. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol. 2008;15:1255–1262. doi: 10.1038/nsmb.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao Y, Thomas JO, Chow RL, Lee GH, Cowan NJ. A cytoplasmic chaperonin that catalyzes beta-actin folding. Cell. 1992;69:1043–1050. doi: 10.1016/0092-8674(92)90622-j. [DOI] [PubMed] [Google Scholar]
- 8.Yaffe MB, et al. TCP1 complex is a molecular chaperone in tubulin biogenesis. Nature. 1992;358:245–248. doi: 10.1038/358245a0. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, et al. Mechanism of folding chamber closure in a group II chaperonin. Nature. 2010;463:379–383. doi: 10.1038/nature08701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kapatai G, et al. All three chaperonin genes in the archaeon Haloferax volcanii are individually dispensable. Mol Microbiol. 2006;61:1583–1597. doi: 10.1111/j.1365-2958.2006.05324.x. [DOI] [PubMed] [Google Scholar]
- 11.Archibald JM, Blouin C, Doolittle WF. Gene duplication and the evolution of group II chaperonins: Implications for structure and function. J Struct Biol. 2001;135:157–169. doi: 10.1006/jsbi.2001.4353. [DOI] [PubMed] [Google Scholar]
- 12.Muñoz IG, et al. Crystal structure of the open conformation of the mammalian chaperonin CCT in complex with tubulin. Nat Struct Mol Biol. 2011;18:14–19. doi: 10.1038/nsmb.1971. [DOI] [PubMed] [Google Scholar]
- 13.Spiess C, Miller EJ, McClellan AJ, Frydman J. Identification of the TRiC/CCT substrate binding sites uncovers the function of subunit diversity in eukaryotic chaperonins. Mol Cell. 2006;24:25–37. doi: 10.1016/j.molcel.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rivenzon-Segal D, Wolf SG, Shimon L, Willison KR, Horovitz A. Sequential ATP-induced allosteric transitions of the cytoplasmic chaperonin containing TCP-1 revealed by EM analysis. Nat Struct Mol Biol. 2005;12:233–237. doi: 10.1038/nsmb901. [DOI] [PubMed] [Google Scholar]
- 15.Amit M, et al. Equivalent mutations in the eight subunits of the chaperonin CCT produce dramatically different cellular and gene expression phenotypes. J Mol Biol. 2010;401:532–543. doi: 10.1016/j.jmb.2010.06.037. [DOI] [PubMed] [Google Scholar]
- 16.Martín-Benito J, et al. The inter-ring arrangement of the cytosolic chaperonin CCT. EMBO Rep. 2007;8:252–257. doi: 10.1038/sj.embor.7400894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cong Y, et al. 4.0-A resolution cryo-EM structure of the mammalian chaperonin TRiC/CCT reveals its unique subunit arrangement. Proc Natl Acad Sci USA. 2010;107:4967–4972. doi: 10.1073/pnas.0913774107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dekker C, et al. The crystal structure of yeast CCT reveals intrinsic asymmetry of eukaryotic cytosolic chaperonins. EMBO J. 2011;30:3078–3090. doi: 10.1038/emboj.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liou AK, Willison KR. Elucidation of the subunit orientation in CCT (chaperonin containing TCP1) from the subunit composition of CCT micro-complexes. EMBO J. 1997;16:4311–4316. doi: 10.1093/emboj/16.14.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalisman N, Levitt M. Insights into the intra-ring subunit order of tric/cct: A structural and evolutionary analysis. Pac Symp Biocomput. 2010;2010:252–259. doi: 10.1142/9789814295291_0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leitner A, et al. Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol Cell Proteomics. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rappsilber J. The beginning of a beautiful friendship: Cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J Struct Biol. 2011;173:530–540. doi: 10.1016/j.jsb.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulz DM, et al. Annexin A2/P11 interaction: New insights into annexin A2 tetramer structure by chemical crosslinking, high-resolution mass spectrometry, and computational modeling. Proteins. 2007;69:254–269. doi: 10.1002/prot.21445. [DOI] [PubMed] [Google Scholar]
- 24.Maiolica A, et al. Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol Cell Proteomics. 2007;6:2200–2211. doi: 10.1074/mcp.M700274-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Chen ZA, et al. Architecture of the RNA polymerase II-TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bohn S, et al. Structure of the 26S proteasome from Schizosaccharomyces pombe at subnanometer resolution. Proc Natl Acad Sci USA. 2010;107:20992–20997. doi: 10.1073/pnas.1015530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dimova K, et al. Structural insights into the calmodulin-Munc13 interaction obtained by cross-linking and mass spectrometry. Biochemistry. 2009;48:5908–5921. doi: 10.1021/bi900300r. [DOI] [PubMed] [Google Scholar]
- 28.Kalkhof S, Sinz A. Chances and pitfalls of chemical cross-linking with amine-reactive N-hydroxysuccinimide esters. Anal Bioanal Chem. 2008;392:305–132. doi: 10.1007/s00216-008-2231-5. [DOI] [PubMed] [Google Scholar]
- 29.Rinner O, et al. Identification of cross-linked peptides from large sequence databases. Nat Methods. 2008;5:315–318. doi: 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sennels L, Bukowski-Wills JC, Rappsilber J. Improved results in proteomics by use of local and peptide-class specific false discovery rates. BMC Bioinformatics. 2009;10:179. doi: 10.1186/1471-2105-10-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seebacher J, et al. Protein cross-linking analysis using mass spectrometry, isotope-coded cross-linkers, and integrated computational data processing. J Proteome Res. 2006;5:2270–2282. doi: 10.1021/pr060154z. [DOI] [PubMed] [Google Scholar]
- 32.Tubb MR, Silva RA, Fang J, Tso P, Davidson WS. A three-dimensional homology model of lipid-free apolipoprotein A-IV using cross-linking and mass spectrometry. J Biol Chem. 2008;283:17314–17323. doi: 10.1074/jbc.M800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chu F, et al. Unraveling the interface of signal recognition particle and its receptor by using chemical cross-linking and tandem mass spectrometry. Proc Natl Acad Sci USA. 2004;101:16454–16459. doi: 10.1073/pnas.0407456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferreyra RG, Frydman J. Purification of the cytosolic chaperonin TRiC from bovine testis. Methods Mol Biol. 2000;140:153–60. doi: 10.1385/1-59259-061-6:153. [DOI] [PubMed] [Google Scholar]
- 35.Shomura Y, et al. Crystal structures of the group II chaperonin from Thermococcus strain KS-1: Steric hindrance by the substituted amino acid, and inter-subunit rearrangement between two crystal forms. J Mol Biol. 2004;335:1265–1278. doi: 10.1016/j.jmb.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 36.Efron B. Bootstrap methods: Another look at the jackknife. Ann Stat. 1979;7:1–26. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.