

Abstract
Activation and regulation of transcription factors (TFs) are the major mechanisms regulating changes in gene expression upon environmental exposure. Tobacco smoke (TS) is a complex mixture of chemicals, each of which could act through different signal cascades, leading to the regulation of distinct TFs and alterations in subsequent gene expression. We proposed that TS exposure affects inflammatory gene expression at the transcriptional level by modulating the DNA binding activities of TFs. To investigate transcriptional regulation upon TS exposure, a protein/DNA array was applied to screen TFs that are affected by TS exposure. This array-based screening allowed us to simultaneously detect 244 different TFs. Our results indicated that multiple TFs were rapidly activated upon TS exposure. DNA-binding activity of differentially expressed TFs was confirmed by EMSA. Our results showed that at least 20 TFs displayed more than twofold expressional changes after smoke treatment. Ten smoke-induced TFs, including NF-κB, VDR, ISRE, and RSRFC4, were involved in MAPK signaling pathways. The NF-κB family, which is involved in inflammation-induced gene activation, was selected for further study to characterize TS exposure-induced transcriptional activation. Western blot analysis and immunofluorescence microscopy indicated that TS exposure induced phosphorylation of IκB and translocation of NF-κB p65/p50 heterodimers into the nucleus. This activity was abrogated by the MAPK inhibitors PD98059 and U0126. Our results confirmed that activation of MAPK signaling pathways by TS exposure increased transcriptional activity of NF-κB. These data provide a potential mechanism for TS-induced inflammatory gene expression.
Keywords: tobacco smoke, transcription factor, MAPK, NF-κB
Transcriptional activation in eukaryotes is a highly regulated process that requires the concerted interactions of DNA-binding transcriptional activators, general transcription factors (TFs), and coactivator proteins to stimulate the recruitment and activity of RNA polymerase II to the appropriate gene promoters in response to biological signals. Most of these signals are generated by extracellular signaling molecules that initiate distinct signaling pathways, consequently terminating in the activation of one or more TFs. Abnormalities in signal transduction and TF activation underlie several inflammatory diseases and the majority of cancers (3, 4). Specifically, chronic inflammatory airway diseases and lung cancer are frequently associated with tobacco smoke (TS) exposure, consistent with the high volume of TS to which the lungs are exposed. However, the molecular mechanisms underlying TS-induced pulmonary diseases are not well understood. To better characterize the mechanisms responsible for TS-induced lung diseases, our initial approach was to investigate TS-regulated TF activation in airway epithelial cells and the potential upstream signaling pathways responsible for this upregulation.
TS is a chemical mixture that contains more than 2,000 chemicals, free radicals, and oxidant molecules (6, 12). Each component potentially stimulates a variety of signal cascades. Previous studies suggested that TS regulates the expression of AP-1, NF-κB, and Smad (1, 6, 16, 17). Unfortunately, this conventional EMSA approach, which confirms DNA binding activity of a single TF at a time, limits our ability to understand the complex signaling interactions that likely occur after TS exposure. To address this issue, we applied protein/DNA array technology to screen multiple TFs that were affected by TS exposure. This array-based screening technology allowed us to detect activation of 244 different TFs simultaneously.
Our data suggest that the majority of TS-induced TFs are regulated by upstream MAPK signaling pathways. MAPKs are members of a ubiquitous protein serine/threonine kinase family responsible for signal transduction in eukaryotic organisms. The major kinase cascades of the MAPK family include ERK, JNK, and p38 MAPK. These signaling cascades commonly affect TF activation involved in cell proliferation, differentiation, and apoptosis. On the basis of reports that ERK 1/2 signaling pathways are involved in transcriptional control of TS-induced gene expression (6, 11, 21), we further examined how inhibition of ERK 1/2 affects the DNA binding activities of TS-regulated TFs. Specifically, we examined how the ERK 1/2 pathway may regulate NF-κB activation and nuclear translocation after TS exposure.
MATERIALS AND METHODS
Cell line and cell culture
A549 cells, obtained from ATCC (19, 20), are a human lung cell line originally established from a human non-small cell lung cancer (adenocarcinoma) that demonstrate some properties similar to type II alveolar epithelial cells. A549 cells were maintained in RPMI 1640 medium supplemented with 2 mM L-glutamine, adjusted to contain 1.5 g/l sodium bicarbonate and 10% fetal bovine serum. Cells were seeded and cultured until 80% confluence and serum starved overnight before experiments were conducted.
Reagents
The chemical inhibitor U0126 was purchased from Promega (Madison, WI). PD98059 was purchased from Axxora (San Diego, CA). All other chemicals were from Sigma (St. Louis, MO). Antibodies specific to ERK 1/2 and NF-κB were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies specific to phospho-ERK 1/2 was purchased from Cell Signaling Technology (Beverly, MA). Anti-GAPDH antibody was purchased from Novus Biologicals (Littleton, CO).
In vitro TS exposure
Research-grade 2R1 cigarettes without filters were obtained from the University of Kentucky Tobacco Research and Development Center (Lexington, KY). Cells were grown in either 12.5-cm2 (for transfections) or 25-cm2 (for protein and RNA isolation) Falcon flasks (Becton Dickinson, Franklin Lakes, NJ) to 80% confluence, serum starved for 16 h, and then exposed to mainstream TS as described previously (18). Briefly, TS (30 ml) was drawn into a syringe-driven device fitted with a tube. TS was delivered via the tube into cell culture flasks that were inverted to expose cells directly to smoke for 3 min. The flasks were restored to their original orientation, so that the cells were covered with culture medium without serum, and the flasks were placed in the 37°C incubator with 5% CO2. Cells were harvested after a 1.5-h TS-treatment period and 3-h recovery period followed by extraction of nuclear proteins.
Nuclear protein extraction
Nuclear protein extraction from cells were prepared by using the NE-PER nuclear and cytoplasmic extraction kit (Pierce Biotechnology, Rockford, IL) according to the user’s manual. Briefly, cells were washed twice with cold phosphate-buffered saline (PBS) and cell pellets were collected by centrifugation at 500 g for 3 min. Ice-cold reagent cytoplasmic extract reagent I (CERI; 200 μl) was added to the pellets and vigorously vortexed for 15 s. After incubation on ice for 10 min, 11 μl of ice-cold CERII was added into the traction and incubated on ice for 1 min. After centrifugation at >16,000 g for 5 min, the supernatant was collected as the cytoplasmic fraction. The insoluble pellet, which contained nuclei, was resuspended in 100 μl ice-cold NER and votexed for 15 s, incubated on ice with repeat votexing every 5 min, for a total of 40 min. After 40 min, the reaction was centrifuged at ~16,000 g, and the supernatants were collected as the nuclear protein extraction.
Protein/DNA array screening
Protein/DNA array were carried out with a TranSignal protein/DNA array kit (Panomics, Redwood City, CA) according to the user’s manual. Briefly, 25 μg of nuclear extract was incubated with biotin-labeled DNA probes mix in binding buffer for 30 min at 15°C. Protein/DNA complexes were loaded on 2% agarose gel and electrophoresed at 120 V in 0.5× Tris-buffered saline (TBS) for 20 min. The DNA probes were recovered from the excised gel and denatured at 95°C for 3 min before being hybridized to array membrane at 42°C overnight. The membrane was then washed twice in 2× SSC/0.5% SDS at 42°C for 20 min and twice in 0.1× SSC/0.5% SDS at 42°C for 20 min. The probes were detected with streptavidin-horseradish peroxidase (HRP) conjugate and visualized with SuperSignal chemiluminescence reagent (Pierce). The results were subjected to quantitative analysis, and those TFs with twofold changes compared with that of control were identified as candidates for confirmation by EMSA.
EMSA
Cells were exposed to TS, nuclear extracts were prepared, and EMSA were performed using biotin-labeled double-stranded consensus probes. The probes sequences were as follows: GATA, CACTTGATAACAGAAAGTGATAACTCT; NF-κB, AGTTGAGGGGACTTTCCCAGGC; PAX5, GAATGGGGCACTGAGGCGTGACCACCG; Smad 3/4, TCGAGAGCCAGACAAAAAGCCAGACATTTAGCCAGACAC; ISRE, CAGTTTCACTTTCCC; ICSBP, TGAGGAAACGAAACCATGAGGAAACGAAACCA; RSRFC4, GGTCTATTTATAGCTT; VDR, AGCTTCAGGTCAAGGAGGTCAGAGAGCT.
Detection of protein/oligonucleotide complex was performed with an EMSA gel-shift kit (Pamonics, CA). Briefly, nuclear protein (5 μg) was incubated with 10 ng/μl of biotin-labeled oligonucleotide for 30 min at room temperature in 5× binding buffer and 100 ng of poly(dI-dC). The specificity of the DNA/protein binding was determined in competition reactions in which a 50-fold molar excess of unlabeled oligonucleotide was added to the binding reaction. Products of binding reactions were resolved by electrophoresis on a 6% polyacrylamide gel by using 0.5× TBE buffer following by electrobloting to a nylon membrane (Schleicher and Schuell, Keene, NH). After incubation in blocking buffer for 15 min at room temperature, the membrane was incubated with streptavidin-HRP conjugate for 30 min at room temperature. The membrane was washed and visualized with SuperSignal chemiluminescence reagent (Pierce).
Immunoblot and immunofluorescence analysis
Cells were exposed to TS for various time points, washed three times with chilled PBS containing 1 mM Na3VO4, and then lysed in an MAPK lysis buffer, and the protein concentrations were determined by the method of Lowry using the Bio-Rad DC assay (Bio-Rad, Hercules, CA). Equal protein amounts (40 μg) were resolved by 12% discontinous SDS-PAGE and blotted onto polyvinylidene difluoride membranes. After being blocked with 5% nonfat milk in TBS-Tween at room temperature for 1 h, the membranes were incubated with a polyclonal antibody against NF-κB (Santa Cruz Biotechnology). Membranes were washed three times with TBS-Tween before being probed with HRP-conjugated secondary antibodies for 1 h at room temperature. Blots were then visualized with SuperSignal chemiluminescence reagent (Pierce). For internal normalization of protein loading, membranes were stripped and probed with anti-GAPDH antibodies.
For immunofluorescence analysis, A549 cells were seeded and cultured on coverslips in a T25 flask. Cells, serum-starved overnight, were either directly exposed to 5 ml of freshly generated smoke for 30 min or pretreated with 10 μM U0126 and 20 μM PD98059 for 2 h and then exposed to 5 ml of freshly generated smoke for 30 min. Cells were washed with PBS, fixed with −20°C methanol for 10 min, washed in PBS (3 × 5 min), and blocked with 10% normal goat serum-1% BSA in PBS for 1 h at room temperature. After one wash with PBS, cells were incubated with primary antibody to the p65 or p50 subunit of NF-κB (Santa Cruz) overnight at 4°C. The coverslips were then washed and incubated with fluorescently labeled secondary antibody diluted in blocking solution for 45 min at room temperature. The nuclei were stained with DAPI. The coverslips were washed, mounted, and observed under the microscope. Cellular response to TS exposure was very similar to all treated/exposed cells in at least 15 fields examined that included at least 50 cells per field.
NF-κB binding ELISA
Activation of NF-κB p50- or p65-dependent DNA binding activity was quantified with an ELISA-based detection method as described previously (14). Briefly, nuclear protein extraction from cells without or with treatments were applied to wells of streptavidin-coated 96-well plates containing an immobilized duplex of biotinylated-consensus sequences (2 pmol of probe/well) for NF-κB (AGTTGAGGGGACTTTCCCAGGC) and its mutant form (AGTTGAGGCCACTTTCCCAGGC) and incubated for an hour with mild agitation. The captured active NF-κB bound to the biotinylated-consensus sequence was then incubated with specific primary antibody (p50 or p65) and followed with a secondary HRP-conjugated antibody. All microwells between experimental steps were washed four times with 200 μl PBS + 0.1% Tween-20. Ultra-3,3′,5,5′-tetramethylbenzadine (TMB) substrate (100 μl; Pierce Biotechnology) was then added to the wells for 15 min at room temperature before 50 μl of 2N H2SO4 stopping solution was added. Optical density was then read at 450 nm, via a Packard microplate reader (Perkin-Elmer, Waltham, MA). The results were expressed after subtraction of the blank values. The blanks were realized following the same procedure as the samples, except that lysis buffer was incubated in the microwells instead of cell lysate. The specificity of NF-κB DNA binding was confirmed by competitions with wild-type and mutant κB binding sequences.
Plasmids and luciferase reporter assays
Dominant negative ERK 1 (Lys71 → Arg) and ERK 2 (Lys52 → Arg) mutants each cloned in pCEP4 vector were generously provided by Dr. Melanie Cobb and had been characterized previously (15). NF-κB-pGL3 luciferase construct was a kind gift from Dr. Takashi Fujita (University of Tokyo, Japan). DNA transfections were performed by use of FuGENE 6 reagent according to the manufacturer’s protocol (Roche Biochemical, Indianapolis, IN). Cells were transfected with 100 ng of promoter luciferase constructs. After 36-h expression, the cells were directly exposed to 3 ml per T12.5 flask of freshly generated smoke for 4 h. Cells were then washed with PBS and cell lyses were collected for luciferase activity measurement using a steady-glow luciferase reagent (Promega, Madison, WI). Luciferase activity of individual samples was normalized to that of β-galactosidase as described previously. All assay samples were performed in triplicate, and each experiment was repeated at least three times.
Stably transfected dominant negative cell lines
Dominant negative (dn) ERK mutant cell lines (dnERK 1-A549, dnERK 2-A549, and control expression vectors pCEPT4-A549) were established by selecting stably transfected (with dnERK 1, dnERK 2, and pCEPT4 expression constructs) in A549 cells using hygromycin. Cells lines were first identified by the overexpression of 6× histidine-tagged proteins, representative clones for dn-ERK 1-A549 and dnERK 2-A549 were selected, and the functional properties of these cell lines were confirmed when phosphorylated ERK 1 or ERK 2 was significantly decreased in PMA-induced ERK 1/2 phosphorylation compared with pCEPT4-A549 cells (supplemental data).1
Statistical methods
Data are expressed as the means ± SE of at least three independent experiments. The statistical significance of the differences between groups was determined by Student’s t-test. A P value of <0.05 was considered significant.
RESULTS
Identification of multiple transcription factors regulated by TS in A549 cells
We proposed that TS exposure will affect the binding and regulation of multiple TFs, which subsequently modulate changes in gene expression. To identify which TFs in lung epithelial cells are involved in TS-induced gene activation, we isolated nuclear protein from A549 cells (see MATERIALS AND METHODS) treated with or without TS exposure and applied them to protein/DNA arrays. Three separate protein/DNA arrays, which identified 244 unique DNA binding proteins, were utilized (54 TFs for array I, 94 TFs for array II, and 96 TFs for array III). Optimal treatment conditions to elicit TS-induced phosphorylation of MAPK signaling pathways, increased transcriptional activity, and subsequent gene expression had been determined prior to this study (data not shown). By comparing untreated and TS-treated cells, we identified 20 TFs that demonstrated greater than a twofold change in their nuclear protein/DNA binding activity (Fig. 1). As shown, not all TFs were detectable via this protein/DNA array system. In addition, the binding activities of a significant number of TFs in A549 cells were unchanged after TS exposure (Fig. 1). The measured changes in binding intensity and the identities of these TFs are summarized in Table 1.
Fig. 1.

Identification of multiple transcriptional factors regulated by tobacco smoke (TS) in A549 cells using protein/DNA array. Overnight serum-starved A549 cells in a T25 flask were directly exposed to 5 ml of freshly generated TS for 1.5 h followed by recovery in fresh medium for 3 h, and nuclear proteins were extracted for protein/DNA array as described in MATERIALS AND METHODS. The representative transcription factors (TFs) regulated by TS were outlined with black boxes and subject to quantitative analysis. These results are representative of 3 independent experiments.
Table 1.
Identification of multiple transcriptional factors regulated by tobacco smoke in A549 cells using protein-DNA arrays I, II and III
| TF Name | Regulation by Smoke | Ratio |
|---|---|---|
| GATA | Increase | * |
| GRE | Increase | * |
| TR(DR4) | Increase | 7.2 |
| NF-κB | Increase | * |
| VDR(DR3) | Increase | 12.1 |
| RXR(DR1) | Increase | 6.9 |
| PAX5 | Increase | 8.8 |
| Stat 4 | Increase | 8.4 |
| c-Myb | Increase | 12.2 |
| NF1 | Increase | 4.7 |
| Smad 3/4 | Increase | 7.2 |
| TFIID | Increase | * |
| AP-2 | Increase | 8.1 |
| ISRE(TransTF) | Decrease | 0.24 |
| Antiox. RE | Decrease | 0.32 |
| HBS/HAS | Decrease | 0.2 |
| RSRFC4 | Increase | 6.5 |
| Skn | Increase | 4.8 |
| ICSBP | Decrease | 0.3 |
| WT1 | Increase | 8.9 |
Ratio values are given compared with control group.
Ratio can not be calculated since signal of the control group was below detectable limits
Confirmation of transcription factors regulated by TS in A549 cells by EMSA analysis
On the basis of the semiquantitative results from the protein/DNA arrays (Table 1), four TS-induced TFs (GATA, NF-κB, PAX5, and Smad 3/4) and two TS-suppressed TFs (ISRE and ICSBP) were selected for further confirmation by EMSA analysis. Parallel experiments in A549 cells were performed to determine whether EMSA experiments confirmed our protein/DNA array data. The binding activities of nuclear proteins GATA, NF-κB, PAX5, and Smad 3/4 to their corresponding consensus DNA sequences were significantly increased in cells treated with TS compared with nuclear protein isolated from untreated cells (Fig. 2, A–D). Similarly, the binding activities of ISRE and ICSBP to their corresponding consensus DNA sequences were significantly decreased after TS treatment compared with untreated controls (Fig. 2, E–F). Intensity of the shifted bands for each examined TF were significantly or completely reduced by their respective cold probes (lane 4 of each panel of Fig. 2), which confirmed TF specificity. Also, no shifted bands were detected in the probe-only lane for all experiments (lane 1 of each panel of Fig. 2). These results were consistent with the results obtained from the protein/DNA arrays and further validated that TS exposure altered the DNA binding activities of multiple TFs.
Fig. 2.

Confirmation of DNA binding activity of TS-regulated transcriptional factors using EMSA. Cells were treated with TS and nuclear proteins were extracted and incubated with biotin-labeled probes corresponding to different TF binding sequences for EMSA as described in MATERIALS AND METHODS. A: GATA. B: NF-κB. C: PAX5. D: Smad 3/4. E: ISRF. F: ICSBP. Lane 1 of each panel indicates free probes. DNA binding activity is represented without (lane 2) and with (lane 3) TS exposure in each panel. Lane 4 of each panel represents binding activity after TS exposure but with 20× excess of cold probes added to the incubation. These results are representative of 3 independent experiments. SMK, smoke.
Screening and identification of TS-regulated transcription factors regulated by MAPK signaling pathways
It is known that ERK 1/2 signaling pathways modulate TS-induced gene expression at the transcriptional level (6, 19). To understand whether ERK 1 and ERK 2 regulate TS-induced gene expression through TF-DNA binding activity changes, A549 cells were pretreated for 2 h with pharmacological inhibitors for ERK 1/2 (PD98059 and U0126) before cells were directly exposed to TS. After TS exposure, cells were harvested for nuclear protein isolation as above. Pretreatment with either PD98059 or U0126 inhibitor did not noticeably affect basal TF-DNA binding activities in cells prior to smoke treatment (data not shown). However, the DNA binding activities of several TS-induced TFs were significantly inhibited by pre-treatment with ERK 1/2 inhibitors (Fig. 3). Although either PD98059 or U0126 similarly affected TS-induced TF binding activities, U0126 at the suggested inhibitory concentration (IC50 = 5 μM) appeared to be more effective than PD98059 (IC50 = 10 μM) in supressing TS-induced DNA binding activities.
Fig. 3.

DNA binding activity of several TS-regulated TFs are susceptible to ERK 1/2 inhibition. A549 cells in a T25 flask were treated with TS as previously described and pretreated without inhibitor (A), with 10 μM U0126 (B), and with 20 μM PD98059 (C) for 2 h. The comparable signal intensity and contrast of the membranes were adjusted according to the alignment spots along the right and bottom edges, respectively. The TS-regulated TFs putatively mediated by ERK 1/2 were outlined with black boxes as indicated in D. These results are representative of 3 independent experiments.
Examination of ERK 1/2-mediated DNA binding activities of TS-regulated TFs by EMSA in A549 cells
To further investigate the effects of ERK 1/2 pathways on the DNA binding activities of TS-regulated TFs, two additional TS-upregulated TFs (VDR and RSRFC4) and two TS-downregulated TFs (ICSBP and ISRE) were selected for confirmatory studies using EMSA. A549 cells were treated with smoke and harvested for nuclear protein as described above. DNA binding activity of VDR or RCRFC4 was partially or completely suppressed when cells were pretreated with the MAPK inhibitors PD98059 and U0126 prior to TS exposure consistent with data from the protein/DNA arrays (Fig. 4, A and B). Similarly, DNA binding activities of ICSBP and ISRE decreased after TS treatment. Of potential importance, pretreatment of cells with U0126 or PD98059 suppressed basal (data not shown) and additionally suppressed TS-induced decreases (Fig. 4, C and D) in ICSBP and ISRE DNA binding activities.
Fig. 4.

Effects of ERK 1/2 inhibitors on TS-regulated DNA binding activity in A549 cells. Cells were treated with and without TS and nuclear proteins were extracted and incubated with biotin-labeled probes corresponding to different TF binding sequences for EMSA as described in MATERIALS AND METHODS. A: VDR. B: RSRFC4. C: ISCSP. D: ISRE. Lane 1 of each panel indicates free probes. DNA binding activity is represented without (lane 2) and with (lane 3) TS exposure in each panel. Lane 4 of each panel represents binding activity after TS exposure but with 20× excess of cold probes added to the incubation. Lane 5 (U0126) and lane 6 (PD98059) are TS-exposed binding in the presence of ERK 1/2 inhibitors. These results are representative of 3 independent experiments. Fp, free probe.
TS-induced nuclear translocation of NF-κB is regulated by MAPK-mediated IκB phosphorylation
Because NF-κB modulates the expression of several important proinflammatory cytokines, we focused our attention on the mechanisms of TS-induced NF-κB activation in A549 cells. This included a detailed analysis of TS-induced IκB phosphorylation and NF-κB nuclear translocation. As shown in Fig. 5, peak phosphorylation of IκB was observed 10 min after TS exposure and steadily decreased at later time points (Fig. 5A). Nuclear p65 protein, p65(NP), increased immediately following a 10-min TS exposure and steadily increased at subsequent time points. In comparison, there was no significant change seen in cytosolic p65 protein, p65(CP), expression during TS exposure. Nuclear translocation of p50 similarly increased after 10 min of TS exposure. However, p50 translocation was variable after the initial 10-min time point.
Fig. 5.

TS-induced nuclear translocation of NF-κB and phosphorylation of IκB are mediated through ERK 1/2 signaling pathways. Overnight serum-starved A549 cells in a T25 flask were either directly exposed to 5 ml of freshly generated smoke for different time points as indicated (A) or pretreated with 10 μM U0126 and 20 μM PD98059 for 2 h followed by exposure to 5 ml of freshly generated smoke for 10 min (B). The membranes were probed with antibodies specific for phosphorylated IκB (pIκB), p65, p50, and GAPDH and developed with a chemiluminescence imaging system as described in MATERIALS AND METHODS. GAPDH intensity was used as internal control to normalize the sample loading. These results are representative of 3 independent experiments.
Because inhibition of the MAPK pathway affected NF-κB binding activity in our initial array (Fig. 3), we further examined whether MAPK was involved in TS-induced IκB phosphorylation and nuclear translocation of NF-κB protein. As shown in Fig. 5B, pretreatment of A549 cells with the MEK1/ERK inhibitors U0126 or PD98059 had no effect on NF-κB signaling components in untreated cells. However, peak TS-induced IκB phosphorylation (10 min) was significantly suppressed after pretreatment with U0126 or PD98059 (Fig. 5B). Similarly, pretreatment of A549 cells with U0126 or PD98059 inhibited peak TS-induced nuclear translocation of p65/p50 (Fig. 5B). Pretreatment of A549 cells with U0126 or PD98059 did not have significant effects on basal IκB phosphorylation or nuclear translocation of p65/p50 in untreated cells (Fig. 5B), which excluded nonspecific toxic effects due to the inhibitors.
TS-induced nuclear translocation of p65 and p50 was further confirmed by immunofluorescent microscopy. The p65/50 was normally expressed in the cytoplasm of untreated cells. After TS exposure, intense nuclear staining of both p65 and p50 was observed, with a concomitant decrease in cytoplasmic staining of each protein (Fig. 6). These data strongly support the notion that TS induced nuclear translocation of NF-κB protein. The observed nuclear translocation of NF-κB (both p65 and p50) was inhibited when cells were pretreated with the MEK1/ERK inhibitors U0126 or PD98059 (Fig. 6). These data were consistent with our Western blot analyses (Fig. 5B).
Fig. 6.
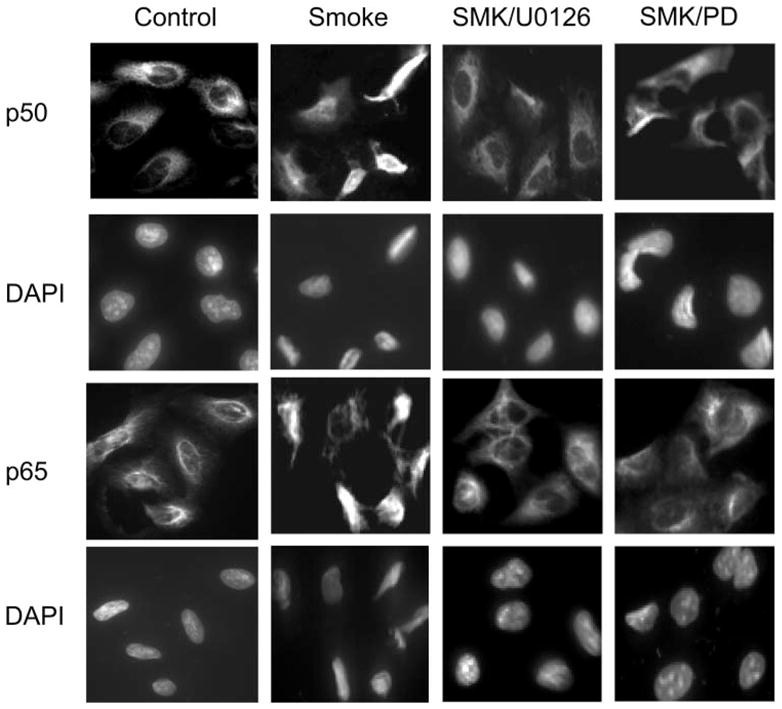
U0126 and PD98059 blocked TS-induced nuclear translocation of NF-κB p50 and p65 subunits. A549 cells (1 × 106/ml) in a T25 flask were treated without or with 10 μM U0126 or 20 μM PD98059 for 2 h at 37°C and then exposed to 5 ml of freshly generated smoke for 10 min. Immunofluorescence analysis was performed as described in MATERIALS AND METHODS. Top 2 rows show cells stained with primary anti-p50 antibody and Alexa Fluor-conjugated second antibody. Bottom 2 rows show cells stained with primary anti-p65 antibody and Alexa Fluor-conjugated second antibody. The TS-induced nuclear translocation of both p50 and p65 proteins were inhibited by either U0126 or PD98059 (PD) (right 2 columns). Nuclei were indicated by DAPI staining.
TS-induced NF-κB DNA binding activity is mediated through MAPK signaling pathways in A549 cells
To further examine whether translocated NF-κB actually resulted in increased functional activity, we performed EMSA to detect changes in NF-κB DNA binding activity after TS exposure. When A549 cells were exposed to TS, NF-κB DNA binding activity was significantly enhanced (Fig. 7A). This activity was significantly suppressed when cells were treated with the MEK1/ERK inhibitors PD98059 or U0126 prior to smoke exposure (Fig. 7A). No effect on basal NF-κB DNA binding was observed with the chemical inhibitors. To exclude the possibility that these observations were due to nonspecific effects of the chemical inhibitors, we also employed a dominant-negative approach. Using cell lines that stably express dominant negative (dn) ERK 1 or ERK 2 (A549-dnERK 1 and A549-dnERK 2), we observed similar inhibition of NF-κB DNA binding activity after TS exposure compared with vector-alone transfected cells (A549-pCEPT4) (Fig. 7B). No effect on basal NF-κB DNA binding was observed with either the chemical inhibitors or the dominant-negative expression vectors. These results strongly suggested that ERK 1 and ERK 2 pathways regulate TS-induced NF-κB DNA binding activity.
Fig. 7.

Phosphorylation of ERK 1- and ERK 2-mediated DNA binding activity of NF-κB. A: serum-starved A549 cells in a T25 flasks pretreated without or with 20 μM PD98059 or 10 μM U0126 for 2 h and then exposed to 3 ml of freshly generated smoke for 1.5 h. B: stably transfected control and dominant negative cells (A549-pCEPT4, A549-dnERK 1, and A549-dnERK 2) were treated without or with TS exposure of 3 ml of freshly generated smoke for 1.5 h as described in MATERIALS AND METHODS. Extracted nuclear proteins were incubated with biotin-labeled probes corresponding to NF-κB binding sequences for EMSA.
To further confirm the increased DNA binding activity of NF-κB was specific for p65 and p50 subunits, we employed an ELISA-based binding activity assay. The specific DNA binding activity of NF-κB consensus sequences to both p65 and p50 increased after TS exposure (Fig. 8, A and B). TS-induced NF-κB DNA binding activity was greatly reduced when an excess of nonbiotinylated probe [5× (5 pmol) or 20× (20 pmol)] containing wild-type NF-κB consensus sequence was added to the incubated cell lysate. However, nonbiotinylated probe containing the mutated NF-κB consensus sequence did not affect basal NF-κB binding activity. TS-induced increases in p65 or p50 NF-κB binding activity was supressed when ERK 1/2 inhibitors were added to cells prior to TS exposure (Fig. 8, A and B).
Fig. 8.

Decrease of TS-induced NF-κB binding and promoter activity by ERK 1/2 inhibitors. A and B: extracted nuclear proteins without or with TS exposure were added into 96-well plates that had prebound NF-κB duplex DNA. Microwells bearing the biotinylated NF-κB DNA probe (1 pmol) were incubated with an increasing excess of the nonbiotinylated probe, containing either the wild-type (wt) or the mutated NF-κB-binding consensus sequence. Specific DNA binding activity for either p65 (A) or p50 (B) was determined by measured activity after incubation with respective primary antibody against p65 or p50. C: A549 cells were transiently transfected with a NF-κB-containing plasmid linked to the pGL3 basic vector and then treated without or with ERK 1/2 inhibitors. After TS exposure, cell supernatants were collected and assayed for luciferase activity as described in MATERIALS AND METHODS. Triplicate samples were harvested from each condition, and results are expressed as relative fold increase in luciferase activity compared with empty vector control. *P < 0.05, compared with non-smoke-treated basal activity; **P < 0.05, compared with TS-treated groups.
Transient transfection experiments with pGL3-NF-κB promoter-luciferase reporter constructs further supported our observations. A549 cells transiently transfected with pGL3-NF-κB plasmids resulted in a 20-fold increase in luciferase activity compared with cells transfected with the pGL3 empty basal vector. TS exposure enhanced luciferase activity of pGL3-NF-κB 35-fold higher than the pGL3 empty basal vector (Fig. 8C). This 1.7-fold increase in TS-induced NF-κB promoter activity paralleled the increase in nuclear translocation of NF-κB protein as shown by Western blots (Fig. 5). The ERK 1/2 inhibitors U0126 and PD98059 both abolished TS-induced NF-κB promoter activity.
DISCUSSION
TS has become a serious public health concern for several decades. Because the respiratory system, and in particular the respiratory epithelium, is in direct contact with the environment, the respiratory system is the most important target for TS exposure. Tremendous research efforts have been focused on better understanding the complex mechanisms responsible for TS-related lung diseases. In this study, we proposed that a global TF expression profile will provide mechanistic understanding of how TS will alter transcriptional activation of gene promoters and subsequent gene expression. We have recognized in our preliminary studies that a 3-min gas-phase TS exposure followed by a 90-min incubation in smoke-conditioned medium is sufficient to increase transcriptional activity of several airway secretory genes including SPLUNC1, MUC5AC, and MUC5B (unpublished observations). We adapted this TS exposure paradigm to examine TS-regulated DNA binding activity of different TFs to their consensus sequences. We identified multiple important TFs that were regulated by TS exposure in A549 lung carcinoma cells and discovered that several of these TS-regulated TFs, including NF-κB, were regulated through the ERK 1/2 signaling pathway.
Through a variety of genomic approaches, scientists can now identify how alterations in a single TF will modulate global gene expression (9). Also, pioneering work with individual TFs such as AP-1 and NF-κB demonstrated that these TFs were modulated after treatment with cigarette smoke condensate (1, 5, 6, 16, 17). However, there are more than 1,000 TFs. We suggest that an approach that focuses on a few TFs at a time will probably provide limited understanding of how TF activation can lead to human disease. The protein/DNA array technology applied in this study allowed us to simultaneously detect 244 different TFs. Most of these TFs have been previously demonstrated to be important for regulating gene expression (8). Using this approach, we were able to identify a manageable number of TS-regulated TFs, including TFs important for tumorigenesis and cell cycle regulation. To the best of our knowledge, this is the first report to screen and identify multiple TS-regulated TFs simultaneously. Our results provided novel information that could lead to better understanding of TS-related disease development and treatment.
In addition to the type of TFs that are induced by TS, we were able to identify a common mechanism responsible for increased TF activation. TFs can be activated through transcriptional, translational, or posttranslational mechanisms (2). For these studies, we focused on protein phosphorylation, which is likely the most common mechanism underlying TF activation after environmental stimulation. Results from our laboratory and others showed that ERK 1/2 signaling pathways are activated upon TS treatment (6, 11, 21). To further understand whether the MAPK/ERK pathway was also involved in TS-induced TF activation, we combined protein/DNA arrays with EMSAs to confirm that TS-regulated TFs were mediated by MAPK signaling cascades.
U0126 and PD98059 are MAPK inhibitors, and their major activity is to block the MAPK phosphorylation process. On the basis of our results from Western-blot analyses and EMSAs, translocation of NF-κB was significantly inhibited after pre-treating cells with U0126 or PD98059. Pretreatment of cells with inhibitors alone did not elicit significant effects on basal level expressions of IκB phosphorylation or p65/p50 activity (Fig. 5). This indicated that TS-induced translocation of NF-κB is a transient event dependent on ERK 1/2 activation. The distribution of NF-κB between the cytoplasm and the nucleus of quiescent cells is relatively balanced, suggesting that resting cells have minimal activation of the NF-κB pathway. Consistent with this notion, inhibitory effects on IκB phosphorylation and p65/p50 translocation in resting cells were not observed. However, inhibition of MAPK signaling pathways in TS-treated cells resulted in significant modulation of IκB phosphorylation, NF-κB nuclear translocation, and NF-κB DNA binding activity. Our results suggest that the MAPK signaling pathways are essential in mediating TS-induced NF-κB activation and subsequent inflammatory gene expression.
It was reported that celecoxib, a cyclooxygenase-2 inhibitor, inhibited cigarette smoke condensate-induced nuclear translocation of p65 while blocking the decrease of cytosolic p65 in human non-small cell lung carcinoma H1299 cells (16). However, we observed substantial translocation of both p65 and p50 into the nucleus upon TS exposure but no significant decrease in cytoplasmic protein expression. It may be due to the high abundance of p65 and p50 protein in quiescent A549 cells, with only a small portion of protein translocated into the nucleus after TS treatment.
Of potential importance, VDR was activated upon TS exposure. VDR has previously been associated with decreased serum levels of the active form of vitamin D and an increased risk for certain cancers (7, 20). In comparison, two interferon-related TFs, ICSBP and ISRE, were shown to decrease their DNA binding activities after TS exposure. On the basis of these findings, the roles of ICSBP and ISRE in TS-induced airway inflammation merit further investigation.
To further establish the biological relevance of our results, 1-kb promoter sequences proximal to the transcriptional initiation site of two TS-responsive genes were analyzed using a bioinformatic approach. On the basis of a literature search, we chose one gene that is reported to be increased by TS exposure in airway epithelium (IL-8) and one gene that is suppressed by TS exposure (eotaxin) (10, 13). The results from our bioinformatics approach were combined with our array results (Table 1) to develop a schematic representation of the predicted TS-regulated TF binding sites present on IL-8 and eotaxin (Fig. 9). A twofold excess of potential TS-regulated TF binding sites were present on the proximal 1-kb promoter of the TS-inducible gene IL-8 compared with the TS-suppressed gene eotaxin. In addition, the proximal 1-kb promoter of eotaxin contains two putative TF binding sites for ICSBP. This TF was suppressed by TS in our studies. These data support our proposition that the array approach provides an efficient means of obtaining a global understanding of TS-induced gene regulation.
Fig. 9.

Schematic representation of putative TS-regulated TF binding sites in TS-responsive cytokines. The TS-induced gene IL-8 contains increased numbers of putative TS-induced TF binding sites compared with the TS-suppressed gene eotaxin within the proximal 1-kb promoter region. In addition, eotaxin contains two TS-suppressed TF binding sites for ICSBP that do not exist in IL-8.
We have shown that high-throughput protein/DNA array technology is a reliable and valid method to identify several important TFs that are modulated by TS. Through these data, we identified a novel NF-κB activation mechanism via ERK 1/ERK 2 MAPK signaling pathways. We also provided an example of how our array data potentially will provide a broader understanding of how multiple genes, such as IL-8 and eotaxin, are differentially regulated by the same stimulus. We suggest that similar studies will be essential to better understand the complex biological signaling networks that occur in response to a particular exposure or treatment.
Acknowledgments
GRANTS
This work was supported in part by grants to Y. P. Peter Di from the National Institute of Environmental Health Sciences (ES-011033), American Heart Association-Pennsylvania Delaware Affiliate (0365327U), Tobacco-Related Disease Research Program (10KT-0261), and Competitive Medical Research Fund (University of Pittsburgh Medical Center) Research Award.
Glossary
- Antiox. RE (ARE)
Antioxidant responsive element
- AP-1 and -2
Activator proteins 1 and 2
- c-Myb
v-myb myeloblastosis viral oncogene homolog
- GATA
GATA binding protein (globin transcription factor)
- GRE
Glucocorticord receptor responsive element
- HBS/HAS (HIF-1)
Hypoxia-inducible factor-1
- ISRE
Interferon-stimulated responsive element
- ICSBP
Interferon consensus sequence-binding protein
- NF-1
Nuclear factor I
- NF-κB
Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1
- PAX5
Paired box gene 5 (B-cell lineage specific activator protein)
- RXR(DR-1)
Retinoid X receptor
- RSRFC4
Related to serum response factor protein C4
- Skn (HHAT)
Hedgehog acyltransferase
- Smad 3/4
MAD, mothers against decapentaplegic homolog 3/4
- Stat4
Signal transducer and activator of transcription 4
- TFIID
Transcription initiation factor IID
- TR
Thyroid hormone receptor
- VDR
Vitamin D receptor
- WT1
Wilms tumor 1
Footnotes
Supplemental data for this article is available online at the American Journal of Physiology Lung Cellular and Molecular Physiology website.
References
- 1.Adcock IM, Ito K, Barnes PJ. Glucocorticoids: effects on gene transcription. Proc Am Thorac Soc. 2004;1:247–254. doi: 10.1513/pats.200402-001MS. [DOI] [PubMed] [Google Scholar]
- 2.Brivanlou AH, Darnell JE., Jr Signal transduction and the control of gene expression. Science. 2002;295:813–818. doi: 10.1126/science.1066355. [DOI] [PubMed] [Google Scholar]
- 3.Campbell IL. Cytokine-mediated inflammation, tumorigenesis, and disease-associated JAK/STAT/SOCS signaling circuits in the CNS. Brain Res. 2005;48:166–177. doi: 10.1016/j.brainresrev.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Ditsworth D, Zong WX. NF-kappaB: key mediator of inflammation-associated cancer. Cancer Biol Ther. 2004;3:1214–1216. doi: 10.4161/cbt.3.12.1391. [DOI] [PubMed] [Google Scholar]
- 5.Du B, Altorki NK, Kopelovich L, Subbaramaiah K, Dannenberg AJ. Tobacco smoke stimulates the transcription of amphiregulin in human oral epithelial cells: evidence of a cyclic AMP-responsive element binding protein-dependent mechanism. Cancer Res. 2005;65:5982–5988. doi: 10.1158/0008-5472.CAN-05-0628. [DOI] [PubMed] [Google Scholar]
- 6.Gensch E, Gallup M, Sucher A, Li D, Gebremichael A, Lemjabbar H, Mengistab A, Dasari V, Hotchkiss J, Harkema J, Basbaum C. Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP-1 and JNK. J Biol Chem. 2004;279:39085–39093. doi: 10.1074/jbc.M406866200. [DOI] [PubMed] [Google Scholar]
- 7.Gong YL, Xie DW, Deng ZL, Bostick RM, Miao XJ, Zhang JH, Gong ZH. Vitamin D receptor gene Tru9I polymorphism and risk for incidental sporadic colorectal adenomas. World J Gastroenterol. 2005;11:4794–4799. doi: 10.3748/wjg.v11.i31.4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang X, Norman M, Roth L, Li X. Protein-DNA array-based identification of transcription factor activities regulated by interaction with the glucocorticoid receptor. J Biol Chem. 2004;279:38480–38485. doi: 10.1074/jbc.M403948200. [DOI] [PubMed] [Google Scholar]
- 9.Kirmizis A, Farnham PJ. Genomic approaches that aid in the identification of transcription factor target genes. Exp Biol Med (Maywood) 2004;229:705–721. doi: 10.1177/153537020422900803. [DOI] [PubMed] [Google Scholar]
- 10.Kubo S, Kobayashi M, Masunaga Y, Ishii H, Hirano Y, Takahashi K, Shimizu Y. Cytokine and chemokine expression in cigarette smoke-induced lung injury in guinea pigs. Eur Respir J. 2005;26:993–1001. doi: 10.1183/09031936.05.00042405. [DOI] [PubMed] [Google Scholar]
- 11.Mercer BA, Kolesnikova N, Sonett J, D’Armiento J. Extracellular regulated kinase/mitogen activated protein kinase is up-regulated in pulmonary emphysema and mediates matrix metalloproteinase-1 induction by cigarette smoke. J Biol Chem. 2004;279:17690–17696. doi: 10.1074/jbc.M313842200. [DOI] [PubMed] [Google Scholar]
- 12.Mossman BT, Lounsbury KM, Reddy SP. Oxidants and signaling by mitogen-activated protein kinases in lung epithelium. Am J Respir Cell Mol Biol. 2006;34:666–669. doi: 10.1165/rcmb.2006-0047SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oltmanns U, Chung KF, Walters M, John M, Mitchell JA. Cigarette smoke induces IL-8, but inhibits eotaxin and RANTES release from airway smooth muscle. Respir Res. 2005;6:74. doi: 10.1186/1465-9921-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, Remacle J. Development of a sensitive multi-well colorimetric assay for active NF-kappaB. Nucleic Acids Res. 2001;29:E21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robbins DJ, Zhen E, Owaki H, Vanderbilt CA, Ebert D, Geppert TD, Cobb MH. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J Biol Chem. 1993;268:5097–5106. [PubMed] [Google Scholar]
- 16.Shishodia S, Aggarwal BB. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates activation of cigarette smoke-induced nuclear factor (NF)-kappaB by suppressing activation of IkappaBalpha kinase in human non-small cell lung carcinoma: correlation with suppression of cyclin D1, COX-2, and matrix metalloproteinase-9. Cancer Res. 2004;64:5004–5012. doi: 10.1158/0008-5472.CAN-04-0206. [DOI] [PubMed] [Google Scholar]
- 17.Springer J, Scholz FR, Peiser C, Groneberg DA, Fischer A. SMAD-signaling in chronic obstructive pulmonary disease: transcriptional down-regulation of inhibitory SMAD 6 and 7 by cigarette smoke. Biol Chem. 2004;385:649–653. doi: 10.1515/BC.2004.080. [DOI] [PubMed] [Google Scholar]
- 18.Sun W, Wu R, Last JA. Effects of exposure to environmental tobacco smoke on a human tracheobronchial epithelial cell line. Toxicology. 1995;100:163–174. doi: 10.1016/0300-483x(95)03087-v. [DOI] [PubMed] [Google Scholar]
- 19.Takeyama K, Jung B, Shim JJ, Burgel PR, Dao-Pick T, Ueki IF, Protin U, Kroschel P, Nadel JA. Activation of epidermal growth factor receptors is responsible for mucin synthesis induced by cigarette smoke. Am J Physiol Lung Cell Mol Physiol. 2001;280:L165–L172. doi: 10.1152/ajplung.2001.280.1.L165. [DOI] [PubMed] [Google Scholar]
- 20.Van Schooten FJ, Hirvonen A, Maas LM, De Mol BA, Kleinjans JC, Bell DA, Durrer JD. Putative susceptibility markers of coronary artery disease: association between VDR genotype, smoking, and aromatic DNA adduct levels in human right atrial tissue. FASEB J. 1998;12:1409–1417. doi: 10.1096/fasebj.12.13.1409. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Q, Adiseshaiah P, Reddy SP. Matrix metalloproteinase/epidermal growth factor receptor/mitogen-activated protein kinase signaling regulate fra-1 induction by cigarette smoke in lung epithelial cells. Am J Respir Cell Mol Biol. 2005;32:72–81. doi: 10.1165/rcmb.2004-0198OC. [DOI] [PubMed] [Google Scholar]