

Abstract
Impaired wound healing states lead to substantial morbidity and cost with treatment resulting in an expenditure of billions of dollars per annum in the USA alone. Both chronic wounds and impaired acute wounds are characterized by excessive inflammation, enhanced proteolysis, and reduced matrix deposition. These confounding factors are exacerbated in the elderly, in part, as we report here, related to increased local and systemic tumor necrosis factor alpha(TNFα) levels. Moreover, we have used a secretory leukocyte protease inhibitor(SLPI) null mouse model of severely impaired wound healing and excessive inflammation, comparable to age-related delayed human healing, to demonstrate that topical application of anti-TNFα neutralizing antibodies blunts leukocyte recruitment and NFκB activation, alters the balance between M1 and M2 macrophages, and accelerates wound healing. Following antagonism of TNFα, matrix synthesis is enhanced, associated with suppression of both inflammatory parameters and NFκB binding activity. Our data suggest that inhibiting TNFα is a critical event in reversing the severely impaired healing response associated with the absence of SLPI, and may be applicable to prophylaxis and/or treatment of impaired wound healing states in humans.
Keywords: wound healing, inflammation, SLPI, TNFα, macrophage
INTRODUCTION
Cutaneous wound healing is a complex process encompassing multiple overlapping events following injury, including coagulation, leukocyte recruitment, matrix deposition, epithelialization, and resolution of inflammation with formation of a mature scar. However, this cascade of events may become disrupted, leading to delayed healing of acute wounds or development of chronic nonhealing wounds/ulcers. Both chronic and slowly-healing acute wounds in the elderly are characterized by excessive leukocytosis, augmented cytokines and chemokines, and subsequently, enhanced degradation of matrix constituents(1). Abundant evidence implicates inflammation as a causative factor in delayed healing, and suggests that, in the absence of infection, the inflammatory response may be inappropriately excessive. Not only does failed healing have profound local impact, but intractable inflammation has been linked to multiple local and systemic disorders, from atherosclerosis to cancer(2).
In specific conditions of inflammation-mediated pathophysiology, exemplified by chronic arthritis, TNFα is recognized as a crucial contributing factor(3-5). Impaired healing in animal models and age-related delayed healing of acute human wounds exhibit raised local(6) and systemic levels of TNFα that may parallel the pro-inflammatory phenotype(6, 7). Despite previous studies suggesting that venous ulcer fluid TNFα may impair fibroblast function, and that increased mast cell TNFα is associated with venous ulcers(8, 9), there is a dearth of literature addressing possible contributions of TNFα to chronic wound states in humans. While beneficial in host defense to pathogens, in excess, TNFα is associated with host damaging effects.
We recently described an animal model of markedly delayed healing, resembling chronic wound healing in elderly humans, which have in common augmented leukocyte infiltration and protease activity during initial stages of injury and tissue destruction(10-12). This pathology results from absence of the anti-inflammatory and anti-proteolytic agent secretory leukocyte protease inhibitor(SLPI) in the SLPI null mouse. SLPI is a 12kDa protein inhibitor of serine proteases(13, 14) and has also been shown to have multiple functions relevant to innate host defense, including anti-microbial and anti-inflammatory activity(3, 15-17). A further role for SLPI is suggested by its inhibition of macrophage TNFα production(18) and induction of SLPI expression by TNFα itself in epithelial cells(19). Intriguingly, NFκB activation in vitro and in vivo is inhibited by SLPI(10, 20), suggesting a feedback loop and implicating the TNFα pro-inflammatory pathway as a potentially critical target of the anti-inflammatory actions of SLPI. Nonetheless, evidence also supports a role for TNFα in promoting healing, in part through driving bone morphogenetic protein(BMP)-2 and partial epithelial-mesenchymal transition underlying epithelial motility(21). Thus, it remains unclear whether TNFα represents a necessary mediator or detractor to optimal tissue repair.
In this study we report that TNFα levels, both systemically and in acute wounds, are significantly increased in humans predisposed to impaired healing, and that elevated local tissue levels decline in parallel with healing. Furthermore, using the SLPI null model, we demonstrate that local TNFα inhibition reverses the impaired healing phenotype, otherwise characterized by a sustained population of classically-activated M1 macrophages. In addition, TNFα blockade enhances collagen deposition even in wild-type mice, suggesting that tissue levels of this cytokine, in the absence of infection, may retard wound healing. TNFα inhibition by antibody or exogenous SLPI is also associated with decreased NFκB activity, consistent with interconnectivity between this transcription factor and TNFα-mediated responses. These data suggest a rational approach to accelerate human impaired healing characterized by excessive inflammation.
MATERIALS AND METHODS
Human tissues and serum
For acute wound healing studies, 4mm punch biopsies in upper inner arms of post-menopausal females(with venous ulceration and healthy age-matched) were performed under local anesthesia and wound areas excised at day 7 post-wounding(1). Local Research Ethics Committee approval (Central Manchester LREC01/218; Project 02018, University of Manchester) was obtained and participants provided written consent. Subjects taking HRT or immunoregulatory drugs were excluded. Chronic venous ulcers from elderly(50-90yrs) males and females were biopsied at presentation using 4mm punches at edge of wound. Biopsies at leading edge of healing ulcers were taken 1 month following standard treatment with 4-layer bandaging(1). Tissue was processed in 10% formalin and paraffin-embedded for histopathology.
Serum was obtained from blood of control subjects with varicose veins(VV) but no history of ulceration(i.e., underlying pathology with no impaired healing)(n=38 male, 43 female) and from subjects with a history of chronic venous ulceration(CU)(n=17 male, 25 female) and stored at -80°C. In addition, serum samples were also collected from individuals whose venous ulcers had healed (n = 6 male, 9 female). Levels of serum TNFα were determined by ELISA (R&D Systems, Minneapolis, MN).
Murine wound healing
SLPI deficient mice(SLPI-/-) were generated and maintained under specific pathogen-free conditions(10). Eight to ten-week old male mice were anesthetized, dorsum shaved, cleaned with alcohol, and 4 equidistant 1cm full-thickness incisional wounds were made through skin and panniculus carnosus muscle and left to heal by secondary intention(10). Wounds were harvested at indicated days and bisected for histology, snap-frozen in liquid nitrogen for RNA analysis/protein extraction, or placed in ex-vivo media(BioWhittaker, Walkersville, MD) with 1% fungizone(Cambrex Biosciences, Walkersville, MD) and PenStrep 50u/ml(Gibco, Gaithersburg, MD). For a subset of animals, immediately prior to wounding(day 0), area to be incised was injected subcutaneously with 100μl of 1-10μg rat anti-mouse TNFα(Centocor, Malvern, PA), PBS, or unmanipulated. Treatments were rotated to ensure no site bias.
Histology, immunocytochemistry and image analysis
Histological sections were stained with H&E, Masson’s Trichrome, Picrosirius Red, or subjected to immunohistochemistry. Tissues were stained with rabbit antibodies to TNFα(anti-TNFα; Abcam ab6671, 1:500), anti-iNOS (Millipore 06-573, 1:100), anti-arginase(Santa Cruz Biotechnology sc-20150, 1:100), Mac2 (1:100, generous gift of Dr. S. Vogel, Univ MD) and anti-phosphoNFκBp65(Cell Signaling, Ser276, #3037, 1:50). Primary antibody was detected using a FITC-labeled secondary antibody(DAKO) or using VECTOR peroxidase kit(VECTOR, Burlingame, CA). H&E stained cross-sections were quantified for the width of the epithelial gap and for the wound area, defined by the inflammatory region under the clot/scar, above the panniculus muscle and fat layers, and flanked by the wound edges, as described using an Optimas program(1, 10, 12). For inflammatory cell counts, sections were stained with Giemsa and six areas (20X) were captured per wound by Nikon DXM1200 camera(10). For immunohistochemistry, images were captured using an AperioT3 Scanscope (Aperio Technologies, Vista, CA).
MMP9 activity assay
Within 1 hr of excision, wound tissue was sectioned into 6-8 pieces and divided between two wells containing 200μl media and cultured at 37°C for 3 days. Each day 100μl of conditioned medium(CM) was removed, frozen at -80°C and 100μl fresh medium added. MMP9 activity was determined in CM by ELISA (Amersham MMP9 Biotrak Activity, GE Healthcare, Piscataway, NJ).
Gel shift and Western blot
Nuclear protein extracts were prepared as described(3). End-labeled oligonucleotide probes(0.5ng; 0.5-105 counts/min) were incubated 10min at room temperature in binding reaction buffer(20mM HEPES, pH7.9; 60mM NaCl; 5mM MgCl2, 1mM DTT; 0.5mM PMSF; 17% glycerol) containing 1μg poly(dI-dC) and 5μg nuclear extracts. DNA-protein complexes were separated by electrophoresis(6% polyacrylamide gels) in 0.25XTris borate-EDTA for phosphorimaging. For IκBα, equal amounts of protein were separated by SDS-PAGE in 10% acrylamide gels, transferred to nitrocellulose membranes(Schleicher & Schuell) and blots incubated with antibody in TBS+0.05% Tween-20(TBST). After polyclonal anti-IκBα(1:1000), blots were incubated 2hr with horseradish peroxidase-conjugated goat anti-rabbit antibody(Santa Cruz,1:2000). Proteins were visualized by Amersham ECL System and band intensity calculated with Scion Image(NIHimage program).
Murine macrophage culture
Macrophages were elicited by intraperitoneal injection of 4% thioglycollate or Biogel. At 4-5days post-injection, cells were harvested by peritoneal lavage, filtered through 70-μm nylon mesh, washed, and resuspended in serum-free DMEM containing gentamicin(10μg/ml) and glutamine(2mM). Cells were added to 6-well plates(4-5×106/ml/well) and after 2hr, medium was supplemented with 5% heat-inactivated-FCS and cultures incubated overnight before removing nonadherent cells. Cells were incubated 1hr with or without recombinant SLPI(10μg/ml, R&D Systems), followed by LPS(30ng/ml) and/or IFNγ(100units/ml, Peprotech), TNFα(50ng/ml) or IL-4(100ng/ml, R&D Systems) for 30min for protein or 2hr for RNA. All animal work was performed in accordance with protocols approved by the National Institute of Dental and Craniofacial Research Animal Care and Use Committee (Animal Study Proposal Number: 10-552).
RT-PCR
Total RNA was isolated from macrophages with RNeasy and tissue with TRIzol (Invitrogen). For conventional RT-PCR, cDNA(2μl) was amplified and sequences for primer pairs(5’-3’) are: GAPDH forward, TGCACCACCAACTGCTTAG, reverse, GATGCAGGGATGATGTTC; TNFα forward, AAAGATGGGGGGCTTCCAGAACTC, reverse AAGATGATCTGAGTGTGAGGGTCTG. Amplified products were analyzed by ethidium bromide staining after agarose(1.8%) gel electrophoresis and bands quantitated using digital imaging system(Alpha Innotech). For real time PCR, cDNA was amplified (ABI7500 sequence Detector, Applied Biosystems) using TaqMan expression assays for TNFα(Mm00443258_m1), ADAM17(Mm00456428_m1), MMP2(Mm00439506_m1), MMP9(Mm00600163_m1), Col1A1(Mm00801666_g1), iNOS(Mm00440485_m1), arginase(Mm00475988_m1), and HPRT(Mm00446968_m1). Results expressed as fold-increase by 2-ΔΔCT method.
Statistical analysis
Statistical differences were determined using the t-test, Wilcoxon-Mann-Whitney U test or ANOVA. A p value of <0.05 was considered significant.
RESULTS
Increased TNFα in human subjects predisposed to chronic wound conditions
In order to invoke a potential role for TNFα in impaired human healing responses, we monitored TNFα levels both systemically and at the tissue level in acute and chronic wound biopsies in aged human subjects. First, serum TNFα levels were significantly increased in aged subjects with active chronic venous ulceration compared to a control population with underlying pathology, but no ulceration(venous hypertension/varicose veins)(Fig. 1A; p<0.0001), independent of gender. Similarly, serum levels of TNFα were significantly (p < 0.05) higher in subjects with an active venous ulcer (61.9 pg/ml in males, 60.7 pg/ml in females) compared with those with healed ulcers (48.0 pg/ml in males, 45.1 pg/ml in females). Of considerable interest however, subjects with healed ulcers showed significantly increased systemic TNFα levels relative to levels measured in the control population (Fig. 1A, 32.4 pg/ml in males, 30.1 pg/ml in females; p < 0.05). These results suggested that heightened serum TNFα was not merely a reflection of spillover by a local non-healing wound, but rather a plausible phenotypic difference between subjects.
Figure 1. TNFα levels are increased locally and systemically in human subjects predisposed to impaired healing.
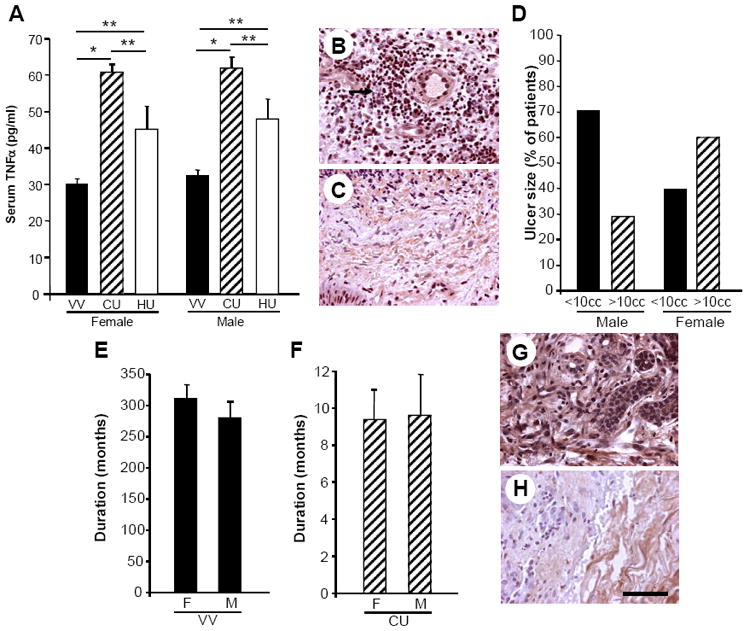
A) Systemic TNFα levels, determined by ELISA on serum samples, are significantly increased in subjects who have previously suffered from, or who currently have, chronic venous ulceration (CU) compared to age-matched controls with underlying pathology (i.e. venous hypertension/varicose veins, VV), but not unresolved inflammation. Female VV n=43; Female CU n=25; Male VV n=38; Male CU n=17. Data represent mean +/- SEM *p < 0.0001. Serum samples were also collected from individuals whose venous ulcer had healed (healed ulcer, HU) (n = 6 male, 9 female) and serum levels of TNFα were significantly (**p < 0.05) higher in subjects with an active venous ulcer (CU) compared with those with healed ulcers (HU). Subjects with healed ulcers (HU) also showed significantly (**p < 0.05) increased systemic TNFα levels compared with those measured in the control population (VV). B) Immunohistochemical analysis revealed local TNFα levels to be high in initial chronic ulcer biopsies (arrow indicates inflammatory cells staining positively for TNFα) and decrease as chronic venous ulcers heal (C). Representative patient biopsy images before and after healing, n=4. D) Ulcer size in male (n=17) and female (n=25) subjects represented as percentage of subjects with ulcers greater or less than 10cc. E) Duration of varicose veins in male and female subjects in months. F) Duration of venous ulcers in male and female subjects in months. G) Acute day 7 wound biopsies from the upper inner arm (site distant from chronic ulcer) show increased TNFα immunostaining in chronic ulcer patients compared to healthy controls (H). Representative images, n=4. Bar = 40 μm.
Based on increased detection of serum TNFα in subjects characterized by non-healing wounds, we next examined their ulcerated tissues for TNFα. Chronic wounds at presentation showed marked TNFα staining (Fig. 1B, representative patient, n=4). However, tissue levels of detectable TNFα decreased substantively as declining infiltrates, resolution and healing of chronic ulcers progressed(Fig. 1C; 30days post-treatment). Although females tended to have larger ulcers(>10cc), no significant differences in duration of either varicose veins or ulcers or gender bias were evident relative to serum or tissue TNFα levels(Fig. 1D-F).
In additional correlative studies, acute wounds were induced at distal sites(upper inner arm) in subjects (n=4) with, or who had previously had, chronic ulceration, and these individuals exhibited raised local levels of TNFα protein, even on day 7 after wounding, compared to age-matched controls without any diagnosed chronic inflammatory disorders who displayed reduced numbers of inflammatory cells and TNFα at this same time interval(Fig. 1G,H). This prolonged elevated expression of TNFα at acutely injured sites in subjects with defective healing was reflective of their augmented blood TNFα, suggesting an association between impaired healing and TNFα, and prompting an assessment of such a causal role for TNFα in aberrant healing in an experimental model.
Enhanced TNFα in SLPI null wounds
Following full-thickness dorsal incisional wounding of mice deficient in SLPI and wild-type(WT) littermates, an impaired rate of healing was readily apparent in SLPI null animals, as reported(10-12). Since SLPI reportedly inhibits macrophage TNFα expression in vitro(18) and SLPI-deficient macrophages generate higher levels of inducible TNFα than WT(Fig. 2A), we reasoned that, in the absence of SLPI, local injured tissue TNFα would escalate. By conventional RT-PCR, TNFα expression was elevated in untreated or PBS-treated wounds in null mice to a greater extent than WT(Fig. 2B), mirrored by enhanced TNFα detection using immunostaining(Fig. 2C,D). In addition to infiltrating inflammatory cells, TNFα staining appeared in keratinocytes of skin adjacent to wounds(Fig. 2C,D). When null mice were treated locally with exogenous SLPI(10), augmented levels of TNFα declined(Fig. 2B; p<0.05), concomitant with accelerated healing. By comparison, exogenous SLPI did not appear to significantly alter TNFα levels in SLPI-replete wounds. Not only does SLPI inhibit macrophage TNFα expression(Fig. 3A, p<0.05), but SLPI’s major protease target is elastase and in a SLPI-deficient milieu, over-abundant elastase activity likely provides a mechanism for augmented TNFα activation(22). Moreover, increased TNFα induces not only more TNFα(Fig. 3B; p<0.05), but in concert, triggers increased TNFα converting enzyme(TACE)/ ADAM17(Fig. 3C, p<0.05), capable of cleaving soluble active 17kDa TNFα from 26kDa membrane-associated precursor(23), setting in motion a persistent cycle of exacerbated inflammatory cell activation and its sequelae.
Figure 2. Increased TNFα in murine model of delayed healing.

A) Thioglycollate-induced peritoneal exudate macrophages (pooled from 3-5 mice) from SLPI null and WT mice were treated with a TLR4 agonist LPS (100ng/ml) for 1 hr and the cells processed for RT-PCR analysis. TLR4 stimulation of SLPI null macrophages induced higher levels of TNFα than for WT macrophages. N=2. B) By conventional RT-PCR, TNFα expression is minimal in control skin and enhanced in wounds from SLPI null mice compared to WT wounds at day 3 post-wounding treated with PBS vehicle. Local treatment with recombinant SLPI reduced TNFα expression in the SLPI null mice wounds to near levels seen in WT and resulted in accelerated healing. N=6 animals. *p < 0.05 comparing null control with null PBS-treated and ** p <0.05 for null PBS-treated vs. null SLPI-treated (black bars). Inset: Representative experiment showing conventional RT-PCR for TNFα expression in control skin and wounds treated locally with PBS vehicle or with SLPI. C,D) Increased intensity of immunostaining for TNFα in day 3 SLPI null skin wounds (D) relative to WT mice (C).
Figure 3. TNFα as a target of SLPI and accelerated healing by TNFα neutralization.

A) Pooled WT peritoneal macrophages were stimulated with LPS in culture in the presence or absence of SLPI (10 μg/ml added 30min prior to LPS) which significantly suppressed TNFα expression determined by real time PCR 1hr after stimulation with LPS. *p<0.05. Representative experiment, N=2. B, C) Exogenous TNFα added to peritoneal macrophages induces significant TNFα expression (B) in parallel with augmented ADAM17/TACE expression (C) which activates TNFα to enhance a cycle of pro-inflammatory activity. *p<0.05. D, E) Local administration of anti-TNFα reverses delayed healing phenotype in SLPI null mice. At the macroscopic level, anti-TNFα antibody treatment (αTNFα) accelerates healing in SLPI null mouse (D) to a greater extent than in the WT (E) (day 3 post-wounding shown). F) Anti-TNFα antibodies applied at the time of wounding significantly reduce cross-sectional wound areas in SLPI null mice compared to control (PBS treated) wounds at day 3 post-wounding. Areas= 105 mm2. Data represent mean +/- SEM * P < 0.05. N=6. G,H) Masson’s Trichrome staining illustrates a marked reduction of wound size in representative SLPI null mouse treated with anti-TNFα antibodies (G) compared to PBS (H). Arrows demarcate wound margins. Bar (H) = 300 μm (G-H).
Neutralization of TNFα accelerates impaired healing
We next explored whether directly antagonizing TNFα could alter aberrant healing responses characteristic of a SLPI-deficient milieu. Neutralization of TNFα at incisional sites accelerated the rate of healing macroscopically(Fig. 3D,E) and in a dose-dependent fashion as visualized microscopically in SLPI null animals at day 3 post-wounding(Fig. 3F-H). Quantification of wounds revealed that TNFα antibodies significantly reduced wound areas in SLPI null animals(Fig. 3F, p=0.05, day 3). Whereas a single application of 10μg/ml anti-TNFα administered at time of wounding significantly accelerated rate of healing in null mice, it also facilitated healing in wildtype, albeit not significantly, and 1μg/ml was insufficient in either wound (Fig. 3F-H).
Aberrant MMP and matrix in null wounds
In analyzing Masson Trichrome-stained wound sections, we noted more abundant collagen in anti-TNFα-treated null wounds relative to delayed repair in unmanipulated or PBS-treated wounds(Fig. 3G,H, blue stain=collagen), suggesting a TNFα-dependent effect on ECM accumulation. The specter of raised TNFα, inflammation, and excessive proteolysis could be reconciled not only by the well-established pro-inflammatory effects of TNFα, but, in addition, by evidence of activation of MMP9(24) seen in PBS-treated wound sites 1-3 days after incision(Fig. 4A). We therefore reasoned that MMP9 dysregulation must contribute to dysfunctional wound healing in the absence of SLPI, and would respond to treatment with neutralizing TNFα antibody. Whereas null wound tissue-culture supernatants showed enhanced MMP9 activity compared to unwounded skin(Fig. 4A), parallel cultured specimens from wounds treated with anti-TNFα had markedly reduced levels of MMP9 activity. These data emphasize that enhanced MMP9 activity, and likely other proteases associated with delayed healing in null mice, occurs as a consequence of a lack of SLPI and/or excess TNFα. In this regard, TNFα directly upregulates MMP9, as well as MMP2(Fig. 4B inset), and other macrophage stimuli linked to classical activation or M1 phenotype also induce MMP expression. In SLPI-depleted macrophages, M1 activation via IFN and LPS resulted in significantly higher MMP, as shown for MMP2(Fig. 4B; p<0.01), whereas alternatively activated/M2 macrophages, i.e., IL-4-stimulated, did not significantly upregulate MMPs(Fig. 4B). These data underscore the role that TNFα, along with M1 macrophages, supercharged in the absence of SLPI, may play in failed resolution of inflammation. Since heightened expression of MMP activity in wounded tissues could be dampened by disrupting one cog, specifically the cytokine TNFα(Fig. 4A), there appears to be a feedback loop involving SLPI and TNFα which influences local inflammation and tissue breakdown. Correspondingly, treatment with anti-TNFα resulted not only in dampened inflammation, but also in enhanced matrix accumulation. Collagen deposition as detected with Picrosirius Red was increased in anti-TNFα-treated wounds, not only in SLPI null(Fig. 4E,F), but also in WT(Fig. 4C,D). Compounded with enhanced tissue destruction via inflammatory proteases, including MMPs, reduced production of collagen, as monitored by COL1A1 RNA following injury(Fig. 4G), contributes to the nonhealing phenotype. Thus, enhanced ECM deposition after treatment likely reflects the ability of TNFα antagonists to suppress TNFα-driven proteases and regulation of synthesis of structural ECM components(25).
Figure 4. MMP is reduced and deposition of matrix protein is increased by anti-TNFα treatment.
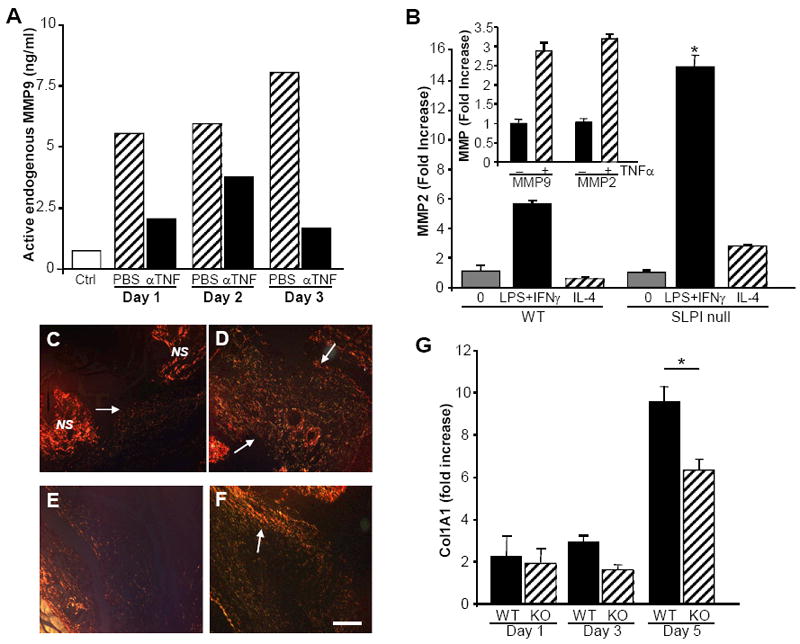
A) Supernatants from pooled and cultured SLPI null wound tissues at days 1-3 post wounding were tested for MMP9 activity by ELISA. Increased levels of active MMP9 were seen in the PBS-treated wounds cultured ex vivo compared to control skin. Treatment of wounds in vivo with anti-TNFα antibodies resulted in reduced levels of active MMP9 generated in parallel cultures from wounds on day 1, 2 or 3. B) Inducible macrophage MMP expression (MMP2 shown) is consistent with classically activated (M1), rather than alternatively activated macrophages (IL-4, M2) and is higher in SLPI null relative to WT peritoneal macrophages. null vs WT *p<0.01. Inset: TNFα independently stimulates MMP2 and MMP9 expression in WT (shown) and null macrophages as determined by RT-PCR. C-F) Picrosirius red staining of tissues at day 3 post-wounding illustrates increased collagen deposition (arrows) in anti-TNFα treated wounds compared to untreated control: C) WT day 3 post-wounding. D) WT treated with anti-TNFα day 3. E) SLPI null day 3 wound. F) SLPI null wound treated with anti-TNFα. NS= normal skin adjacent to wound. Bar (F) = 150 μm (C-F). G) Wound tissues at indicated intervals post wounding were evaluated by RT-PCR for type I collagen (Col1A1) gene expression. *p<0.05
Blockade of TNFα modulates local inflammation
Not only were wounds macroscopically and microscopically enlarged in SLPI null mice, but substantial increases in cells per unit area compared to WT reflects a prolonged accumulation of neutrophils and macrophages, as previously reported (10-12)(Fig. 5A). In this regard, TNFα antagonism significantly decreased inflammatory cell infiltrates in SLPI null wounds(Fig. 5A) as well as fewer, but not significantly so, in WT wounds. Because TNFα and MMP9 are elevated in pathological responses in SLPI null mice and because both are linked to M1 macrophage populations, we evaluated wounded tissues for a potential disproportionate representation of proinflammatory M1 macrophages compared to M2 populations, which putatively favor resolution and wound healing(26). In the absence of SLPI, evidence was indicative of a predominant infiltration of M1-like macrophages, in that detection of the M1 prototypic molecule iNOS by RT-PCR was higher in null wounds, particularly on day 3 post-wounding, but persisting at elevated levels (Fig. 5B). When interrogated further by immunohistochemistry, iNOS+ macrophages were pronounced as determined by Mac2+ staining in iNOS+ regions, but iNOS staining was also evident in epithelial and endothelial cells(Fig. 5B,C). By comparison, arginase, considered a marker for M2 macrophages was also typically more evident and persistent in wounds of null mice(Fig. 5D, E). Despite abundant neutrophils early after wounding, expression of arginase likely reflects infiltrating macrophages, since murine neutrophils lack arginase protein(26). Following anti-TNFα, numbers of inflammatory cells declined(Fig. 5A), including arginase+ cells(Fig. 5D) and wound size was reduced(Fig. 3F, 5B inset). To explore this relationship further, we examined whether SLPI differentially influenced M1/M2 polarization in vitro. SLPI suppressed M1 activation by LPS/IFNγ monitored by iNOS expression (Fig. 5F, p=0.03), whereas it did not suppress M2(IL-4) macrophages using arginase as output(Fig. 5G, p=NS). This differential SLPI-dependent regulation of M1 activity may represent an underlying basis for exacerbation of inflammation with an abundance of TNFα and MMP9, despite increases in arginase+ cells, which reportedly favor repair, although macrophages likely receive mixed signals within the wound milieu and are not strictly M1 or M2, but bear traits of both(26).
Figure 5. Increased inflammatory cell infiltrate reflecting M1 bias in SLPI deficient mice.
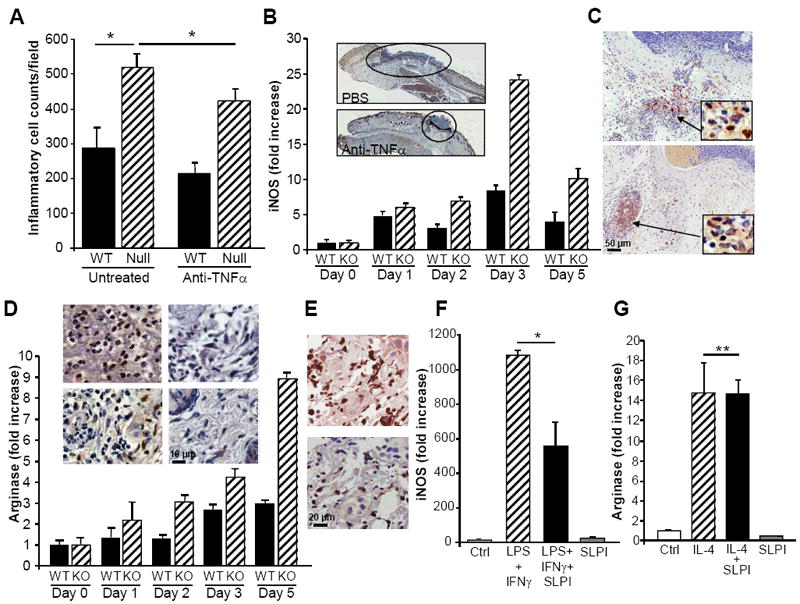
A) Inflammatory cell quantification demonstrates significantly increased inflammatory cells per unit area in SLPI null compared to wild-type (WT) wounds at day 3, as described (1). Treatment with anti-TNFα significantly reduced inflammatory infiltrate. Data represent mean+/- SEM *P<0.05. B) Wound tissues were assessed by RT-PCR for expression levels of iNOS as a marker for M1 macrophages, which were elevated and sustained in null relative to WT wounds. Inset: PBS treated(top) and anti-TNFα treated wounds showing reduced wound size with treatment. Day 3, original magnification 5X. C) Reduced iNOS staining in anti-TNFα treated wounds(bottom) relative to untreated(top) wounds, consistent with a decrease in M1 pro-inflammatory cells. Insets: Mac2 staining on serial sections showing macrophages within infiltrated region of iNOS staining. Representative staining, n=4. D) Arginase as detected by RT-PCR and protein(inset) reveal increasing expression of arginase in SLPI null wounds over time as compared to parallel wounds in WT mice. Inset: With TNFα inhibition, arginase expressing inflammatory cells by immunohistochemistry decrease as inflammation resolves and fibroblasts appear more numerous in SLPI null mice(top two panels of inset: left panel day 3 wound; right panel day 3 post anti-TNFα) and in WT (bottom two panels of inset: left panel day 3 wound; right panel day 3 post anti-TNFα). E) Day 3 wound serial tissue sections stained for arginase (top panel) and Mac2 (bottom) identify areas enriched in macrophages that are positive for detection of arginase, original magnification 40X. F) Thioglycollate-induced peritoneal macrophages were treated in culture with IFNγ (10ng/ml) and LPS(100ng/ml) to induce M1 phenotype and expression of iNOS monitored in presence or absence of SLPI(10μg/ml). SLPI inhibited M1 iNOS expression, *p=0.03. G) Thioglycollate-induced macrophages were treated with IL-4(10ng/ml) to induce M2 phenotype and arginase expression determined by RT-PCR in presence or absence of SLPI(10μg/ml). SLPI did not suppress arginase (n=3). **P=NS.
It is well established that M1 activation is driven largely by NFκB and in the absence of SLPI, an NFκB inhibitor(10, 20) that forestalls IκB degradation as shown for macrophages in Fig. 6A, NFκB activation is enhanced in total wound tissue specimens as determined by EMSA(Fig. 6B). Tight regulation of NFκB depends upon levels of its endogenous inhibitor IκB, and following IκB phosphorylation and degradation, liberated NFκB translocates to the nucleus to drive target genes. Since TNFα activates NFκB to potentiate chemokines, adhesion molecules, MMPs, and pro-inflammatory mediators including TNFα itself(3, 15), blockade of TNFα at the time of wounding reduced NFκB activity in SLPI null wounds(Fig. 6B), consistent with less tissue NFκB phospho-p65 staining and inflammatory cells in WT compared to null (Fig. 6C,D). Consequently, this cyclic TNFα-dependent proinflammatory loop and maintenance of an increased M1/M2 ratio deters the ability of the host to resolve inflammation, orchestrate repair and furthermore, provide evidence for an unappreciated and evolving role of SLPI in regulation of macrophage polarization.
Figure 6. Enhanced NFκB binding activity in the absence of SLPI is reduced in anti-TNFα treated wounds.

A) Peritoneal macrophages from WT and SLPI null mice were stimulated with LPS for indicated intervals and NFκB activation monitored by Western blot (p65) showing increased activation of this transcription factor in the absence of SLPI. In parallel, by Western blot, IκB, high in unstimulated macrophages, is degraded within minutes after exposure to LPS, and then the levels return to normal in the WT, whereas a slower recovery of the inhibitor occurs in the absence of SLPI. B) NFκB DNA binding is increased in nuclear extracts from SLPI null (KO) normal skin compared to wild-type (WT) normal skin tissues. Increased activity at day 1 post-wounding, particularly in the SLPI null wounds was reduced by anti-TNFα treatment (+). (− = PBS, no anti-TNFα). N = 6. C, D) NFκB phospho-p65 staining in KO (C) and WT (D) wounds at day 3 showing enhanced numbers of inflammatory cells and increased p65 staining in SLPI null wounds. Inset: Higher magnification (original magnification 40X) showing nuclear staining.
DISCUSSION
Age-impaired wound healing conditions lead to increased morbidity and mortality, and the problem is likely to increase with the ongoing expansion of the elderly population. In elderly patients with venous ulcers, we find that both systemic and local levels of TNFα are raised relative to subjects with underlying pathology, but lacking a non-healing phenotype. In those patients with a delayed healing phenotype, newly initiated acute wounds in distant sites were typically replete with TNFα relative to age-matched subjects lacking aberrant healing responses. Even acute wound healing in otherwise healthy elderly subjects is characterized by excessive inflammation and raised local TNFα levels compared to younger populations(27). Our data implicate both systemic and local increases in TNFα in the pathogenesis of, and predisposition to, chronic venous ulcers, and age-related increases in systemic TNFα may represent a unifying basis for the pathogenesis of a number of pro-inflammatory states in the aged(6, 7).
In an animal model of impaired healing resembling an age-related phenotype, raised TNFα levels are also characteristic. This appears to be relatively specific, since mRNA levels of other key modulators of inflammation, such as MIF and TGF-β1, are not significantly influenced by prevailing levels of SLPI(10). SLPI, composed of two cysteine-rich domains with a protease inhibitory site situated at leucine 72 in the carboxyl terminal domain(14), is released by activated inflammatory cells, and also by keratinocytes(10) to moderate inflammation and exert antimicrobial properties. A key mechanism by which depletion of SLPI catapults the pathogenesis of impaired wound healing, beyond failed protease inhibition, involves a marked increase in the inflammatory response and up-regulation of pro-inflammatory cytokines, typified by TNFα. In corroboration of these data, we have shown that neutralization of TNFα dampened excessive inflammation in the SLPI null animals and significantly accelerated the rate of wound healing. When viewed in the context of a chronic wound in which inflammation, raised TNFα levels, and bacterial infection contribute to delayed healing, TNFα antagonists, including SLPI, represent potential candidates for influencing multiple aspects of the healing response.
In WT mice, the early inflammatory infiltrate precedes extracellular matrix deposition, neovascularization, and collagenous scarring, whereas the SLPI-deficient mice exhibit delayed matrix accumulation(10). Elastase and MMP9 activity have been implicated as critical factors in a number of pathophysiological processes involving inflammation and tissue destruction/remodeling(5, 28). Impaired acute wound healing in the elderly is also associated with significantly increased tissue levels of inflammatory cell-derived MMPs, and chronic wound healing states are linked with raised wound fluid levels of MMP9 and reduced protease inhibitors(5, 29, 30). Multiple levels of regulation in the expression and activation of MMPs are known which suggest tight control during normal physiological processes. Besides its direct inhibition of elastase and consequently, inhibition of elastase-mediated MMP activation, SLPI is a potent suppressor of monocyte MMP9 via a prostaglandin E2 pathway, indicating that dysregulation of MMP9 may be a harbinger of impaired healing in age-related pathogenesis and/or in the absence of SLPI(15). Since TNFα induces expression of MMP9 in a variety of cell types, we reasoned that the beneficial effects of blocking TNFα in our delayed healing model would be paralleled by modulated MMP9. Our data demonstrate that anti-TNFα treatment reduces MMP9 activity in SLPI null wound tissue, implicating MMP9 as an integral component of the SLPI/TNFα axis in vivo. Levels of collagen(COL1A1) were diminished in SLPI null wounds consonant with the negative impact of TNFα on collagen gene expression(25, 31), and delayed accumulation of matrix-generating fibroblasts due to disproportionate numbers of inflammatory cells occupying the wound bed. Together with MMP-dependent degradation of new matrix, there is a net loss of ECM accumulation in the SLPI null relative to WT wounds. Antagonizing TNFα was associated with increased matrix deposition not only in the SLPI null animals but also in WT, indicating that modulation of TNFα levels in vivo may influence both pathologic and ‘physiological’ wound healing responses.
Engagement of TNFR1p55/TNFR2p75 by TNFα recruits adaptor proteins to initiate downstream signaling pathways that culminate in transcription of inflammatory genes. TNFα activates NFκB, which in turn induces gene expression of a plethora of pro-inflammatory cytokines including TNFα itself and proteases, such as MMP and also TACE to release soluble TNFα, thereby potentiating the effects of this inflammatory cytokine(32). We demonstrated that NFκB activity, increased in early wound tissue in the SLPI null compared to WT mice, was suppressed by anti-TNFα. Such data underscore a role for SLPI in modulating the positive feedback loop that exists between TNFα and NFκB activity. Both in vitro and in vivo SLPI inhibits TNFα production, most likely through a block on NFκB activation involving retention of its inhibitor, IκB. Whereas both p50 and p65 homodimers and heterodimers are critical to pro-inflammatory gene activation by TNFα in human monocytes, primarily p65 regulates ICAM-1 expression by endothelial cells(33). In this regard, it is interesting to note that normal/uninjured skin from SLPI null mice shows evidence of constitutive activation of the p65 homodimer. One consequence of this may be enhanced adhesion molecule expression in SLPI null mice, effectively priming vessels for leukocyte infiltration upon injury.
Although recent data suggest that physiologic wound macrophages exhibit a complex phenotype which does not segregate into distinct M1 and M2 populations, but rather include traits reflective of both populations in an environment devoid of M2 polarizing IL-4 or IL-13(26), our data in a nonphysiologic delayed healing model suggest an abundance of macrophages expressing iNOS, MMP9 and TNFα, likely related to unrestricted NFκB activation, all of which exacerbate the inflammatory response to tissue injury and in a vicious cycle appear to provide an enforcing feedback to recruit and activate additional inflammatory M1-biased macrophages. M2/alternatively-activated macrophages expressing higher levels of arginase are also abundant in the wound bed. These cells are thought to be important in resolution of inflammation(34), although new evidence suggests that this population may, in some circumstances, actually impede fibrosis(35). Interruption of the activation cycle with exogenous SLPI(10) or antagonizing TNFα as shown here, has the beneficial effect of enabling resolution of the exaggerated inflammation with restoration of healing pathways, implicating TNFα as a master regulator of impaired wound healing in vivo.
While comparisons between murine models and human pathogenesis are fraught with discrepancies, our parallel data concerning elevated TNFα expression in human chronic wounds both locally and systemically warrant further consideration. Importantly, we suggest that the beneficial effects of antagonizing TNFα could be extrapolated to human impaired wound healing conditions, whether by neutralizing antibodies, soluble receptors, receptor fusion proteins directly, or via gene transfer or other strategies(4, 5, 25, 36-38). Even though TNFα antagonists have not yet been explored thoroughly in chronic nonhealing wounds, as a prelude, a series of case studies demonstrated a beneficial impact of topical TNFα antibodies(39) and it has recently been noted that rheumatoid arthritis patients receiving anti-TNFα agents have not had compromised healing or negative consequences in healing of postoperative sites(40). Particularly relevant may be the ability of locally and temporally delivered TNFα antagonists, without the need for systemic or prophylactic treatment, to improve aberrant healing states, which may delimit untoward negative consequences of systemic delivery of anti-TNFα therapy.
Acknowledgments
The authors are grateful to Calley Grace for editorial assistance, Azita Adli, Nancy Marinos, and Alec Waite for histological assistance and expertise with immunocytochemistry and Dr. S. Vogel, U MD School of Medicine, Baltimore for Mac2 antibody. Gillian Ashcroft was funded under a Wellcome Senior Fellowship in Clinical Science. This research was supported in part by the Intramural Research Program of the NIH, National Institute of Dental and Craniofacial Research.
Footnotes
The authors have no conflicting financial interests.
References
- 1.Ashcroft GS, Greenwell-Wild T, Horan MA, Wahl SM, Ferguson MW. Topical estrogen accelerates cutaneous wound healing in aged humans associated with an altered inflammatory response. Am J Pathol. 1999;155(4):1137–46. doi: 10.1016/S0002-9440(10)65217-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9(8):628–38. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- 3.Song X, Zeng L, Jin W, Thompson J, Mizel DE, Lei K, Billinghurst RC, Poole AR, Wahl SM. Secretory leukocyte protease inhibitor suppresses the inflammation and joint damage of bacterial cell wall-induced arthritis. J Exp Med. 1999;190(4):535–42. doi: 10.1084/jem.190.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feldmann M, Williams RO, Paleolog E. What have we learnt from targeted anti-TNF therapy? Ann Rheum Dis. 2010;69(Suppl 1):i97–9. doi: 10.1136/ard.2009.117143. [DOI] [PubMed] [Google Scholar]
- 5.Chan JM, Villarreal G, Jin WW, Stepan T, Burstein H, Wahl SM. Intraarticular gene transfer of TNFR:Fc suppresses experimental arthritis with reduced systemic distribution of the gene product. Mol Ther. 2002;6(6):727–36. doi: 10.1006/mthe.2002.0808. [DOI] [PubMed] [Google Scholar]
- 6.Bruunsgaard H, Pedersen BK. Age-related inflammatory cytokines and disease. Immunol Allergy Clin North Am. 2003;23(1):15–39. doi: 10.1016/s0889-8561(02)00056-5. [DOI] [PubMed] [Google Scholar]
- 7.Bruunsgaard H. Effects of tumor necrosis factor-alpha and interleukin-6 in elderly populations. Eur Cytokine Netw. 2002;13(4):389–91. [PubMed] [Google Scholar]
- 8.Mendez MV, Raffetto JD, Phillips T, Menzoian JO, Park HY. The proliferative capacity of neonatal skin fibroblasts is reduced after exposure to venous ulcer wound fluid: A potential mechanism for senescence in venous ulcers. J Vasc Surg. 1999;30(4):734–43. doi: 10.1016/s0741-5214(99)70113-8. [DOI] [PubMed] [Google Scholar]
- 9.Huttunen M, Aalto ML, Harvima RJ, Horsmanheimo M, Harvima IT. Alterations in mast cells showing tryptase and chymase activity in epithelializating and chronic wounds. Exp Dermatol. 2000;9(4):258–65. doi: 10.1034/j.1600-0625.2000.009004258.x. [DOI] [PubMed] [Google Scholar]
- 10.Ashcroft GS, Lei K, Jin W, Longenecker G, Kulkarni AB, Greenwell-Wild T, Hale-Donze H, McGrady G, Song XY, Wahl SM. Secretory leukocyte protease inhibitor mediates non-redundant functions necessary for normal wound healing. Nat Med. 2000;6(10):1147–53. doi: 10.1038/80489. [DOI] [PubMed] [Google Scholar]
- 11.Angelov N, Moutsopoulos N, Jeong MJ, Nares S, Ashcroft G, Wahl SM. Aberrant mucosal wound repair in the absence of secretory leukocyte protease inhibitor. Thromb Haemost. 2004;92(2):288–97. doi: 10.1160/TH03-07-0446. [DOI] [PubMed] [Google Scholar]
- 12.Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, Ding A. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111(6):867–78. doi: 10.1016/s0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
- 13.Franzke CW, Baici A, Bartels J, Christophers E, Wiedow O. Antileukoprotease inhibits stratum corneum chymotryptic enzyme. Evidence for a regulative function in desquamation. J Biol Chem. 1996;271(36):21886–90. doi: 10.1074/jbc.271.36.21886. [DOI] [PubMed] [Google Scholar]
- 14.Eisenberg SP, Hale KK, Heimdal P, Thompson RC. Location of the protease-inhibitory region of secretory leukocyte protease inhibitor. J Biol Chem. 1990;265(14):7976–81. [PubMed] [Google Scholar]
- 15.Zhang Y, DeWitt DL, McNeely TB, Wahl SM, Wahl LM. Secretory leukocyte protease inhibitor suppresses the production of monocyte prostaglandin H synthase-2, prostaglandin E2, and matrix metalloproteinases. J Clin Invest. 1997;99(5):894–900. doi: 10.1172/JCI119254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McNeely TB, Dealy M, Dripps DJ, Orenstein JM, Eisenberg SP, Wahl SM. Secretory leukocyte protease inhibitor: a human saliva protein exhibiting anti-human immunodeficiency virus 1 activity in vitro. J Clin Invest. 1995;96(1):456–64. doi: 10.1172/JCI118056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma G, Greenwell-Wild T, Lei K, Jin W, Swisher J, Hardegen N, Wild CT, Wahl SM. Secretory leukocyte protease inhibitor binds to annexin II, a cofactor for macrophage HIV-1 infection. J Exp Med. 2004;200(10):1337–46. doi: 10.1084/jem.20041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin FY, Nathan C, Radzioch D, Ding A. Secretory leukocyte protease inhibitor: a macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell. 1997;88(3):417–26. doi: 10.1016/s0092-8674(00)81880-2. [DOI] [PubMed] [Google Scholar]
- 19.Devoogdt N, Revets H, Kindt A, Liu YQ, De Baetselier P, Ghassabeh GH. The tumor-promoting effect of TNF-alpha involves the induction of secretory leukocyte protease inhibitor. J Immunol. 2006;177(11):8046–52. doi: 10.4049/jimmunol.177.11.8046. [DOI] [PubMed] [Google Scholar]
- 20.Taggart CC, Cryan SA, Weldon S, Gibbons A, Greene CM, Kelly E, Low TB, O’neill SJ, McElvaney NG. Secretory leucoprotease inhibitor binds to NF-kappaB binding sites in monocytes and inhibits p65 binding. J Exp Med. 2005;202(12):1659–68. doi: 10.1084/jem.20050768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan C, Grimm WA, Garner WL, Qin L, Travis T, Tan N, Han YP. Epithelial to Mesenchymal Transition in Human Skin Wound Healing Is Induced by Tumor Necrosis Factor-{alpha} through Bone Morphogenic Protein-2. Am J Pathol. 2010;176(5):2247–58. doi: 10.2353/ajpath.2010.090048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer-Hoffert U. Neutrophil-derived serine proteases modulate innate immune responses. Front Biosci. 2009;14:3409–18. doi: 10.2741/3462. [DOI] [PubMed] [Google Scholar]
- 23.Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Becherer JD, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385(6618):733–6. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 24.Han YP, Tuan TL, Hughes M, Wu H, Garner WL. Transforming growth factor-beta - and tumor necrosis factor-alpha -mediated induction and proteolytic activation of MMP-9 in human skin. J Biol Chem. 2001;276(25):22341–50. doi: 10.1074/jbc.M010839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buck M, Houglum K, Chojkier M. Tumor necrosis factor-alpha inhibits collagen alpha1(I) gene expression and wound healing in a murine model of cachexia. Am J Pathol. 1996;149(1):195–204. [PMC free article] [PubMed] [Google Scholar]
- 26.Daley JM, Brancato SK, Thomay AA, Reichner JS, Albina JE. The phenotype of murine wound macrophages. J Leukoc Biol. 2010;87(1):59–67. doi: 10.1189/jlb.0409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ashcroft GS, Mills SJ. Androgen receptor-mediated inhibition of cutaneous wound healing. J Clin Invest. 2002;110(5):615–24. doi: 10.1172/JCI15704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price NM, Gilman RH, Uddin J, Recavarren S, Friedland JS. Unopposed matrix metalloproteinase-9 expression in human tuberculous granuloma and the role of TNF-alpha-dependent monocyte networks. J Immunol. 2003;171(10):5579–86. doi: 10.4049/jimmunol.171.10.5579. [DOI] [PubMed] [Google Scholar]
- 29.Wysocki AB, Staiano-Coico L, Grinnell F. Wound fluid from chronic leg ulcers contains elevated levels of metalloproteinases MMP-2 and MMP-9. J Invest Dermatol. 1993;101(1):64–8. doi: 10.1111/1523-1747.ep12359590. [DOI] [PubMed] [Google Scholar]
- 30.Bullen EC, Longaker MT, Updike DL, Benton R, Ladin D, Hou Z, Howard EW. Tissue inhibitor of metalloproteinases-1 is decreased and activated gelatinases are increased in chronic wounds. J Invest Dermatol. 1995;104(2):236–40. doi: 10.1111/1523-1747.ep12612786. [DOI] [PubMed] [Google Scholar]
- 31.Verrecchia F, Mauviel A. TGF-beta and TNF-alpha: antagonistic cytokines controlling type I collagen gene expression. Cell Signal. 2004;16(8):873–80. doi: 10.1016/j.cellsig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Sano C, Shimizu T, Tomioka H. Effects of secretory leukocyte protease inhibitor on the tumor necrosis factor-alpha production and NF-kappaB activation of lipopolysaccharide-stimulated macrophages. Cytokine. 2003;21(1):38–42. doi: 10.1016/s1043-4666(02)00485-4. [DOI] [PubMed] [Google Scholar]
- 33.True AL, Rahman A, Malik AB. Activation of NF-kappaB induced by H(2)O(2) and TNF-alpha and its effects on ICAM-1 expression in endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2000;279(2):L302–11. doi: 10.1152/ajplung.2000.279.2.L302. [DOI] [PubMed] [Google Scholar]
- 34.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 35.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30(3):245–57. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heinrich SA, Messingham KA, Gregory MS, Colantoni A, Ferreira AM, Dipietro LA, Kovacs EJ. Elevated monocyte chemoattractant protein-1 levels following thermal injury precede monocyte recruitment to the wound site and are controlled, in part, by tumor necrosis factor-alpha. Wound Repair Regen. 2003;11(2):110–9. doi: 10.1046/j.1524-475x.2003.11206.x. [DOI] [PubMed] [Google Scholar]
- 37.Maish GO, 3rd, Shumate ML, Ehrlich HP, Cooney RN. Tumor necrosis factor binding protein improves incisional wound healing in sepsis. J Surg Res. 1998;78(2):108–17. doi: 10.1006/jsre.1998.5315. [DOI] [PubMed] [Google Scholar]
- 38.Mori R, Kondo T, Ohshima T, Ishida Y, Mukaida N. Accelerated wound healing in tumor necrosis factor receptor p55-deficient mice with reduced leukocyte infiltration. FASEB J. 2002;16(9):963–74. doi: 10.1096/fj.01-0776com. [DOI] [PubMed] [Google Scholar]
- 39.Streit M, Beleznay Z, Braathen LR. Topical application of the tumour necrosis factor-alpha antibody infliximab improves healing of chronic wounds. Int Wound J. 2006;3(3):171–9. doi: 10.1111/j.1742-481X.2006.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirano Y, Kojima T, Kanayama Y, Shioura T, Hayashi M, Kida D, Kaneko A, Eto Y, Ishiguro N. Influences of anti-tumour necrosis factor agents on postoperative recovery in patients with rheumatoid arthritis. Clin Rheumatol. 2010;29(5):495–500. doi: 10.1007/s10067-009-1346-1. [DOI] [PubMed] [Google Scholar]