

Abstract
Encapsulation of drugs within nanocarriers that selectively target malignant cells promises to mitigate side effects of conventional chemotherapy and to enable delivery of the unique drug combinations needed for personalized medicine. To realize this potential, however, targeted nanocarriers must simultaneously overcome multiple challenges, including specificity, stability, and a high capacity for disparate cargos. Here we report porous nanoparticle-supported lipid bilayers (protocells) that synergistically combine properties of liposomes and nanoporous particles. Protocells modified with a targeting peptide that binds to human hepatocellular carcinoma (HCC) exhibit a 10,000-fold greater affinity for HCC than for hepatocytes, endothelial cells, and immune cells. Furthermore, protocells can be loaded with combinations of therapeutic (drugs, siRNA, and toxins) and diagnostic (quantum dots) agents and modified to promote endosomal escape and nuclear accumulation of selected cargos. The enormous capacity of the high-surface-area nanoporous core combined with the enhanced targeting efficacy enabled by the fluid supported lipid bilayer allow a single protocell loaded with a drug cocktail to kill a drug-resistant HCC cell, representing a 106-fold improvement over comparable liposomes.
Targeted delivery of drugs encapsulated within nanocarriers1-2 can overcome problems exhibited by conventional ‘free’ drugs, including poor solubility, limited stability, rapid clearing, and, in particular, lack of selectivity, which results in non-specific toxicity to normal cells3 and prevents the dose escalation necessary to eradicate malignant cells4. Passive targeting schemes rely on the enhanced permeability of tumor vasculature and the decreased draining efficacy of tumor lymphatics (the so-called enhanced permeability and retention, or EPR, effect)5-6 to direct accumulation of nanocarriers at tumor sites, but the lack of cell-specific interactions needed to induce nanocarrier internalization decreases therapeutic efficacy and can result in drug expulsion and induction of multiple drug resistance (MDR)7. Furthermore, not all tumors exhibit the EPR effect5-6, and passively-targeted nanocarriers are no more effective at treating blood cancers than free drugs8. Selective targeting strategies employ ligands that specifically interact with receptors expressed on the cell surface of interest to promote nanocarrier binding and internalization9. This strategy requires that receptors are highly over-expressed by cancer cells (104-105 copies/cell) relative to normal cells in order to maximize selectivity and therapeutic efficacy1. Multiple copies of a targeting ligand can be conjugated to the nanocarrier surface to promote multivalent binding effects10, which result in enhanced affinity11 and more efficient drug delivery through receptor-mediated internalization pathways that help circumvent MDR efflux mechanisms12. However, high ligand densities can promote non-specific interactions with endothelial and other non-cancerous cells and increase immunogenicity, resulting in opsonization-mediated clearance of nanocarriers13. Modifying the nanocarrier surface with hydrophilic polymers, such as polyethylene glycol (PEG), increases circulation times by reducing interactions with serum proteins and mitigating uptake by phagocytic cells; such strategies invariably reduce targeting specificity, however13. The major challenge for targeted nanocarriers is to simultaneously achieve high targeting specificity and delivery efficiency, while avoiding non-specific binding and entrapment by the body's defenses.
Here we report a new class of nanocarrier that synergistically combines features of mesoporous silica particles14-19 and liposomes20-22 to address the multiple challenges of targeted delivery. Fusion of liposomes to a spherical, high-surface-area, nanoporous silica core23-26, followed by modification of the resulting supported lipid bilayer (SLB) with multiple copies of a targeting peptide, a fusogenic peptide, and PEG results in a nanocarrier construct (the ‘protocell’) that, compared to liposomes, the most extensively-studied class of nanocarriers20-22, improves upon capacity, selectivity, and stability and enables targeted delivery and controlled release of high concentrations of multicomponent cargos within the cytosol of cancer cells (see Fig. 1 and Supplementary Methods for experimental details). Specifically, due to its high surface area (> 1000 m2/g), the nanoporous silica core (Fig. 2a) possesses a higher capacity for therapeutic and diagnostic agents than similarly-sized liposomes. Furthermore, due to substrate-membrane adhesion energy, the core suppresses large-scale bilayer fluctuations (see Supplementary Fig. 3a and references 27-32), resulting in greater stability than unsupported liposomal bilayers. Interestingly, the nanoporous support also results in enhanced lateral bilayer fluidity compared to that of either liposomes or SLBs formed on non-porous particles. As we will demonstrate, this synergistic combination of materials and biophysical properties enables high delivery efficiency and enhanced targeting specificity with a minimal number of targeting ligands, features crucial to maximizing specific binding, minimizing non-specific binding, reducing dosage, and mitigating immunogenicity.
Figure 1. Schematic illustration of the nanoporous particle-supported lipid bilayer, depicting the disparate types of therapeutic and diagnostic agents that can be loaded within the nanoporous silica core, as well as the ligands that can be displayed on the surface of the SLB.

Targeting and fusogenic peptides are chemically conjugated to phosphatidylethanolamine (DOPE or DPPE), present in the SLB at 1-5 wt%, via a heterobifunctional crosslinker with a polyethylene glycol (PEG) spacer arm (n = 24). The SLB, composed of either fluid (DOPC) or non-fluid (DPPC) zwitterionic lipids with 30 wt% cholesterol, is further modified with 5 wt% PEG-2000 PE to enhance colloidal stability and decrease non-specific interactions.
Figure 2. Physical and biophysical characteristics of protocells.

(a) Cryogenic TEM image of the protocell, showing the nanoporous core and the SLB (∼4-nm thick). Particle sizes reflect those naturally generated by the aerosol-assisted self-assembly process26; particles were separated into a narrow distribution centered around ∼100-nm for all surface binding, internalization, and delivery experiments (see Supplementary Fig. 1). Scale bar = 25 nm. (b) Temperature-dependent FRAP of NBD-labeled DPPC bilayers (green) supported on nanoporous (○) or solid (●) spherical silica particles. Inset: normalized fluorescence recovery in the photobleached region (blue circle) was determined by dividing the fluorescence intensity (FI) in region of interest 1 (ROI1) by the FI in ROI2 to account for photobleaching that occurred during the recovery period. Scale bar = 5 μm.
Protocells are synthesized via liposome fusion to high-surface-area spherical silica particles characterized by an isotropic, worm-like nanoporosity (see Fig. 2a and Supplementary Fig. 1). To demonstrate that SLBs formed on particles with surface-accessible nanopores have unique long-range fluidity, we performed temperature-dependent fluorescence recovery after photobleaching (FRAP) of DPPC bilayers supported on either a nanoporous or solid (i.e. non-porous) silica particle (see Fig. 2b). We observe that fluorescence in the photobleached region begins to recover abruptly at 35°C (± 1°C) for the SLB formed on a nanoporous particle, as compared to 41°C (± 1°C) for the SLB formed on a solid particle; 41°C is the gel-to-fluid transition temperature (Tm) of DPPC, as well as the Tm reported for unilamellar DPPC liposomes33. These data indicate that the nanoporous support results in a substantial reduction (6°C) in Tm. We reason that this melting point suppression and the resulting enhancement in bilayer fluidity, also observed for nanoporous particle-supported DOPC bilayers (see Supplementary Fig. 3b), are consequences of unique physical constraints that exist at the interface between the bilayer and the nanoporous support. The underlying 3D porosity and corresponding periodic roughness of the particle surface, which is composed of nanoscopic patches of silica and water, generate localized, nanoscale gradients in adhesion and lateral tension that enhance long-range, in-plane fluidity without introducing roughness or appreciably changing the SLB's average packing density (determined by us previously via neutron reflectivity of lipid bilayers supported on planar nanoporous supports34). This conclusion is reinforced by previous experimental and theoretical studies, which found that the support suppresses all but nanoscopic, out-of-plane bilayer fluctuations35-36, as well as small angle neutron scattering (SANS) data, which indicate that the protocell SLB perfectly conforms to the underlying nanoporous silica support (see Supplementary Fig. 3a). Furthermore, based on simple thermodynamic arguments, we expect particle curvature to influence bilayer fluidity only for R ≪ (κ/2ε)1/2, where R is the particle radius, κ is the bending modulus, and ε is the adhesion energy. Given that κ = 1020 J for DOPC or DPPC and ε = 10-3 – 10-5 J/m2, this condition is only met when R ≪ 100-nm, as demonstrated by recent studies that report very slight increases in the fluidity of bilayers supported on nanowires less than 50-nm in diameter37. Overall, our data provide experimental evidence for previous theoretical predictions of the effect that nanoscale topography has on supported bilayer conformations32,38. As described below, the enhanced fluidity of nanoporous particle-supported lipid bilayers enables protocells modified with a minimal number of targeting peptides to selectively bind to and become internalized by cancer cells, while their enhanced stability vis-à-vis liposomes prevents drug leakage upon exposure to simulated body fluids.
The schematic in Figure 3 depicts the mechanism by which targeted protocells deliver encapsulated cargo specifically to a cancer cell of interest; successive steps of binding (step 1), internalization (step 2), endosomal escape (step 3), and nuclear targeting of desired cargo(s) (step 4) are individually described below. Protocells are synthesized via fusion of liposomes to spherical, nanoporous silica cores (100-150 nm in diameter after size separation; see Fig. 1, Fig. 2a, and Supplementary Fig. 1a and 1d) that are pre-loaded via simple immersion in a solution of the desired cargos. Based on optimization studies (see Supplementary Fig. 5) that aimed to maximize colloidal stability and cargo retention in simulated body fluids and minimize non-specific interactions with serum proteins and non-cancerous cells, we utilized the following SLB composition in all surface-binding, internalization, and cargo delivery experiments: DOPC (or DPPC) with 5 wt% DOPE (or DPPE), 30 wt% cholesterol, and 5 wt% 18:1 (or 16:0) PEG-2000 PE (see Fig. 1 and Supplementary Fig. 4 for lipid structures). Using a heterobifunctional crosslinker with a PEG (n = 24) spacer, SP94 peptides (H2N-SFSIILTPILPLGGC-COOH, identified via filamentous phage display to have an affinity for unknown receptor(s) expressed by human HCC39) were covalently conjugated to DOPE (or DPPE) moieties in the SLB (see Fig. 1) at concentrations ranging from 0.002 wt% (1 peptide per particle, on average) to 5.0 wt% (2048 peptides per particle, on average – see Supplementary Table I). 120-nm liposomes with identical bilayer compositions were synthesized for comparative purposes.
Figure 3. Schematic depicting the successive steps of multivalent binding and internalization of targeted protocells, followed by endosomal escape and nuclear localization of protocell-encapsulated cargo.

DOPC protocells [1] bind to HCC with high affinity due to recruitment of SP94 targeting peptides (magenta) to the cell surface, [2] become internalized via receptor-mediated endocytosis, and [3] release their cargo into the cytosol upon endosome acidification and protonation of the H5WYG fusogenic peptide (blue). Cargos modified with a NLS are transported through the nuclear pore complex and become concentrated in the nucleus [4].
Dissociation constants (Kd, where Kd is inversely related to affinity) were used to quantify surface binding of SP94-targeted protocells and liposomes to HCC cells (Hep3B), normal hepatocytes, endothelial cells, and immune cells. All Kd values were determined at 4°C to prevent nanocarrier internalization (see Supplementary Fig. 6 and Supplementary Methods). Figures 4a and 4b plot Kd values of SP94-targeted protocells and liposomes for Hep3B and hepatocytes as a function of average peptide density. Protocells with SLBs composed largely of DOPC (in a fluid state at 4°C) have a high specific affinity (Kd < 1 nM) for Hep3B, and, over the range of 6 to 2048 peptides per particle, their Kd values are consistently low (0.94 – 0.08 nM) and relatively independent of peptide density. This trend is not observed for DOPC liposomes, where Kd values strongly depend on peptide density and are more than 10-fold greater than those of comparable DOPC protocells. Similarly, protocells and liposomes with bilayers composed of DPPC (in a gel-like state at 4°C) have Kd values that are more than 10-fold greater than corresponding DOPC protocells and exhibit a strong dependence on peptide density. We attribute the ability of DOPC protocells to bind to HCC with high affinity at low peptide densities to recruitment of multiple SP94 peptides to the cancer cell surface. Peptide recruitment is enabled by the fluid SLB and promotes multivalent interactions between the protocell and the target cancer cell. For DPPC protocells and liposomes, multivalent binding and correspondingly high specific affinity can only be realized at high peptide densities because non-fluid bilayers impart kinetic constraints on the lateral mobility of targeting peptides. The importance of SLB fluidity in promoting the peptide recruitment process is vividly illustrated in Figure 4c. DOPC or DPPC liposomes were fused to planar nanoporous substrates (with a 3D pore structure comparable to that of the protocell core40-41), and the resulting SLBs were modified with a low density (∼0.015 wt%, equivalent to ∼6 peptides per particle) of SP94 peptides. Upon addition of Hep3B to the supported planar bilayers, we observed rapid recruitment of SP94 to the cancer cell surface when peptides were displayed on a fluid SLB but no measurable recruitment when peptides were displayed on a non-fluid SLB. This result explains the 100-fold lower Kd value of DOPC protocells versus DPPC protocells, when both display ∼6 peptides per particle (see Fig. 4a and the following discussion).
Figure 4. Selective binding and internalization characteristics of SP94-targeted protocells.

(a) and (b) Dissociation constants (Kd) of SP94-targeted protocells and liposomes for Hep3B (a) and hepatocytes (b) as a function of the average number of SP94 peptides per particle (average SP94 wt% is in parentheses). All surface binding experiments were conducted at 4°C to prevent internalization of targeted protocells and liposomes. All error bars in (a) and (b) represent 95% confidence intervals (1.96 σ) for n = 5. (c) Recruitment of Alexa Fluor® 647-labeled SP94 peptides (white) to the surface of a Hep3B cell when peptides are displayed on a NBD-labeled SLB (green) composed of DOPC (○) or DPPC (●). These data were collected at 4°C to replicate the conditions used to determine Kd values in (a) and (b). Hep3B cells were labeled with CellTracker™ Red CMTPX (red) and Hoechst 33342 (blue). Inset scale bars = 5 μm. (d) and (e) Confocal fluorescence microscopy images of Hep3B (d) and hepatocytes (e) incubated with SP94-targeted protocells for 1 hour at 37°C. Protocells were prepared with Texas Red®-labeled DHPE (red) and Alexa Fluor® 647-labeled nanoporous cores (white); cells were stained with CellTracker™ Green CMFDA (green) and Hoechst 33342 (blue). Cells shown in (d) and (e) are representative of the entire cell population (see Supplementary Table II for population-based internalization data); single cells were selected to enable 3D imaging. Plan (left and center images) and cross-sectional (right image) views of the 3D projection are shown for (d), while the plan view alone is shown for (e). For (d), the merged plan view (left) is shown without the green channel (center) to enable better visualization of lipid (red) and silica (white) moieties. It is important to note that plan views of collapsed projections superimpose all slices in the z-direction, giving the misleading appearance of protocells in the nucleus of (d); this is not the case, however, as is evident in an orthogonal view of the projection (image not shown). All scale bars = 10 μm.
The ability of targeting peptides, when displayed in low densities on a fluid SLB, to be recruited and multivalently bind to surface receptor(s) is crucial to enhance specific affinity, reduce non-specific interactions, and direct receptor-mediated endocytosis of nanocarriers, all of which maximize selective delivery of cargo. Concerning this point, it is important to note the influence of bilayer fluidity and stability on the peptide density-dependent affinity of SP94-targeted protocells and liposomes for HCC (Fig. 4a) and normal hepatocytes (Fig. 4b). Non-fluid DPPC protocells and liposomes have a low affinity (Kd ≥ 1 μM) for hepatocytes at high SP94 densities. However, their affinity for Hep3B (Kd = ∼ 1 – 100 nM) is substantially lower than that of DOPC protocells (Kd < 1 nM) at all peptide densities, and their Kd values for Hep3B more rapidly increase with decreasing peptide density. DOPC protocells and liposomes have a similar affinity for hepatocytes at all SP94 densities (see Fig. 4b), but the Kd values of DOPC liposomes for Hep3B are between 10 and 200 times greater than those of DOPC protocells modified with the same number of peptides (see Fig. 4a). We attribute these observations to the enhanced fluidity of nanoporous particle-supported DOPC bilayers, which enables multivalent peptide recruitment to the Hep3B surface, combined with the ability of the nanoporous core to suppress the large-scale bilayer fluctuations that, for DOPC liposomes especially, appear to act as a steric barrier to high avidity binding. The result is that DOPC protocells modified with ∼6 copies of the SP94 peptide have a differential Kd value (HCC/hepatocytes) of 2.25 × 104, which exceeds that of SP94-targeted DPPC protocells, DPPC liposomes, and DOPC liposomes by > 102. DOPC protocells, additionally, have a 104-fold higher affinity for HCC than for other control cells, including human endothelial cells, mononuclear cells, B lymphocytes, and T lymphocytes (see Supplementary Fig. 7). Also, the Kd value of DOPC protocells for Hep3B is 200-fold lower than that of free SP94 for Hep3B and nearly 50,000-fold lower than that of unmodified protocells for Hep3B (see Supplementary Fig. 7). If sub-nanomolar affinity is undesirable (e.g. results in reduced tumor penetration), the Kd values of SP94-targeted protocells can be precisely modulated by incorporating various amounts of fluid and non-fluid lipids into the SLB (see Supplementary Fig. 8).
DOPC protocells are uniquely able to target HCC at low peptide densities, and their dramatic differential affinity for HCC translates into selective internalization when the experimental temperature is raised from 4°C to 37°C. DOPC protocells modified with a low density of SP94 peptides (∼0.015 wt%) are efficiently endocytosed by Hep3B but not by hepatocytes, as demonstrated by the representative confocal fluorescence microscopy images shown in Figures 4d and 4e; see also Supplementary Table II, which lists average numbers of SP94-targeted protocells and liposomes internalized by Hep3B and hepatocytes. The efficacy with which targeted protocells are internalized by Hep3B depends largely on binding affinity, which can be modulated by changing bilayer fluidity and ligand density. However, it also depends on nanocarrier size (see Supplementary Fig. 9), with 50-nm protocells being most efficiently internalized (∼1800 particles per cell). This result provides evidence that internalization occurs via an endocytotic pathway, given that membrane wrapping occurs most efficiently for particles 30-nm to 60-nm in diameter11. Despite this observation, we use protocells 100-150-nm in diameter for targeted delivery, since the increased cargo capacity, which we measure to be proportional to the cube of the particle radius, more than compensates for the slightly reduced internalization efficiency.
To demonstrate that high affinity surface binding followed by receptor-mediated endocytosis enables targeted delivery of multicomponent cargos, we loaded four fluorescently-labeled surrogates, similar in size and charge to common therapeutic and diagnostic agents, within the protocell core. Figure 5a shows simultaneous encapsulation of a low molecular weight drug mimic (calcein), a siRNA mimic (dsDNA), a protein toxin mimic (RFP), and a model nanoparticle (water-soluble CdSe/ZnS quantum dots), all within a fluorescently-labeled porous silica particle that is completely encased in a fluorescently-labeled DOPC bilayer; a protocell 10-μm in diameter was employed in this experiment to enable optical imaging. The confocal slice (z = 5 μm) demonstrates that the multiple cargos are uniformly distributed throughout the silica core and that the SLB is intact and coherent.
Figure 5. Targeted delivery of multicomponent cargos to the cytosol and nuclei of HCC cells.
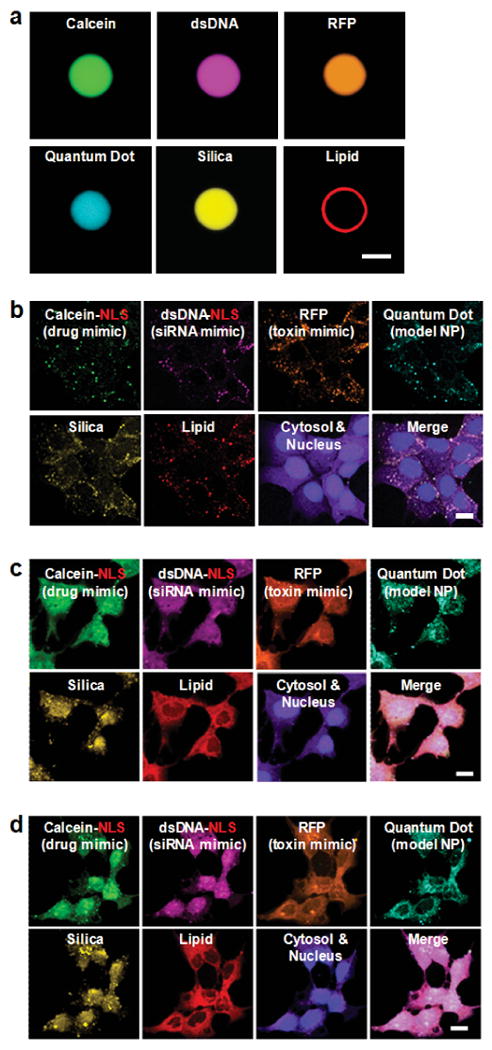
Alexa Fluor® 532-labeled nanoporous silica cores (yellow) were loaded with a multicomponent mixture of four surrogate cargos: calcein (green), an Alexa Fluor® 647-labeled double-stranded DNA oligonucleotide (magenta), red fluorescent protein (orange), and CdSe/ZnS quantum dots (teal). Cargos were sealed in the cores via fusion of Texas Red®-labeled DOPC liposomes (red) that contained 30 wt% cholesterol and 5 wt% PEG-2000 PE, and the resulting SLBs were modified with 0.015 wt% SP94 and 0.500 wt% H5WYG. Protocells were incubated with Hep3B cells (labeled with CellTracker™ Violet BMQC and Hoechst 33342) for 15 minutes, 4 hours, or 12 hours (respectively) at 37°C to collect the images shown in (b) – (d). (a) Hyperspectral confocal fluorescence microscopy slice (z = ∼5 μm) of a 10-μm protocell, demonstrating uniform loading of the nanoporous silica core and complete encapsulation of the core and cargos within the SLB. Particles 100 times larger than those used for all surface-binding, internalization, and delivery studies were used in this experiment to enable optical imaging and have a 2.5 × 105-fold higher capacity for the multicomponent mixture than protocells (100-150 nm in diameter) used to collect the images shown in (b) – (d). Scale bar = 5 μm. (b) – (d): Hyperspectral confocal fluorescence microscopy was employed to individually track the lipid and silica moieties of DOPC protocells (100-150-nm multimodal core), as well as the four surrogate cargos within the cytosol (purple) and nuclei (blue) of Hep3B cells as a function of time. (b) Within 15 minutes of exposing Hep3B to protocells loaded with the multicomponent mixture, the lipid, silica, and cargo moieties have a punctate appearance, indicating that protocells are localized within endosomes. (c) Within 4 hours, the H5WYG peptide promotes endosomal escape, thereby releasing the lipid, silica, and cargos into the cytosol of the Hep3B cells. (d) Within 12 hours, calcein and the dsDNA oligonucleotide, both of which are modified with a NLS, become concentrated in the nucleus, while the RFP and quantum dots (not modified with a NLS) remain largely localized in the cytosol. Protocells used to collect the images shown in (b) – (d) have a high capacity for the multicomponent mixture: 1010 protocells encapsulate 425 μM of calcein, 7.6 μM of the dsDNA oligonucleotide, 945 nM of RFP, and 1.98 × 1013 quantum dots. Scale bars = 20 μm.
As illustrated schematically in Figure 3 and confirmed by hyperspectral confocal fluorescence microscopy (Figs. 5b - 5d), delivery of encapsulated cargo to HCC using SP94-targeted DOPC protocells is achieved via the following successive steps: (1) multivalent binding of SP94 to HCC surface receptor(s) initiates receptor-mediated endocytosis, an internalization pathway that helps to circumvent MDR42. Peptide recruitment to the cell surface promotes the multivalent effects that enhance specificity. (2) As evidenced by the appearance of punctuate regions containing co-localized lipid, silica, and cargo in Figure 5b, protocells are rapidly endocytosed (t½ = 15 minutes) by Hep3B cells and reach a saturating intracellular concentration (∼500 protocells per Hep3B cell; see Supplementary Table II) within an hour. Given that the SP94 peptide directs protocells to lysosomes upon endocytosis by Hep3B (see Supplementary Fig. 10), we further modified the SLB with 0.500 wt% of a histidine-rich fusogenic peptide (H5WYG, H2N-GLFHAIAHFIHGGWHGLIHGWYGGGC-COOH43), which, in addition to preventing degradation of sensitive cargos in endolysosomes, promotes endosomal escape of protocells and cytosolic dispersion of encapsulated cargos (see Supplementary Fig. 11). (3) Endosome acidification destabilizes the SLB (see Supplementary Fig. 12), enabling encapsulated cargo to diffuse out of the nanoporous core. Additionally, protonation of imidazole moieties (pKa = 6.0) in the fusogenic peptide initiates osmotic swelling and membrane destabilization of endosomes via the ‘proton sponge’ mechanism44. As shown in Figure 5c, these events allow the four surrogate cargos, along with lipid and silica moieties of the protocell to become distributed throughout the cytosol within 4 hours. (4) Cargos modified with a nuclear localization sequence45 become concentrated in the nucleus, since the NLS promotes transport through the nuclear pore complex. Figure 5d demonstrates that NLS-modified calcein and dsDNA become localized in the nuclei of Hep3B cells within 12 hours, whereas RFP and quantum dots (not modified with the NLS) remain concentrated in the cytosol.
We have utilized the above sequence of events to deliver high payloads of various cytotoxic agents to HCC, including drugs and drug cocktails, siRNA cocktails (see Supplementary Figs. 13 and 14), and protein toxins (see Supplementary Figs. 15 and 16) without affecting the viability of hepatocytes and other control cells. Figure 6 compares the cargo capacity, time-dependent release characteristics, and selective cytotoxicity of SP94-targeted protocells and liposomes loaded with the chemotherapeutic drug, doxorubicin (DOX). Protocells, due to the high surface area and porosity of their nanoporous cores, have a 1000-fold higher capacity for DOX than similarly-sized liposomes (loaded via an ammonium phosphate gradient-based approach46) and can be engineered to release nearly 90% of their encapsulated DOX in a bioactive form upon endocytosis by HCC (see ‘Effective Capacity’ in Fig. 6a, left axis). Additionally, DOPC protocells exhibit long-term stability when maintained in a simulated body fluid (pH 7.4) at 37°C, whereas DOPC liposomes leak 90% of their encapsulated DOX within 72 hours and have a release profile comparable to that of the nanoporous core with no SLB. Thus, the fluid lipids that enable selective targeting at low peptide densities cannot be used in liposomal drug formulations, since pre-mature release of encapsulated cargo results in undesired toxicity to non-cancerous cells. Stable formulations of liposomal drugs require the use of fully saturated, high Tm lipids (e.g. DSPC, Tm = 55°C) and high concentrations of cholesterol, which act cooperatively to increase the lipid packing density and limit diffusion of the drug across the bilayer47. Even the stability of ‘gold standard’ liposomal doxorubicin (e.g. DSPC with 30 wt% cholesterol and 5 wt% PEG) remains limited, however, as up to 25% of the drug is released within 72 hours when exposed to a simulated body fluid at 37°C (see ‘DSPC Liposomes’ in Fig. 6b).
Figure 6. Cargo capacity, time-dependent release profiles, and concentration-dependent cytotoxicity of SP94-targeted protocells and liposomes that encapsulate chemotherapeutic drugs.

(a) Cargo capacity and cytotoxicity of protocells and liposomes loaded with doxorubicin (DOX). Left axis: the absolute and effective capacities of DOPC protocells, DOPC liposomes, and DSPC liposomes for DOX. Absolute capacity is defined as the concentration of DOX that can be physically encapsulated within 1010 particles, while effective capacity is the concentration of DOX that is released upon endocytosis by Hep3B in a form capable of intercalating nuclear DNA. DOPC protocells, when loaded with a cocktail of DOX, 5-fluorouracil (5-FU), and cisplatin, retain their high absolute and effective capacities. The liposome cocktail is composed of equal volumes of DOX-loaded, 5-FU-loaded, and cisplatin-loaded DSPC liposomes. DSPC liposomes that encapsulate 5-FU have an absolute capacity of 765 nM (per 1010 particles) and were prepared using the reverse-phase evaporation method described by B. Elorza, et al.52. DSPC liposomes that encapsulate cisplatin have an absolute capacity of 980 nM (per 1010 particles) and were prepared using the technique described by T. Peleg-Shulman, et al.53. Right axis: the number of DOX-loaded protocells or liposomes that must be added to 106 MDR1+ Hep3B cells to kill 90% of the cells in the population (LC90) within 24 hours. (b) The time-dependent release of DOX from DOPC protocells, DSPC liposomes, DOPC liposomes, and nanoporous silica cores when exposed to a simulated body fluid (pH 7.4) at 37°C for 21 days. (c) The time-dependent release of DOX from DOPC protocells, DSPC liposomes, and DOPC liposomes when exposed to a pH 5 citric acid buffer at 37°C for 12 hours. Acidic conditions, which mimic those of the endosome, destabilize the SLB and promote release of DOX from the protocell's nanoporous core. (d) Left axis: the number of MDR1+ Hep3B and hepatocytes that remain viable after exposure to 9.6 μM of free DOX, protocell-encapsulated DOX, or liposomal DOX for 24 hours at 37°C. 9.6 μM is the LC90 value of free DOX when exposed to Hep3B with induced MDR (MDR1+ phenotype) and was, therefore, selected as the standardized drug concentration. Cells were exposed to drugs and drug-loaded nanocarriers for 24 hours since the typical doubling time of HCC is 24-36 hours. Right axis: the number of MDR1+ Hep3B that remain viable after exposure to 2.4 μM of free DOX, protocell-encapsulated DOX, or liposomal DOX for 24 hours at 37°C; 2.4 μM is the LC50 value of free DOX. Sytox® Green nucleic acid stain and Alexa Fluor 647®-labeled annexin V were used to distinguish viable (double-negative) from non-viable (single- or double-positive) cells. All error bars represent 95% confidence intervals (1.96 σ) for n = 3.
Exposing protocells to a pH 5.0 buffer, which simulates the endosomal environment and destabilizes the SLB (see Supplementary Fig. 12), promotes rapid release of drugs loaded within the nanoporous core; DOPC protocells release 99% of their encapsulated DOX within 12 hours (see Fig. 6c). DSPC and DOPC liposomes release nearly all of their encapsulated DOX upon exposure to a pH 5.0 buffer for 4 hours (see Fig. 6c). Differences in absolute cargo capacities must be taken into account, however, to accurately compare the drug delivery capabilities of targeted protocells and liposomes. DOPC protocells release ∼50% of their encapsulated DOX within 4 hours, which corresponds to a drug concentration of nearly 500 μM when the protocell concentration is maintained at 1010 particles/mL. In comparison, 1010 liposomes release only ∼ 1 μM of DOX in the same period of time. It is important to note that the DSPC liposomes referred to in Figure 6 have a similar capacity for DOX (∼1.1 μM per 1010 particles, which corresponds to a drug:lipid ratio of 0.113:1) as other PEGylated liposomal doxorubicin formulations, including Doxil® (drug:lipid ratio of 0.125:1)47.
The unique properties of drug-loaded DOPC protocells modified with a minimal number of targeting peptides solve the conundrum of simultaneously achieving high targeting specificity, high cytotoxicity to the target cell, and low collateral damage to non-cancerous cells. Figure 6a (right axis) plots the number of DOX-loaded DOPC protocells, DSPC liposomes, and DOPC liposomes needed to kill 90% of Hep3B (LC90) with an induced MDR1 phenotype. We find that 105 fewer DOX-loaded protocells are necessary to achieve this LC90 value when compared to DOX-loaded DSPC or DOPC liposomes. Figure 6d (left axis) plots the percentage of Hep3B and hepatocytes that remain viable after exposure to either free DOX or to DOX encapsulated within DOPC protocells, DSPC liposomes, or DOPC liposomes for 24 hours at 37°C; here the total DOX concentration was normalized to 9.6 μM, which is the concentration of free DOX necessary to kill 90% of MDR1+ Hep3B within 24 hours. We observe that DOX-loaded DOPC protocells maintain greater than 90% hepatocyte viability, while killing nearly 97% of MDR1+ Hep3B. In comparison, DOX-loaded DSPC and DOPC liposomes are less efficient at killing HCC and cause significant cytotoxicity to non-cancerous cells. Figure 6d (right axis) shows the number of MDR1+ Hep3B that remain viable after incubation with a lower concentration (2.3 μM, the LC50 value of free DOX) of free DOX, DOX-loaded protocells, or DOX-loaded liposomes. This data is included to clearly demonstrate the enhanced killing efficacy of DOX-loaded protocells when compared to both free DOX and DOX-loaded liposomes, an observation that is further supported by the fact that DOX-loaded protocells decrease the LC90 value of free DOX (9.6 μM) to ∼145 nM. We attribute the striking differences shown in Figures 6a (right axis) and 6d to the 1000-fold higher capacity (Fig. 6a, left axis), the enhanced binding affinity (Fig. 4a), and the greater long-term stability (Fig. 6b) of DOPC protocells. These factors synergistically combine to provide dramatic improvements in selective cytotoxicity of cancer, while limiting undesired toxicity to normal hepatocytes. Protocells can, furthermore, be easily loaded with multicomponent cargos by simply soaking the nanoporous core in a solution of the desired cargos prior to fusion of the SLB. Figures 6a (right axis) and 6d show that, when loaded with a cocktail of DOX, 5-fluorouracil, and cisplatin (a chemotherapeutic drug cocktail known to be particularly effective against drug-resistant HCC48), as few as one SP94-targeted DOPC protocell is sufficient to kill a Hep3B cell with an induced MDR1 phenotype while maintaining > 90% hepatocyte viability. Similar results cannot be achieved using DOPC and DSPC liposomes, since liposomes cannot be loaded with drug cocktails using transmembrane pH gradient-based loading strategies. A cocktail of DSPC liposomes that individually encapsulate DOX, 5-FU, or cisplatin was employed as a control but failed to substantially improve upon the selective cytotoxicity of DOX-loaded DSPC liposomes (see Figs. 6a and 6d).
We have demonstrated that targeted protocells possess the high specificity, enhanced cargo capacity, and long-term stability necessary to deliver a variety of chemically disparate therapeutic and diagnostic agents to cancer cells with minimal non-specific binding and toxicity to normal cells. We have, furthermore, shown that the nanoporous core can be adapted to release encapsulated cargo within 24 hours or over the course of several weeks (see Supplementary Fig. 2) and that the SLB can be modified with a variety of ligands, including peptides, antibodies, and glycoproteins, in order to promote specific affinity for a target cell (see section 2 in Supplementary Figures and Legends). To date, no other nanoparticle-based delivery vehicle has been reported that possesses all of these attributes, making protocells the first example of a nanocarrier that simultaneously addresses the complex requirements of targeted, multicomponent delivery. Perhaps the most striking feature of protocells is their ability to deliver high concentrations of diverse cargos and ‘cocktails’ of chemically disparate components. For example, Supplementary Figures 13 and 14 report preliminary data regarding the killing efficacy of SP94-targeted protocells loaded with a siRNA cocktail that silences expression of epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor-2 (VEGFR-2), and platelet-derived growth factor receptor-α (PDGFR-α). Protocells encapsulate 1000-fold more siRNA than similarly-sized liposomes with the same bilayer composition and, when targeted with the SP94 peptide, induce apoptosis in 50% of Hep3B within 36 hours without affecting the viability of hepatocytes. Another distinctive characteristic of protocells is that the enhanced fluidity and stability of the SLB support multivalent peptide recruitment to surface receptors expressed by the target cell, which suggests that displaying two or more types of ligands on the protocell surface might enable complex binding interactions. We, therefore, expect that modifying the protocell SLB with ligand(s) that bind to surface receptor(s) uniquely or over-expressed by the target cell along with a ligand that promotes internalization (e.g. the octaarginine peptide, which stimulates macropinocytosis49) would enable both selective targeting and intracellular delivery for cancers where cell-specific receptors are not normally endocytosed.
Methods Summary
Nanoporous silica particles were synthesized and characterized as described previously26,50 and as detailed in Supplementary Figure 1 and the Supplementary Methods section. Particles larger than ∼150-nm in diameter were removed via differential centrifugation or size-exclusion chromatography (see Supplementary Figs. 1a and 1d). Protocells were formed by fusing ∼120-nm liposomes to the nanoporous core as reported previously23-25, and the composition of the SLB was optimized to reduce non-specific binding associated with cationic and, to a lesser extent, anionic lipids51 (see Supplementary Fig. 5). Zwitterionic lipids (DOPC or DPPC) with 5 wt% phosphatidylethanolamine (DOPE or DPPE, respectively), 5 wt% PEG-2000 PE (18:0 or 16:0, respectively), and 30 wt% cholesterol were used in all further studies; PEGylated lipids were incorporated into the liposomes used for fusion and are, therefore, expected to be present on both the inner and outer leaflets of the SLB. The size of the nanoporous core was also optimized to attain a balance between achievable cargo capacity and the rate of protocell internalization (see Supplementary Fig. 9); nanoparticles 100- to 150-nm in diameter were employed in the delivery of drugs, drug cocktails, siRNA cocktails, and protein toxins. The nanoporous cores were soaked in a 10 mM solution of cargo(s) for 1-12 hours prior to liposome fusion; individual components of the surrogate cargo mixture (Fig. 5) and the drug cocktail (Fig. 6) were loaded into nanoporous cores simultaneously (as opposed to sequentially). The rates of cargo release were optimized by incorporating various percentages of AEPTMS, an amine-containing silane, into the sol used to form nanoporous cores (see Supplementary Fig. 2). Particles containing 15 wt% AEPTMS were used to deliver drugs and drug cocktails (Fig. 6), while particles containing 20 wt% AEPTMS were used to deliver the multicomponent mixture (Fig. 5), the siRNA cocktail (Supplementary Figs. 13 and 14), and diphtheria toxin A-chain (Supplementary Figs. 15 and 16).
Supplementary Material
Acknowledgments
This work was supported by the NIH/Roadmap for Medical Research under grant PHS 2 PN2 EY016570B; NCI Cancer Nanotechnology Platform Partnership grant 1U01CA151792-01; the Air Force Office of Scientific Research grant FA 9550-07-1-0054/9550-10-1-0054; the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering; the Sandia National Laboratories' Laboratory Directed Research and Development (LDRD) program; the President Harry S. Truman Fellowship in National Security Science and Engineering at Sandia National Laboratories (C.E.A.); the UCLA Center for Nanobiology and Predictive Toxicology (NIEHS grant 1U19ES019528-01); and the NSF ERC Center for Environmental Implications of Nanotechnology at UCLA (NSF:EF-0820117). C.E.A. was supported by IGERT Fellowship Grant NSF DGE-0504276, and E.C.C. and N.J.C. were supported by NSF IGERT grant DGE- 0549500. T.N.H. was supported by NSF Nanoscience and Microsystems REU program (grant DMR-0649132) at the University of New Mexico Center for Micro-Engineered Materials. N.J.C and D.N.P. were supported by NSF PREM/DMR 0611616. Rebecca Lee provided guidance for imaging protocols and FRAP experiments, Mona Aragon created schematics, Robin Sewell performed nitrogen sorption experiments, and Ying-Bing Jiang performed TEM imaging. Cryogenic TEM was performed at Baylor College of Medicine (Houston, TX) by Caroline Jia-Yin Fu, Htet Khant, and Wah Chiu. Some images in this paper were generated in the University of New Mexico Cancer Center Fluorescence Microscopy Facility supported by NCRR, NSF, and NCI as detailed at http://hsc.unm.edu/crtc/microscopy/Facility.html. Data was generated in the Flow Cytometry Shared Resource Center supported by the University of New Mexico Health Sciences Center and the University of New Mexico Cancer Center. Sandia is a multiprogram laboratory operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Company, for the United States Department of Energy's National Nuclear Security Administration under contract DE-AC04-94AL85000.
Footnotes
Authors' Contributions Statement. C.E.A. engineered protocells for targeted delivery, performed most experiments, analyzed data, and wrote the manuscript; E.C.C. assisted with experiment coordination, data analysis, and manuscript preparation; G.K.P. performed confocal fluorescence microscopy imaging; D.P. synthesized and characterized multimodal particles; P.A.B. performed FRAP experiments; T.N.H. assisted with doxorubicin capacity and release studies; J.L. contributed to the development of the original protocell construct; N.C. developed the emulsion processing necessary to synthesize multimodal particles; B.P. and M.B.C. performed flow cytometry experiments; X.J. synthesized unimodal particles; D.R.D. performed SANS experiments and analyzed nitrogen sorption data; D.N.P. supervised development of the multimodal particles; D.G.E. supervised FRAP experiments; A.P. suggested the FRAP experiment and aided in its interpretation; P.N.D., C.L.W., B.C., W.W., and D.S.P. provided intellectual oversight for delivery experiments involving drugs, siRNA, and protein toxins; C.J.B. conceived of the protocell construct, provided overall intellectual guidance, performed final edits of the manuscript, and is PI of the major supporting grants.
References
- 1.Peer D, et al. Nanocarriers as an emerging platform for cancer therapy. Nature Nanotechnology. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 2.Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotech. 2006;24:1211–1217. doi: 10.1038/nbt1006-1211. [DOI] [PubMed] [Google Scholar]
- 3.Nel AE, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 4.Ferrari M. Cancer nanotechnology: Opportunities and challenges. Nature Reviews Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 5.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. Journal of Controlled Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 6.Matsumura Y, Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Research. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 7.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nature Reviews Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 8.Kohlschütter J, Michelfelder S, Trepel M. Drug delivery in acute myeloid leukemia. Expert Opinion on Drug Delivery. 2008;5:653–663. doi: 10.1517/17425247.5.6.653. [DOI] [PubMed] [Google Scholar]
- 9.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 10.Rai P, et al. Statistical pattern matching facilitates the design of polyvalent inhibitors of anthrax and cholera toxins. Nat Biotech. 2006;24:582–586. doi: 10.1038/nbt1204. [DOI] [PubMed] [Google Scholar]
- 11.Jiang W, KimBetty YS, Rutka JT, ChanWarren CW. Nanoparticle-mediated cellular response is size-dependent. Nat Nano. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 12.Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6:559–565. doi: 10.1038/nrc1891. [DOI] [PubMed] [Google Scholar]
- 13.Ferrari M. Nanogeometry: Beyond drug delivery. Nat Nano. 2008;3:131–132. doi: 10.1038/nnano.2008.46. [DOI] [PubMed] [Google Scholar]
- 14.Giri S, Trewyn BG, Stellmaker MP, Lin VSY. Stimuli-Responsive Controlled-Release Delivery System Based on Mesoporous Silica Nanorods Capped with Magnetic Nanoparticles. Angewandte Chemie International Edition. 2005;44:5038–5044. doi: 10.1002/anie.200501819. [DOI] [PubMed] [Google Scholar]
- 15.Lai CY, et al. A Mesoporous Silica Nanosphere-Based Carrier System with Chemically Removable CdS Nanoparticle Caps for Stimuli-Responsive Controlled Release of Neurotransmitters and Drug Molecules. Journal of the American Chemical Society. 2003;125:4451–4459. doi: 10.1021/ja028650l. [DOI] [PubMed] [Google Scholar]
- 16.Liong M, et al. Multifunctional Inorganic Nanoparticles for Imaging, Targeting, and Drug Delivery. ACS Nano. 2008;2:889–896. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen TD, et al. A reversible molecular valve. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10029–10034. doi: 10.1073/pnas.0504109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel K, et al. Enzyme-Responsive Snap-Top Covered Silica Nanocontainers. Journal of the American Chemical Society. 2008;130:2382–2383. doi: 10.1021/ja0772086. [DOI] [PubMed] [Google Scholar]
- 19.Vallet-Regí M, Balas F, Arcos D. Mesoporous Materials for Drug Delivery. Angewandte Chemie International Edition. 2007;46:7548–7558. doi: 10.1002/anie.200604488. [DOI] [PubMed] [Google Scholar]
- 20.Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 21.Gordon AN, et al. Recurrent Epithelial Ovarian Carcinoma: A Randomized Phase III Study of Pegylated Liposomal Doxorubicin Versus Topotecan. J Clin Oncol. 2001;19:3312–3322. doi: 10.1200/JCO.2001.19.14.3312. [DOI] [PubMed] [Google Scholar]
- 22.Peer D, Zhu P, Carman CV, Lieberman J, Shimaoka M. Selective gene silencing in activated leukocytes by targeting siRNAs to the integrin lymphocyte function-associated antigen-1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:4095–4100. doi: 10.1073/pnas.0608491104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu JW, Jiang XM, Ashley C, Brinker CJ. Electrostatically Mediated Liposome Fusion and Lipid Exchange with a Nanoparticle-Supported Bilayer for Control of Surface Charge, Drug Containment, and Delivery. Journal of the American Chemical Society. 2009;131:7567–+. doi: 10.1021/ja902039y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu JW, Stace-Naughton A, Brinker CJ. Silica nanoparticle supported lipid bilayers for gene delivery. Chemical Communications. 2009:5100–5102. doi: 10.1039/b911472f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu JW, Stace-Naughton A, Jiang XM, Brinker CJ. Porous Nanoparticle Supported Lipid Bilayers (Protocells) as Delivery Vehicles. Journal of the American Chemical Society. 2009;131:1354–+. doi: 10.1021/ja808018y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu YF, et al. Aerosol-assisted self-assembly of mesostructured spherical nanoparticles. Nature. 1999;398:223–226. [Google Scholar]
- 27.Evans E. Entropy-driven tension in vesicle membranes and unbinding of adherent vesicles. Langmuir. 1991;7:1900–1908. [Google Scholar]
- 28.Komura S, Shimokawa N, Andelman D. Tension-Induced Morphological Transition in Mixed Lipid Bilayers. Langmuir. 2006;22:6771–6774. doi: 10.1021/la053135x. [DOI] [PubMed] [Google Scholar]
- 29.Lipowsky R. The conformation of membranes. Nature. 1991;349:475–481. doi: 10.1038/349475a0. [DOI] [PubMed] [Google Scholar]
- 30.Mutz M, Helfrich W. Unbinding transition of a biological model membrane. Physical Review Letters. 1989;62:2881. doi: 10.1103/PhysRevLett.62.2881. [DOI] [PubMed] [Google Scholar]
- 31.Netz RR, Lipowsky R. Stacks of fluid membranes under pressure and tension. Europhysics Letters. 1995;29:345–350. [Google Scholar]
- 32.Swain PS, Andelman D. Supported membranes on chemically structured and rough surfaces. Physical Review E. 2001;63:051911. doi: 10.1103/PhysRevE.63.051911. [DOI] [PubMed] [Google Scholar]
- 33.Bothun GD, Knutson BL, Strobel HJ, Nokes SE. Liposome Fluidization and Melting Point Depression by Pressurized CO2 Determined by Fluorescence Anisotropy. Langmuir. 2004;21:530–536. doi: 10.1021/la0496542. [DOI] [PubMed] [Google Scholar]
- 34.Doshi DA, et al. Neutron Reflectivity Study of Lipid Membranes Assembled on Ordered Nanocomposite and Nanoporous Silica Thin Films. Langmuir. 2005;21:2865–2870. doi: 10.1021/la0471240. [DOI] [PubMed] [Google Scholar]
- 35.Daillant J, et al. Structure and fluctuations of a single floating lipid bilayer. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:11639–11644. doi: 10.1073/pnas.0504588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malaquin L, Charitat T, Daillant J. Supported bilayers: Combined specular and diffuse X-ray scattering. European Physical Journal E. 2010;31:285–301. doi: 10.1140/epje/i2010-10578-2. [DOI] [PubMed] [Google Scholar]
- 37.Huang SCJ, et al. Formation, Stability, and Mobility of One-Dimensional Lipid Bilayers on Polysilicon Nanowires. Nano Letters. 2007;7:3355–3359. doi: 10.1021/nl071641w. [DOI] [PubMed] [Google Scholar]
- 38.Swain PS, Andelman D. The Influence of Substrate Structure on Membrane Adhesion. Langmuir. 1999;15:8902–8914. [Google Scholar]
- 39.Lo A, Lin CT, Wu HC. Hepatocellular carcinoma cell-specific peptide ligand for targeted drug delivery. Molecular Cancer Therapeutics. 2008;7:579–589. doi: 10.1158/1535-7163.MCT-07-2359. [DOI] [PubMed] [Google Scholar]
- 40.Chen Z, et al. DNA translocation through an array of kinked nanopores. Nature Materials. 2010;9:667–675. doi: 10.1038/nmat2805. [DOI] [PubMed] [Google Scholar]
- 41.Lu YF, et al. Continuous formation of supported cubic and hexagonal mesoporous films by sol gel dip-coating. Nature. 1997;389:364–368. [Google Scholar]
- 42.Goren D, et al. Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clinical Cancer Research. 2000;6:1949–1957. [PubMed] [Google Scholar]
- 43.Midoux P, Kichler A, Boutin V, Maurizot JC, Monsigny M. Membrane Permeabilization and Efficient Gene Transfer by a Peptide Containing Several Histidines. Bioconjugate Chemistry. 1998;9:260–267. doi: 10.1021/bc9701611. [DOI] [PubMed] [Google Scholar]
- 44.Behr JP. The Proton Sponge: a Trick to Enter Cells the Viruses Did Not Exploit. CHIMIA International Journal for Chemistry. 1997;51:34–36. [Google Scholar]
- 45.Subramanian A, Ranganathan P, Diamond SL. Nuclear targeting peptide scaffolds for lipofection of nondividing mammalian cells. Nat Biotech. 1999;17:873–877. doi: 10.1038/12860. [DOI] [PubMed] [Google Scholar]
- 46.Fritze A, Hens F, Kimpfler A, Schubert R, Peschka-Süss R. Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2006;1758:1633–1640. doi: 10.1016/j.bbamem.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 47.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing Liposomes for Delivery of Chemotherapeutic Agents to Solid Tumors. Pharmacol Rev. 1999;51:691–744. [PubMed] [Google Scholar]
- 48.Lee JO, et al. Combination chemotherapy with capecitabine and cisplatin for patients with metastatic hepatocellular carcinoma. Annals of Oncology. 2009;20:1402–1407. doi: 10.1093/annonc/mdp010. [DOI] [PubMed] [Google Scholar]
- 49.Khalil IA, Kogure K, Futaki S, Harashima H. High Density of Octaarginine Stimulates Macropinocytosis Leading to Efficient Intracellular Trafficking for Gene Expression. Journal of Biological Chemistry. 2006;281:3544–3551. doi: 10.1074/jbc.M503202200. [DOI] [PubMed] [Google Scholar]
- 50.Carroll NJ, Pylypenko S, Atanassov PB, Petsev DN. Microparticles with Bimodal Nanoporosity Derived by Microemulsion Templating. Langmuir. 2009 doi: 10.1021/la900988j. [DOI] [PubMed] [Google Scholar]
- 51.Xia T, et al. Comparison of the Mechanism of Toxicity of Zinc Oxide and Cerium Oxide Nanoparticles Based on Dissolution and Oxidative Stress Properties. ACS Nano. 2008;2:2121–2134. doi: 10.1021/nn800511k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elorza B, Elorza MA, Frutos G, Chantres JR. Characterization of 5-fluorouracil loaded liposomes prepared by reverse-phase evaporation or freezing-thawing extrusion methods: study of drug release. Biochimica et Biophysica Acta. 1993;1153:135–142. doi: 10.1016/0005-2736(93)90398-j. [DOI] [PubMed] [Google Scholar]
- 53.Peleg-Shulman T, Gibson D, Cohen R, Abra R, Barenholz Y. Characterization of sterically stabilized cisplatin liposomes by nuclear magnetic resonance. Biochimica et Biophysica Acta. 2001;1510:278–291. doi: 10.1016/s0005-2736(00)00359-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.