

Abstract
Prohibitin is an essential mitochondrial protein that has been implicated in a wide variety of functions in many cell types, but its role in neurons remains unclear. In a proteomic screen of rat brains in which ischemic tolerance was induced by electrical stimulation of the cerebellar fastigial nucleus, we found that prohibitin is upregulated in mitochondria. This observation prompted us to investigate the role of prohibitin in neuronal death and survival. We found that prohibitin is upregulated also in the ischemic tolerance induced by transient ischemia in vivo, or oxygen–glucose deprivation in neuronal cultures. Cell fractionation and electron-microscopic immunolabeling studies demonstrated that prohibitin is localized to neuronal mitochondria. Upregulation of prohibitin in neuronal cultures or hippocampal slices was markedly neuroprotective, whereas prohibitin gene silencing increased neuronal vulnerability, an effect associated with loss of mitochondrial membrane potential and increased mitochondrial production of reactive oxygen species. Prohibitin upregulation was associated with reduced production of reactive oxygen species in mitochondria exposed to the complex I inhibitor rotenone. In addition, prohibitin protected complex I activity from the inhibitory effects of rotenone. These observations, collectively, establish prohibitin as an endogenous neuroprotective protein involved in ischemic tolerance. Prohibitin exerts beneficial effects on neurons by reducing mitochondrial free radical production. The data with complex I activity suggest that prohibitin may stabilize the function of complex I. The protective effect of prohibitin has potential translational relevance in diseases of the nervous system associated with mitochondrial dysfunction and oxidative stress.
Introduction
Mitochondrial failure is a key contributor to the pathobiology of ischemic brain injury (Moskowitz et al., 2010). Mitochondria are a major source of postischemic reactive oxygen species (ROS), the production of which is triggered by excessive Ca2+ accumulation and electron transfer impairment (Fiskum et al., 2004). Damaged mitochondria fail to produce ATP leading to bioenergetic failure (Nicholls, 2008). In addition, mitochondrial Ca2+ overload induces permeability transition (Halestrap et al., 2002; Starkov et al., 2004; Chinopoulos and Adam-Vizi, 2006; Halestrap, 2006), an event that causes the collapse of the mitochondrial membrane potential (Δψ), failure of ATP synthesis, and activation of cell death pathways (Chan, 2004; Danial and Korsmeyer, 2004; Jemmerson et al., 2005).
Mitochondria are involved in the powerful neuroprotective effect exerted by ischemic preconditioning (PC), a phenomenon in which ischemia not sufficient to produce extensive cell death protects the brain from subsequent lethal ischemia (Gidday, 2006; Dirnagl et al., 2009; Iadecola and Anrather, 2011). In addition to hypoxia–ischemia, PC can be induced by a wide variety of interventions including, anesthetics, toxins, and electrical stimulation of the cerebellar fastigial nucleus (FN) (Reis et al., 1997; Gidday, 2006). Several lines of evidence point to mitochondria as key players in the mechanisms of ischemic tolerance (Chen et al., 2007; Halestrap et al., 2007). Thus, PC preserves oxidative phosphorylation (Dave et al., 2001), improves mitochondrial resistance to Ca2+-induced depolarization (Cho et al., 2005), and improves the respiration of synaptic mitochondria (Dave et al., 2008). However, the mitochondrial proteins that mediate this protective effect have yet to be identified.
Prohibitin (PHB), a highly conserved protein, is involved in multiple cellular processes including suppression of cell growth, mitochondrial function, and protection from senescence (Mishra et al., 2006, 2010; Artal-Sanz and Tavernarakis, 2009; Merkwirth and Langer, 2009). In human-derived cell lines, PHB is associated with respiratory chain complex I in the inner mitochondrial membrane and contributes its assembly and stability (Bourges et al., 2004). PHB is upregulated in the rat hippocampus in exercise-induced neuroplasticity and in cardiac cells after ischemic–hypoxic preconditioning (Ding et al., 2006; Kim et al., 2006; Muraguchi et al., 2010). Furthermore, PHB downregulation in endothelial cells increases mitochondrial ROS production and promotes a senescent phenotype (Schleicher et al., 2008). These observations raise the possibility that PHB is an endogenous protective protein, but its role in brain remains to be established.
In a proteomic screening for proteins involved in the ischemic tolerance induced by FN stimulation, we found that PHB is upregulated in mitochondria. This observation prompted us to investigate whether PHB is present in brain mitochondria and protects neurons against injury. We found that PHB is localized in neuronal mitochondria and, in addition to FN stimulation, is also upregulated in a number of PC paradigms. Furthermore, PHB expression reduced neuronal death in different injury modalities, whereas its downregulation increased neuronal susceptibility to injury. The protective effect of PHB was associated with suppression of mitochondrial ROS production and preservation of complex I function. The findings identify PHB as a powerful endogenous neuroprotective protein, which acts by supporting mitochondrial function and may have translational potential in several brain diseases.
Materials and Methods
Materials.
Tissue culture reagents (medium, serum, and salt solutions), antibody for cytochrome oxidase subunit IV (COX IV), and the indicators MitoSOX Red, Mitotracker Red, dihydroethidium (DHE), and DAPI were purchased from Invitrogen. Staurosporine, Percoll, and other chemicals were from Sigma-Aldrich. Anti-PHB antibodies were from NeoMarkers. Antibodies for histone H3 and calreticulin were from Cell Signaling. The GAPDH antibody was from Millipore.
In vivo and in vitro PC models.
Experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee. Surgical procedures for electrical stimulation of the FN in male Sprague Dawley rats (body weight, 220–250 g) have been described in detail previously (Reis et al., 1991; Zhou et al., 2001). Briefly, rats were anesthetized with isoflurane (2–3% in 100% O2) and placed on a stereotaxic frame (Kopf). An electrode was placed in the FN (stereotaxic coordinates: 5 mm rostral, 0.8 mm lateral, and 2 mm above the level of the obex) and stimuli were delivered (100 μA; 50 Hz; 1 s on/1 s off) for 60 min (Reis et al., 1991). Stimulation of the dentate nucleus (DN) with the same parameters, a procedure that does not induce preconditioning (Reis et al., 1991), was used as a control. Three days later, a time when PC is well established (Reis et al., 1998), brains were removed and processed for mitochondrial isolation (see below). For ischemic PC, C57BL/6 male mice were anesthetized with isoflurane (2–3%) and subjected to three episodes of bilateral common carotid artery occlusion (BCCAO), each lasting 1 min followed by 5 min reperfusion, a procedure that results in induction of tolerance (Cho et al., 2005). Brains were harvested 24 h later and processed for mitochondrial fractionation (see below).
For PC in vitro, medium in neuronal cultures (12 d in culture) was replaced with buffered salt solution without glucose and subjected to oxygen–glucose deprivation (OGD) for 60 min in a hypoxia chamber (Billups-Rothenberg) by flushing the chamber with 95% N2 and 5% CO2, as previously described (Tauskela et al., 2003). This short OGD protocol (PC-OGD) does not result in cell death, as assessed by morphological criteria, but induces tolerance to subsequent exposure to a lethal OGD challenge of 4 h duration. Sham cultures were treated with buffer containing glucose under normoxia and served as controls. Cells were washed and lysed 24 h later for PHB protein level examination. In some studies, preconditioned and sham-treated cultures were subjected to OGD for 4 h to produce cell damage, and cell viability was assessed by morphological criteria 24 h later as described previously (Zhou et al., 2005a).
Mitochondria isolation and subcellular fractionation.
Brains from FN- or DN-stimulated rats were removed, and mitochondria were isolated from cortex by differential centrifugation and purified by Percoll gradient as described previously (Zhou et al., 2005b). Mitochondria from PC12 cells that were transfected with PHB and vector plasmids were isolated with the same procedure, but without the gradient purification step. Subcellular fractionation of mouse brain was performed using a commercial kit (Calbiochem) following the manufacturer's instructions. Proteins for the mitochondrial, nuclear, membrane, and cytosolic fractions were loaded on to SDS gels in equal amounts, and the presence of PHB was assessed by Western blot.
Two-dimensional gel electrophoresis.
Two-dimensional (2-D) gel electrophoresis of mitochondrial proteins isolated from FN- or DN-stimulated rats was performed with a Bio-Rad Mini-Protean 2-D system. Mitochondrial proteins (60 μg) were electrofocused in the first dimension based on difference in isoelectric points of proteins and separated on second dimension (12% SDS-slab gel) based on difference in molecular weight. After the second dimension separation, the protein spots in the gel were visualized by staining with SYPRO Ruby Red (Invitrogen). Gel images were captured and analyzed by a Kodak digital imaging station 2000R (Kodak). The stained protein spots were excised and submitted for protein identification by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (performed by Biotechnology Core Facility of Cornell University, Ithaca, NY).
Western blot.
After determination of protein concentration with DC reagents (Bio-Rad), equal amount of proteins were gel-separated and transferred to PVDF membranes. Membranes were incubated with specific antibodies in appropriate dilutions after blocking with 5% dry milk in PBS for 1 h at room temperature. The membranes were washed three times with PBST (PBS plus 0.1% Tween 20), incubated with appropriate secondary antibodies for 1 h, washed, and developed by an enhanced chemiluminescence system (ECL; GE Healthcare). Band intensity was analyzed by using a Kodak Digital Imaging Station.
Plasmids construction.
Two different human PHB expression plasmid constructs were used in this study. The first contained the human PHB mRNA coding sequence and was made by inserting a PCR amplified human PHB sequence into pCDNA-3 vector. The primer sequences used in the PCR were as follows: forward primer, 5′-GCGGATCCGCCACCatgGCTGCCAAAGTGTTTGAGTCC; reverse primer, 5′-GCGAATTCACTGGGGCAGCTGGAGGAGC. This construct was used to study the effect of PHB expression in primary neurons. The second construct contains a 10 aa c-Myc tag at the C terminus of PHB in the first construct. The primer sequence for generating the Myc-tagged PHB is as follows: 5′-GCGAATTCACAGGTCTTCCTCACTGATCAGCTTCTGTTCCTGGGGCAGCTGGAGGAGCACGGAC. All the constructs were checked for sequence integrity and orientations by DNA sequencing on both strands. The plasmids were purified using QIAGEN kits.
DNA and siRNA transfection into PC12 cells and primary neurons.
PC12 cells were transfected with PHB and vector control constructs using the Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Three days after transfection, transfected cells were selected with G418 and the stable transfected cells were expanded for experiments. To overexpress PHB in primary neurons, PHB plasmid was cotransfected with the green fluorescent protein (GFP) plasmid by either Lipofectamine 2000 reagent (Invitrogen) or by electroporation (Nucleofector; Amaxa) with transfection efficiency of 3–5% for Lipofectamine and up to 15% for Nucleofector. The primary neurons were prepared from cortices of 15–16 d C57BL/6 mouse embryos. Five days after transfection, the cultures were used for viability experiments after exposure to staurosporine (STS) or xanthine/xanthine oxidase (X/XO) (see below). siRNA was introduced into primary neurons by electroporation. Two premade siRNAs specific for mouse PHB were purchased from Ambion. The target sequences were as follows: GGGACUCAUUUCCUCAUCCtt (siPHB-1); GGAGUUCACAGAGGCAGUAtt (siPHB-2). siRNAs that do not have homolog to known mammalian sequence from both Ambion and QIAGEN were used to serve as negative controls. After electroporation-assisted transfection, primary neurons were plated into 24-well plates and cultured for 5 or 13 d before being used for experiments.
Neuronal cell death induction and analysis.
Five days after transfection, neuronal cultures were treated with STS (0.25 μm) or X/XO (150 μm/15 mU) for 1 h. The cells were washed, fixed (4% paraformaldehyde), and stained with the nuclear dye DAPI (0.5 μg/ml) 24 h after treatment. Cellular and nuclear morphology were viewed with an inverted fluorescence microscope (Nikon), and frames containing GFP+ cells were recorded through a CCD camera. For each GFP+ cell-containing frame, three images were taken: one bright field, one for the GFP+ cells through a green filter, and the third one for DAPI-stained nuclei through a blue filter. To score dead and live cells that were GFP+, the green and blue images were merged and the cell debris were excluded in comparison with the bright-field images. The cells with fragmented and condensed nuclei were scored as dead, while cells with normal nuclei were scored as alive (Zhou et al., 2005a). For cells transfected with siRNA, the viability was assayed either using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) reagent (Promega) or by trypan blue staining.
Organotypic hippocampal slice culture and OGD treatment.
All procedures involving newborn mice for hippocampal slices cultures were approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee. Hippocampal slice cultures were prepared according to the procedure of Muller et al. (2001) as previously described (Kawano et al., 2006; Kim et al., 2007; Yin et al., 2010). Slices were maintained in a humidified chamber and subjected to 45 min of OGD 14 d later. Procedures for OGD were identical with those used in neuronal cultures and described above. Cell death in the CA1 region of the hippocampus was assessed by propidium iodide (PI) staining 24 h after OGD as described previously (Kawano et al., 2006; Kim et al., 2007; Yin et al., 2010).
Mitochondrial membrane potential measurements.
Changes in mitochondrial membrane potential (Δψ) in neurons were assessed using JC-1 as an indicator (Reers et al., 1991). Neurons transfected with PHB siRNA were first treated with X/XO. After 5 h, culture was incubated with JC-1 (3 μm) for 30 min at 37°C. The cells were washed and the intensity of red (emission, 590 nm) and green (emission, 530 nm) fluorescence was measured using a microplate reader (PerkinElmer HTS 7000 Plus). The ratio of red to green was calculated and expressed as percentage of controls (Reers et al., 1991).
ROS measurement in neurons.
ROS production in neurons was assessed with MitoSOX Red or DHE as an indicator. Primary neuronal cultures cotransfected with PHB and GFP were incubated with MitoSOX Red (4 μm) or DHE (5 μm) in the medium according to the manufacturer's instructions. MitoSOX was washed out after 10 min, whereas DHE was left in throughout the fluorescence measurement procedure. Identical cultures cotransfected with empty vector and GFP served as controls. Using an inverted fluorescence microscope, baseline images of MitoSOX or DHE (red) fluorescence were captured at 1 min intervals for 5 min. Then, the mitochondrial complex I inhibitor rotenone was added (5 μm) and images of the same cells were acquired for 30 min (MitoSOX) or 40 min (DHE) at 1 min intervals. Fluorescence intensity of each GFP+ cell was measured using IPLab software, and the results were expressed as percentage change over initial fluorescence units for MitoSOX or as relative fluorescence intensity increase from the baseline for DHE.
Adenoassociated virus-mediated PHB expression in hippocampal slice cultures.
Recombinant viral vectors were constructed by ligating human PHB cDNA tagged with a Myc fragment into an adenoassociated virus (AAV)-IRES-eGFP plasmid (Stratagene). The pAAV-IRES-eGFP vector contained a dicistronic expression cassette in which the recombinant green fluorescent protein (eGFP) is expressed from a second open reading frame on the same transcript as PHB. Therefore, eGFP fluorescence can be used as an indicator of infection efficiency. The integrity of the viral vector construct was verified by DNA sequencing and restriction digestion. rAAV virus vectors were produced by University of Iowa Gene Transfer Vector Core. The empty vector without PHB sequence was used as controls.
Mitochondrial complex I activity assay.
All assays were performed at 30°C. Before analysis, samples were freeze–thawed and gently shaken three times to ensure mitochondrial lysis was complete. Complex I activity was determined as described previously (Birch-Machin and Turnbull, 2001), following the oxidation of NADH at 340 nm using ubiquinone-1 as the electron acceptor.
Electron-microscopic immunolabeling.
Sprague Dawley rats (180–200 g; Charles River) were deeply anesthetized by intraperitoneal injection of sodium pentobarbital (150 mg/kg) before fixation of their brain tissue by vascular perfusion with a solution of 3.75% acrolein and 2% paraformaldehyde. The fixation procedure and electron-microscopic immunolabeling have been described in detail previously (Chan et al., 1990). Briefly, coronal sections (40 μm thick) of the aldehyde-fixed brain tissue were cut through the somatosensory cortex using a Leica Vibratome (Leica Microsystems). These sections were freeze–thawed to enhance penetration and placed in 1% sodium borohydride in 0.1 m PB to remove active aldehyde before processing for immunoperoxidase labeling. For this, the tissue sections were sequentially incubation at room temperature in mouse anti-prohibitin (1 μg/ml; 24 h), biotinylated goat anti-mouse (IgG; 1:200) (Jackson ImmunoResearch Laboratories), and Vectastain ABC Elite kit (Vector Laboratories). All incubations were separated by washes in 0.1 m PBS The product was visualized by reaction in DAB (3,3′-diaminobenzadine) (Sigma-Aldrich) and hydrogen peroxide. Controls consisted of processing the tissue for immunoperoxidase with omission of the primary antibody. For electron-microscopic analysis, the tissue was postfixed in 2% osmium tetroxide and embedded in plastic using conventional methods. Images were collected with a digital camera on a CM10 electron microscope (FEI).
Statistical analysis.
Data are expressed as means ± SEM. Two-group differences were evaluated by the two-tailed paired or unpaired Student's t test, as appropriate.
Results
PHB is upregulated in mitochondria by PC stimuli
Mitochondrial proteins from rats in which the FN was electrically stimulated were resolved by 2-D gel electrophoresis. Rats in which the DN was stimulated, a procedure that does not induce ischemic tolerance (Reis et al., 1991), served as controls. Analysis of the 2-D gel from mitochondria revealed two protein spots that had consistently higher intensity in FN-stimulated brain (n = 6) than in controls (n = 6) (Fig. 1A). One spot corresponded to a 30 kDa protein identified as murine PHB with 100% probability (Fig. 1A). The other spot corresponded to a 26 kDa protein identified as annexin V binding protein with 60% probability (Fig. 1A). We focused subsequent analyses on PHB because of the presumed cytoprotective properties of this protein (Mishra et al., 2010). The upregulation of PHB was confirmed by Western blotting of protein samples from mitochondria isolated from FN-stimulated rat brain (Fig. 1B,C). Therefore, FN stimulation, a potent PC stimulus, upregulates PHB in mitochondria. Next, we sought to determine whether PHB is also upregulated by other PC stimuli. Ischemic PC produced by episodes of transient BCCAO in mice leads to substantial protection against focal cerebral ischemic injury (Cho et al., 2005). BCCAO increased PHB levels in brain mitochondria 24 h later (Fig. 2A,B), a time frame that coincides with neuroprotection in this model (Cho et al., 2005). We also examined whether PHB is upregulated in an in vitro PC model. Sublethal OGD rendered primary neuronal cultures more resistant to lethal OGD administered 24 h later (Fig. 2E), an effect accompanied by an increase of PHB levels (Fig. 2C,D). Thus, PHB is upregulated in mitochondria by PC stimuli both in vitro and in vivo.
Figure 1.

Identification of PHB by 2-D gel electrophoresis and mass spectrometry. A, Mitochondrial proteins from FN- and DN-stimulated rat brain were separated on 2-D gels, and protein spots were visualized by Ruby Red staining. The open arrow indicates the spot that corresponds to PHB with a probability of 100%. The closed arrow indicates a spot that corresponds to annexin V binding protein with 60% probability. B, Western blot of mitochondrial proteins from FN- and DN-stimulated rat brain confirming that PHB is upregulated in mitochondria of brains preconditioned with FN stimulation. The mitochondrial marker cytochrome c oxidase (COX IV) was used to assure equal gel loading. Each lane contains 10 μg of mitochondria. C, Quantification of band intensity in B. *p < 0.05; n = 7/group. Error bars indicate SEM.
Figure 2.

PHB is upregulated in models of ischemic preconditioning in vivo and in vitro. A, PHB is upregulated in mitochondria of brains preconditioned by BCCAO. B, Quantification of band intensity in A (*p < 0.05; n = 7). C, PHB is upregulated in cell lysates from neuronal cultures preconditioned with OGD. D, Quantification of band intensity in C (*p < 0.05; n = 7/group). E, Neurons preconditioned with a nonlethal OGD episode are more resistant to damage produced by lethal OGD. Cell viability was assessed by morphological criteria 24 h after lethal OGD (*p < 0.05; n = 4/group with >1000 cells/group). Error bars indicate SEM.
PHB localization in neuronal mitochondria
The cellular location of PHB depends on the species and cell type. In yeast and plant, PHB has been localized to mitochondria (Ahn et al., 2006), whereas in mammalian cells, in addition to mitochondria (Ikonen et al., 1995), PHB was also found in nuclei (Wang et al., 2002), and cytoplasmic membrane (Terashima et al., 1994). Therefore, we examined PHB localization in brain tissue and neurons. As a first approach, we analyzed PHB cellular distribution by subcellular fractionation of mouse brain. PHB was found mainly in mitochondrial fractions but was not detectable in either nuclear or cytosol fractions (Fig. 3A). To further study the localization of PHB in neurons, we used electron-microscopic immunoperoxidase labeling in rat neocortex. In favorably cut mitochondria, PHB immunoreactivity was distributed along inner membranes but was more commonly localized to outer membranes having the greatest access to immunoreagents (Fig. 3B,C). The immunolabeled mitochondria were seen in somatic as well as in dendritic and axonal compartments of neurons as well as in glia. In addition, the PHB immunoreactivity was often seen along membranes of smooth endoplasmic reticulum and selective portions of both neuronal (Fig. 3B) and glial membranes (Fig. 3C). Finally, we determined the localization of PHB immunoreactivity in primary neuronal cultures. Neurons were double-labeled with anti-PHB antibody and the mitochondrial marker Mitotracker Red (Poot et al., 1996). The staining pattern for PHB matched well with that of Mitotracker Red (Fig. 3D), suggesting a mitochondrial localization of PHB. These observations, collectively, indicate that PHB is expressed in the rat brain and cultured neuronal cells, where it localizes prevalently in mitochondria.
Figure 3.
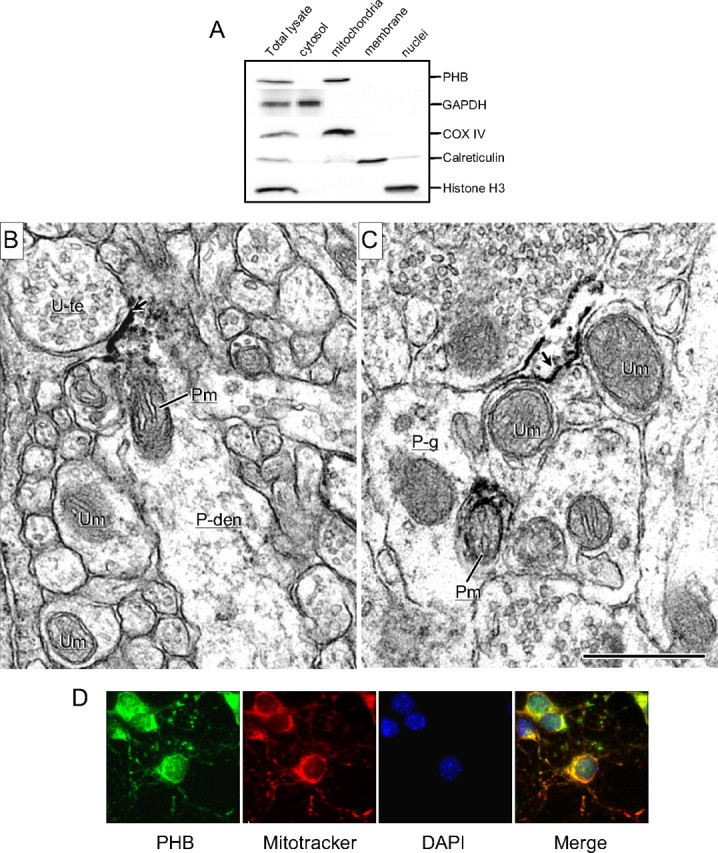
Neuronal localization of PHB. A, Subcellular fractionation of mouse neocortex. PHB is present mainly in the mitochondrial fraction and is not observed in the nuclear or cytosolic fraction. COX IV, histone H3, GAPDH, and calreticulin were used as mitochondrial, nuclear, cytosolic, and membrane markers, respectively. Results shown are representative from three separate experiments. B, C, Electron micrographs showing the immunoperoxidase localization of PHB in neuronal and glial profiles in the rat somatosensory cortex. B, PHB immunoreactivity (ir) is associated with inner and outer membranes of a mitochondrion (Pm) in a dendrite (P-den), located near an intensely labeled plasma membrane (arrow) contacted by an unlabeled terminal (U-te). Other neuronal processes within the neuropil contain unlabeled mitochondria (Um). C, PHB labeling is located on the plasmalemma (arrow) of a glial process (P-g) that sheaths small neuronal process containing mitochondria, which are either PHB-labeled (P-m) or unlabeled (U-m). Scale bar, 500 nm. D, In neuronal cultures, PHB immunoreactivity is colocalized with the mitochondrial marker MitoTracker Red.
Expression of PHB in neurons exerts a protective effect
Next, we sought to determine whether PHB protects neurons from injurious stimuli. PHB cDNA and GFP were cotransfected in primary neuronal cultures and the effect of PHB expression on cell viability was assessed in different injury paradigm. Neurons cotransfected with the vector and GFP were used as control. Because of the low transfection efficiency of primary neurons, we first identified the GFP-positive cells in both PHB and vector cotransfected cells, and then assessed cell viability based on nuclear morphology revealed by DAPI staining. In GFP-positive neurons cotransfected with vector control plasmid, 1 h incubation with the apoptotic inducer STS led to substantial cell death 24 h later (Fig. 4A). GFP-positive neurons cotransfected with PHB showed a significant improvement in viability (Fig. 4A; p < 0.001 from vector). Similarly, exposure of the cultures to oxidative stress produced by X/XO resulted in better survival in neurons expressing PHB than control vector (p < 0.001; Fig. 4B). To provide further evidence for a protective role of PHB, we used AAV-mediated gene transfer to investigate the effect of PHB expression in organotypic hippocampal slice cultures subjected to OGD. PHB expression in hippocampal slices was robust and involved all hippocampal sectors (Fig. 4C,D). The OGD-induced hippocampal damage, assessed by PI staining, was less marked in slices infected with PHB AAV than in slices infected with the empty vector, indicating improved survival (Fig. 4E,F). Therefore, PHB expression increases neuronal viability in cultures as well as hippocampal slices.
Figure 4.
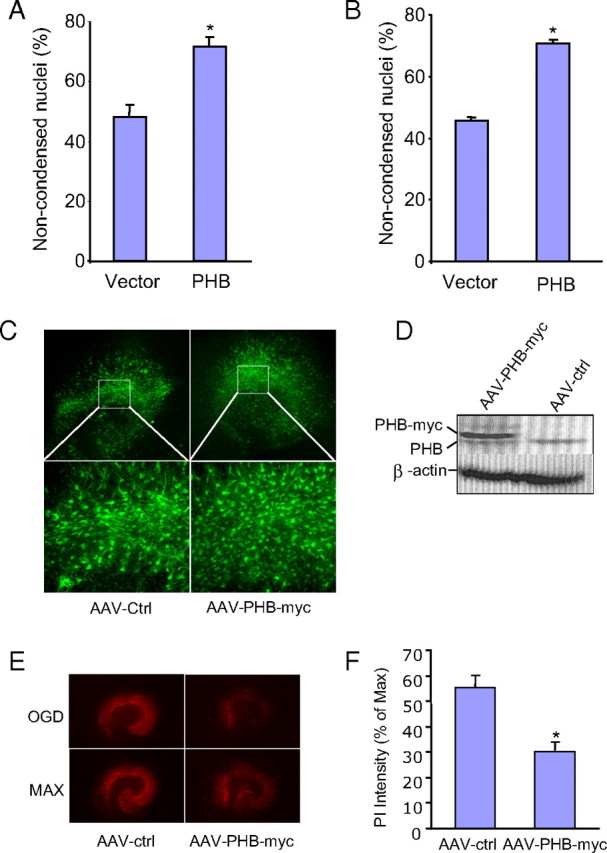
Expression of human PHB is neuroprotective. A, PHB expression improves viability of neurons exposed to STS (0.25 μm for 1 h). Noncondensed nuclei identify viable neurons (*p < 0.05; n = 4/group, 250 cells/group). B, PHB expression improves viability in neurons exposed to X/XO (X, 150 μm; XO, 15 mU for 1 h) (*p < 0.05; n = 4/group, 250 cells/ group). C, AAV mediated PHB-myc-GFP expression in hippocampal slices. D, Myc-tagged PHB is expressed in high level in the slices infected with AAV-PHB-myc but not control vector (AAV-ctrl). Anti-PHB antibody was used to detect the PHB-myc tag fusion protein. E, OGD-induced cell death assessed by PI staining is lower in slices that expressed PHB than in controls. Slices infected with control (AAV-ctrl) and PHB (AAV-PHB-myc) viruses were treated with OGD and imaged. The same slices were then treated with high dose of NMDA to induce maximum cell death (MAX) (see Materials and Methods for details). F, Measurement of PI intensity in slices after OGD (*p < 0.05; n = 3/group, 15–20 slices/group). Error bars indicate SEM.
siRNA-mediated PHB downregulation increases neuronal vulnerability to cell death
To examine the role of endogenous PHB in neuronal injury, we treated neuronal culture with PHB siRNA. Of the two PHB siRNA sequences tested, si-PHB2 attenuated PHB protein level by 75%, assessed by immunoblotting 5 d after transfection (Fig. 5A,B). The siRNA devoid of silencing activity (si-PHB1) was used as one of the controls. STS treatment resulted in elevation of caspase-3 activity in neurons transfected with control siRNA (si-Ctrl and si-PHB1) (Fig. 5C). However, caspase-3 activity was higher in neurons treated with si-PHB2, which had reduced endogenous PHB (Fig. 5C). When cultures were treated with X/XO to induce oxidative stress, si-PHB2-treated cultures exhibited reduced viability compared with those treated with control siRNA (Fig. 5D; p < 0.005). Under basal conditions, si-PHB2-treated neurons appeared to have lower mitochondrial potential than controls, assessed by JC1 staining, but this effect did not reach statistical significance (Fig. 5E). However, X/XO reduced mitochondrial membrane potential more in siPHB2-treated neurons than in controls, reflecting increased mitochondrial vulnerability (Fig. 5E). These data demonstrate that the loss of PHB increases neuronal susceptibility to injury.
Figure 5.

Downregulation of endogenous PHB by RNAi increases neuronal vulnerability. A, si-PHB-2 downregulates endogenous PHB expression, while si-control and si-PHB-1 have no effect. B, si-PHB-2 decreases PHB protein band intensity by >75% (*p < 0.05; n = 4). C, “Knockdown” of PHB by si-PHB-2 enhances the increase in caspase-3 activity induced by STS compared with si-Ctrl and si-PHB-1 (*p < 0.05 from respective vehicle; #p < 0.05 from si-PHB-1 and si-Ctrl; n = 6/group). D, si-PHB-2 reduces the viability of neuronal cultures exposed to X/XO, assessed by trypan blue exclusion (*p < 0.05 from si-PHB-1; n = 4/group, 2500 cells/group). E, si-PHB2 enhances the decrease in relative membrane potential (JC1 fluorescence ratio) induced by X/XO (*p < 0.05; n = 5). Error bars indicate SEM.
siRNA-mediated PHB downregulation increases mitochondrial ROS production and neuronal susceptibility to excitotoxicity
Next, we investigated whether reducing endogenous PHB levels exacerbates the neurotoxicity of glutamate, a process that involves oxidative stress (Wang et al., 2003). Neurons were transfected with si-PHB2 or control siRNA and neurotoxicity to glutamate was assessed 13 d later. At this time, the silencing effect of si-PHB2 was still present and endogenous PHB protein was reduced by 55% (Fig. 6A,B). Neuronal cultures were treated with glutamate (25 μm) and mitochondrial ROS production was assessed with the redox-sensitive dye MitoSOX 5 h later. Neurons transfected with si-PHB2 showed a marked increase in ROS production compared with neurons transfected with control vector (Fig. 6C,D). Furthermore, cell death, assessed by the MTS assay 24 h after glutamate, was enhanced in neurons transfected with si-PHB2 (Fig. 6C–E). These data suggest that PHB downregulation enhances ROS production and cell death following exposure to glutamate.
Figure 6.

siRNA-induced PHB downregulation enhances glutamate neurotoxicity. A, Endogenous PHB downregulation in neuronal cultures is present 13 d after si-PHB-2 treatment. B, si-PHB-2 reduces PHB band intensity by 55% (*p < 0.01; n = 3 separate experiments). C, D, si-PHB-2 increases ROS production induced by glutamate (25 μm for 5 h) and assessed by MitoSOX (*p < 0.05; n = 3 separate experiments). Magnification: 200×. E, si-PHB-2 enhances glutamate induced neuronal death and assessed by the MTS reduction assay (*p < 0.01; n = 3 separate experiments). Error bars indicate SEM.
PHB expression suppresses rotenone-induced ROS production and preserves complex I activity in mitochondria
In some cell lines, PHB is associated with complex I (Bourges et al., 2004), raising the possibility of a functional interaction with this respiratory chain complex. Therefore, we investigated whether PHB expression could modulate mitochondrial ROS production induced by the complex I inhibitor rotenone (Li et al., 2003). Primary neurons were cotransfected with PHB and GFP and challenged with rotenone (5 μm). Neurons cotransfected with vector and GFP served as controls. Neurons transfected with PHB exhibited a reduction in rotenone-induced mitochondrial ROS production, assessed by MitoSOX (Fig. 7A) and DHE (Fig. 7B), compared with controls. In contrast, PHB overexpression did not affect the ROS increase induced by the complex III inhibitor antimycin A (1 μm) (Fig. 7C). Next, we sought to determine whether PHB expression has a direct effect on complex I activity. Reliable biochemical measurements of complex I activity require amounts of mitochondria larger than those obtainable from primary neuronal cultures transfected with PHB. Therefore, we used PC12 cells stably transfected with a human PHB cDNA construct or empty vector control. We found that PHB expression did not alter baseline complex I activity (Fig. 7D). Rotenone attenuated complex I activity in a dose-dependent manner in both PHB- and vector-transfected cells (Fig. 7D). However, with increasing concentration of rotenone complex I activity was better preserved in PHB-transfected cells than in controls (Fig. 7D), indicating that PHB could partially counteract the inhibition of complex I induced by rotenone.
Figure 7.

Expression of PHB in neurons suppresses ROS production induced by complex I, but not complex III, inhibition and preserves complex I activity. A, The increase in MitoSOX fluorescence induced by rotenone (5 μm) is attenuated in neurons expressing PHB. n = 5 /group, 120 cells/group. B, Similar results were obtained when rotenone-induced ROS production was assessed by DHE (5 μm). n = 4 /group, 85 cells/group. C, PHB expression in neurons has no effect on the ROS production induced by the complex III inhibitor antimycin A (1 μm). n = 5 /group, 95 cells/group. D, Effect of rotenone on complex I activity in mitochondria isolated from PC12 cells transfected with PHB or control vector. Complex I activity is resistant to rotenone inhibition in PHB-expressing cells (*p < 0.05; n = 4/group). The inset illustrates PHB upregulation in transfected PC12 cells. Error bars indicate SEM.
Discussion
We have demonstrated that PHB is upregulated in PC induced by FN stimulation or cerebral ischemia in vivo, and by OGD in vitro. PHB is present in neuronal mitochondria and its expression increases the resistance of neuronal cultures to STS or oxidative stress, and hippocampal slices to OGD. Conversely, downregulation of endogenous PHB increases the susceptibility of neurons to injury and increases the production of mitochondrial ROS in neurons exposed to toxic glutamate concentrations. PHB dampens the mitochondrial ROS production evoked by the complex I inhibitor rotenone and protects complex I activity from the effect of this inhibitor. These findings provide the first evidence that PHB is an endogenous protective protein expressed in neuronal mitochondria, which exerts beneficial effect by preserving mitochondrial function.
Subcellular localization of PHB in neurons and its functional implications
PHB is localized in different subcellular compartments in different cell types, underlying its pleiotropic function. In lower organisms, such as yeast, PHB is localized in the inner membrane of mitochondria and acts as a regulator of mitochondrial metabolism (Nijtmans et al., 2000; Tatsuta et al., 2005). In eukaryotic cells, in addition to mitochondria, PHB is also present in the nucleus and lipid rafts, and has been implicated in a wide variety of biological processes including transcriptional regulation, cell signaling, mitochondrial function, as well as development and senescence (Wang et al., 2002; Chen et al., 2005; Mielenz et al., 2005; Rajalingam and Rudel, 2005; Rajalingam et al., 2005). In addition, PHB released into the circulation by adipocytes binds the complement component C3 and may play a role in innate immunity (Mishra et al., 2007). Although PHB was known to be present in brain cells (http://mouse.brain-map.org/brain/Phb.html) (Smalla et al., 2008), there was little available information about its subcellular localization and function in the CNS. Our results with cell fractionation and electron microscopic immunolabeling show that PHB is present in neuronal mitochondria. However, in addition to mitochondria, the ultrastructural analysis revealed a striking plasmalemmal and endomembrane distribution of PHB in selective postsynaptic dendrites and in glial processes ensheathing small axons and axon terminals. These findings are in agreement with a previous study in which PHB was observed in rat forebrain preparations of postsynaptic densities (Smalla et al., 2008). Therefore, in neurons as in other cells, PHB could be involved not only in mitochondrial function but also in synaptic transmission, a possibility to be explored in future studies.
PHB expression and ROS production
The effect of PHB on ROS production may be related to its ability to maintain the function of complex I, a major source of ROS in compromised mitochondria (Liu et al., 2002). In human cell lines, PHB interacts with complex I and has been postulated to play a role in assembly and degradation of the complex (Bourges et al., 2004). Rotenone is a complex I inhibitor that binds to its ND1 and ND4 subunits (Degli Esposti, 1998) and blocks the electron transfer from NADH to ubiquinol and ubiquinone (Schuler et al., 1999). The ROS production triggered by rotenone has been shown to induce cell death in many model systems (Höglinger et al., 2005; Panov et al., 2005; Radad et al., 2006). We found that PHB protects complex I from the inhibitory action of rotenone and suppresses the associated ROS production, providing novel evidence of a functional link between PHB and complex I in neurons. PHB had no effect on ROS generated by complex III, suggesting that effect of PHB is restricted to complex I-derived ROS. However, it is not clear how PHB dampens the ROS production induced by rotenone. PHB could interact directly with complex I and prevent the binding of rotenone by modifying the conformation of the complex, but this possibility seems unlikely because PHB attenuates ROS produced by agents other than rotenone. Another possibility is that PHB influences the assembly and activity of complex I, and that the protective effect of PHB is not limited to rotenone inhibition, but involves a more global effect on complex I function. Finally, considering the critical role that PHB plays in the structural assembly and functional compartmentalization of mitochondria (Merkwirth et al., 2008), it is conceivable that the effect of PHB may also involve other electron transport complexes. Additional studies will have to determine which of these possibilities is more likely. However, regardless of the mechanisms of the effect, our findings unveil a previously unrecognized functional interaction between PHB and complex I in mammalian neurons.
PHB expression also suppresses ROS production induced by glutamate in neurons. Recent data suggest that NADPH oxidase, not mitochondria, is the major source of ROS production induced by NMDA receptor activation (Brennan et al., 2009; Girouard et al., 2009). However, it is not known whether PHB also attenuates ROS produced by NADPH oxidase. In neurons, NADPH oxidase is located in close proximity to mitochondria (Girouard et al., 2009) and it is conceivable that during exposure to glutamate NADPH oxidase-derived ROS induce ROS production in mitochondria (Kimura et al., 2005; Wenzel et al., 2008). Further studies in which mitochondria- and NADPH oxidase-derived ROS are selectively blocked are needed to address this issue.
Endogenous neuroprotection and translational relevance of PHB
Consistent with its protective role in models of neuronal injury, we found that PHB is upregulated in mitochondria in several PC paradigms. Ischemic PC renders the brain remarkably tolerant to cerebral ischemia (Gidday, 2006; Dirnagl et al., 2009). The powerful protective effect of PC results from the concerted action of multiple mechanisms, which aim to maintain cerebral perfusion, reduce the energy demands of the brain, counteract the pathogenic factors of the ischemic cascade, and promote tissue restoration. Multiple intracellular pathways activated by PC converge on the mitochondria to maintain their integrity, thereby protecting the brain from metabolic failure, oxidative stress, and proapoptotic mediators (Dave et al., 2001, 2008; Cho et al., 2005; Dirnagl and Meisel, 2008; Racay et al., 2009). Therefore, PC-induced upregulation of PHB is likely to represent one of the effector mechanisms by which preconditioned mitochondria maintain neuronal viability. In addition, our findings with siRNA-mediated downregulation indicate that the endogenous level of PHB is an important determinant of neuronal viability. In agreement with our studies in primary neurons, increased susceptibility to injury by PHB “knockdown” has also been reported in endothelial cells and in a neuroblastoma cell line (Schleicher et al., 2008; Park et al., 2010). Therefore, PHB is likely to represent an endogenous neuroprotective protein that is part of the cellular defense response to stress (Iadecola and Anrather, 2011). PHB is increased in the brain of schizophrenic patients, in Parkinson's disease, and in a rat model of white matter disease (Ferrer et al., 2007; Smalla et al., 2008; Kuwamura et al., 2011), suggesting a possible homeostatic role to counter the disease process, although maladaptive effects of the upregulation have been postulated in schizophrenia (Smalla et al., 2008) and in capsaicin-induced apoptosis (Kuramori et al., 2009). However, reduced levels of PHB have been implicated in the mechanisms of intestinal epithelial cell death in Crohn's disease and in the brain dysfunction induced by the fetal alcohol syndrome (Theiss et al., 2007; Sari et al., 2010), suggesting a pathogenic role of PHB deficiency. These findings, collectively, point to a critical involvement of PHB in cell survival and related adaptive responses. Furthermore, the beneficial effects of PHB suggest a therapeutic role for this protein in brain diseases caused by acute or chronic neurodegeneration, in which mitochondrial dysfunction and oxidative stress play a prominent role (Adibhatla and Hatcher, 2003, 2010; Gibson et al., 2010). However, assessment of the translational potential of modulating PHB levels will require a deeper understanding of the role of PHB in neurons, including its involvement in both mitochondrial function and synaptic transmission.
Conclusions
PHB is an essential mitochondrial protein well described in many cell types, but its function in neurons is not known. We found that PHB is upregulated in brain mitochondria in several PC paradigms well known to result in neuroprotection, including FN stimulation and hypoxia–ischemia. In the normal brain, PHB was observed in neuronal mitochondria as well as on selective, postsynaptic dendrites and glial processes ensheathing axons. Expression of PHB increases the resistance to injury of neuronal culture and of brain slices, an effect associated with reduced ROS production. Conversely, downregulation of endogenous PHB renders neurons more vulnerable to injury and increases ROS production. In addition, PHB attenuates the ROS production associated with complex I inhibition by rotenone, and increases the resistance of this complex to inhibition, suggesting stabilization of its function. These observations identify PHB as an endogenous neuroprotective protein that exerts its beneficial actions by suppressing ROS production. The functional interaction with complex I suggests a critical role of PHB in the activity of this complex. Although the molecular mechanisms of its effect on complex I and its potential role in synaptic function remain to be defined, PHB emerges as a powerful endogenous neuroprotective protein that is linked to ischemic PC and may have translational relevance in brain diseases associated with oxidative stress and mitochondrial dysfunction.
Footnotes
This work was supported by NIH Grants NS34179 (C.I.), HL96571 (C.I., V.P., P.Z.), and NS67078 (P.Z.). We thank Dr. Eldon R. Jupe, Oklahoma Medical Research Foundation, for providing a plasmid containing the cDNA sequence for human prohibitin.
This paper is dedicated to the memory of Drs. Donald J. Reis and Hong Wang, whose invaluable contributions made these studies possible.
References
- Adibhatla RM, Hatcher JF. Citicoline decreases phospholipase A2 stimulation and hydroxyl radical generation in transient cerebral ischemia. J Neurosci Res. 2003;73:308–315. doi: 10.1002/jnr.10672. [DOI] [PubMed] [Google Scholar]
- Adibhatla RM, Hatcher JF. Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2010;12:125–169. doi: 10.1089/ars.2009.2668. [DOI] [PubMed] [Google Scholar]
- Ahn CS, Lee JH, Reum Hwang A, Kim WT, Pai HS. Prohibitin is involved in mitochondrial biogenesis in plants. Plant J. 2006;46:658–667. doi: 10.1111/j.1365-313X.2006.02726.x. [DOI] [PubMed] [Google Scholar]
- Artal-Sanz M, Tavernarakis N. Prohibitin couples diapause signalling to mitochondrial metabolism during ageing in C. elegans. Nature. 2009;461:793–797. doi: 10.1038/nature08466. [DOI] [PubMed] [Google Scholar]
- Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- Bourges I, Ramus C, Mousson de Camaret B, Beugnot R, Remacle C, Cardol P, Hofhaus G, Issartel JP. Structural organization of mitochondrial human complex I: role of the ND4 and ND5 mitochondria-encoded subunits and interaction with prohibitin. Biochem J. 2004;383:491–499. doi: 10.1042/BJ20040256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J, Aoki C, Pickel VM. Optimization of differential immunogold-silver and peroxidase labeling with maintenance of ultrastructure in brain sections before plastic embedding. J Neurosci Methods. 1990;33:113–127. doi: 10.1016/0165-0270(90)90015-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem Res. 2004;29:1943–1949. doi: 10.1007/s11064-004-6869-x. [DOI] [PubMed] [Google Scholar]
- Chen JC, Jiang CZ, Reid MS. Silencing a prohibitin alters plant development and senescence. Plant J. 2005;44:16–24. doi: 10.1111/j.1365-313X.2005.02505.x. [DOI] [PubMed] [Google Scholar]
- Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol Cell Physiol. 2007;292:C137–C147. doi: 10.1152/ajpcell.00270.2006. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Adam-Vizi V. Calcium, mitochondria and oxidative stress in neuronal pathology. Novel aspects of an enduring theme. FEBS J. 2006;273:433–450. doi: 10.1111/j.1742-4658.2005.05103.x. [DOI] [PubMed] [Google Scholar]
- Cho S, Park EM, Zhou P, Frys K, Ross ME, Iadecola C. Obligatory role of inducible nitric oxide synthase in ischemic preconditioning. J Cereb Blood Flow Metab. 2005;25:493–501. doi: 10.1038/sj.jcbfm.9600058. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Dave KR, Saul I, Busto R, Ginsberg MD, Sick TJ, Pérez-Pinzón MA. Ischemic preconditioning preserves mitochondrial function after global cerebral ischemia in rat hippocampus. J Cereb Blood Flow Metab. 2001;21:1401–1410. doi: 10.1097/00004647-200112000-00004. [DOI] [PubMed] [Google Scholar]
- Dave KR, DeFazio RA, Raval AP, Torraco A, Saul I, Barrientos A, Perez-Pinzon MA. Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase Cε. J Neurosci. 2008;28:4172–4182. doi: 10.1523/JNEUROSCI.5471-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degli Esposti M. Inhibitors of NADH-ubiquinone reductase: an overview. Biochim Biophys Acta. 1998;1364:222–235. doi: 10.1016/s0005-2728(98)00029-2. [DOI] [PubMed] [Google Scholar]
- Ding Q, Vaynman S, Souda P, Whitelegge JP, Gomez-Pinilla F. Exercise affects energy metabolism and neural plasticity-related proteins in the hippocampus as revealed by proteomic analysis. Eur J Neurosci. 2006;24:1265–1276. doi: 10.1111/j.1460-9568.2006.05026.x. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Meisel A. Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning? Neuropharmacology. 2008;55:334–344. doi: 10.1016/j.neuropharm.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Perez E, Dalfó E, Barrachina M. Abnormal levels of prohibitin and ATP synthase in the substantia nigra and frontal cortex in Parkinson's disease. Neurosci Lett. 2007;415:205–209. doi: 10.1016/j.neulet.2007.01.026. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Rosenthal RE, Vereczki V, Martin E, Hoffman GE, Chinopoulos C, Kowaltowski A. Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. J Bioenerg Biomembr. 2004;36:347–352. doi: 10.1023/B:JOBB.0000041766.71376.81. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF. Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim Biophys Acta. 2010;1802:122–134. doi: 10.1016/j.bbadis.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- Girouard H, Wang G, Gallo EF, Anrather J, Zhou P, Pickel VM, Iadecola C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J Neurosci. 2009;29:2545–2552. doi: 10.1523/JNEUROSCI.0133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34:232–237. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie. 2002;84:153–166. doi: 10.1016/s0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. 2007;1767:1007–1031. doi: 10.1016/j.bbabio.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglinger GU, Lannuzel A, Khondiker ME, Michel PP, Duyckaerts C, Féger J, Champy P, Prigent A, Medja F, Lombes A, Oertel WH, Ruberg M, Hirsch EC. The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J Neurochem. 2005;95:930–939. doi: 10.1111/j.1471-4159.2005.03493.x. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. Stroke research at a crossroad: asking the brain for directions. Nat Neurosci. 2011;14:1363–1368. doi: 10.1038/nn.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen E, Fiedler K, Parton RG, Simons K. Prohibitin, an antiproliferative protein, is localized to mitochondria. FEBS Lett. 1995;358:273–277. doi: 10.1016/0014-5793(94)01444-6. [DOI] [PubMed] [Google Scholar]
- Jemmerson R, Dubinsky JM, Brustovetsky N. Cytochrome c release from CNS mitochondria and potential for clinical intervention in apoptosis-mediated CNS diseases. Antioxid Redox Signal. 2005;7:1158–1172. doi: 10.1089/ars.2005.7.1158. [DOI] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Kim N, Lee Y, Kim H, Joo H, Youm JB, Park WS, Warda M, Cuong DV, Han J. Potential biomarkers for ischemic heart damage identified in mitochondrial proteins by comparative proteomics. Proteomics. 2006;6:1237–1249. doi: 10.1002/pmic.200500291. [DOI] [PubMed] [Google Scholar]
- Kim Y, Zhou P, Qian L, Chuang JZ, Lee J, Li C, Iadecola C, Nathan C, Ding A. MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J Exp Med. 2007;204:2063–2074. doi: 10.1084/jem.20070868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Suzuki T, Maeta H, Abe Y. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 2005;45:860–866. doi: 10.1161/01.HYP.0000163462.98381.7f. [DOI] [PubMed] [Google Scholar]
- Kuramori C, Azuma M, Kume K, Kaneko Y, Inoue A, Yamaguchi Y, Kabe Y, Hosoya T, Kizaki M, Suematsu M, Handa H. Capsaicin binds to prohibitin 2 and displaces it from the mitochondria to the nucleus. Biochem Biophys Res Commun. 2009;379:519–525. doi: 10.1016/j.bbrc.2008.12.103. [DOI] [PubMed] [Google Scholar]
- Kuwamura M, Inumaki K, Tanaka M, Shirai M, Izawa T, Yamate J, Franklin RJ, Kuramoto T, Serikawa T. Oligodendroglial pathology in the development of myelin breakdown in the dmy mutant rat. Brain Res. 2011;1389:161–168. doi: 10.1016/j.brainres.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Langer T. Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Löwer B, Wunderlich FT, von Kleist-Retzow JC, Waisman A, Westermann B, Langer T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielenz D, Vettermann C, Hampel M, Lang C, Avramidou A, Karas M, Jäck HM. Lipid rafts associate with intracellular B cell receptors and exhibit a B cell stage-specific protein composition. J Immunol. 2005;174:3508–3517. doi: 10.4049/jimmunol.174.6.3508. [DOI] [PubMed] [Google Scholar]
- Mishra S, Murphy LC, Murphy LJ. The Prohibitins: emerging roles in diverse functions. J Cell Mol Med. 2006;10:353–363. doi: 10.1111/j.1582-4934.2006.tb00404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Moulik S, Murphy LJ. Prohibitin binds to C3 and enhances complement activation. Mol Immunol. 2007;44:1897–1902. doi: 10.1016/j.molimm.2006.09.025. [DOI] [PubMed] [Google Scholar]
- Mishra S, Ande SR, Nyomba BL. The role of prohibitin in cell signaling. FEBS J. 2010;277:3937–3946. doi: 10.1111/j.1742-4658.2010.07809.x. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, Toni N, Buchs P, Parisi L, Stoppini L. Interface organotypic hippocamlpal slice cultures. In: Fedoroff S, Richardson A, editors. Protocols for neural cell culture. Ed 3. Totowa, NJ: Humana; 2001. pp. 13–27. [Google Scholar]
- Muraguchi T, Kawawa A, Kubota S. Prohibitin protects against hypoxia-induced H9c2 cardiomyocyte cell death. Biomed Res. 2010;31:113–122. doi: 10.2220/biomedres.31.113. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Oxidative stress and energy crises in neuronal dysfunction. Ann N Y Acad Sci. 2008;1147:53–60. doi: 10.1196/annals.1427.002. [DOI] [PubMed] [Google Scholar]
- Nijtmans LG, de Jong L, Artal Sanz M, Coates PJ, Berden JA, Back JW, Muijsers AO, van der Spek H, Grivell LA. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000;19:2444–2451. doi: 10.1093/emboj/19.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov A, Dikalov S, Shalbuyeva N, Taylor G, Sherer T, Greenamyre JT. Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J Biol Chem. 2005;280:42026–42035. doi: 10.1074/jbc.M508628200. [DOI] [PubMed] [Google Scholar]
- Park B, Yang J, Yun N, Choe KM, Jin BK, Oh YJ. Proteomic analysis of expression and protein interactions in a 6-hydroxydopamine-induced rat brain lesion model. Neurochem Int. 2010;57:16–32. doi: 10.1016/j.neuint.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Poot M, Zhang YZ, Krämer JA, Wells KS, Jones LJ, Hanzel DK, Lugade AG, Singer VL, Haugland RP. Analysis of mitochondrial morphology and function with novel fixable fluorescent stains. J Histochem Cytochem. 1996;44:1363–1372. doi: 10.1177/44.12.8985128. [DOI] [PubMed] [Google Scholar]
- Racay P, Chomova M, Tatarkova Z, Kaplan P, Hatok J, Dobrota D. Ischemia-induced mitochondrial apoptosis is significantly attenuated by ischemic preconditioning. Cell Mol Neurobiol. 2009;29:901–908. doi: 10.1007/s10571-009-9373-7. [DOI] [PubMed] [Google Scholar]
- Radad K, Rausch WD, Gille G. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem Int. 2006;49:379–386. doi: 10.1016/j.neuint.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Rajalingam K, Rudel T. Ras-Raf signaling needs prohibitin. Cell Cycle. 2005;4:1503–1505. doi: 10.4161/cc.4.11.2142. [DOI] [PubMed] [Google Scholar]
- Rajalingam K, Wunder C, Brinkmann V, Churin Y, Hekman M, Sievers C, Rapp UR, Rudel T. Prohibitin is required for Ras-induced Raf-MEK-ERK activation and epithelial cell migration. Nat Cell Biol. 2005;7:837–843. doi: 10.1038/ncb1283. [DOI] [PubMed] [Google Scholar]
- Reers M, Smith TW, Chen LB. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 1991;30:4480–4486. doi: 10.1021/bi00232a015. [DOI] [PubMed] [Google Scholar]
- Reis DJ, Berger SB, Underwood MD, Khayata M. Electrical stimulation of cerebellar fastigial nucleus reduces ischemic infarction elicited by middle cerebral artery occlusion in rat. J Cereb Blood Flow Metab. 1991;11:810–818. doi: 10.1038/jcbfm.1991.139. [DOI] [PubMed] [Google Scholar]
- Reis DJ, Golanov EV, Galea E, Feinstein DL. Central neurogenic neuroprotection: central neural systems that protect the brain from hypoxia and ischemia. Ann N Y Acad Sci. 1997;835:168–186. doi: 10.1111/j.1749-6632.1997.tb48628.x. [DOI] [PubMed] [Google Scholar]
- Reis DJ, Kobylarz K, Yamamoto S, Golanov EV. Brief electrical stimulation of cerebellar fastigial nucleus conditions long-lasting salvage from focal cerebral ischemia: conditioned central neurogenic neuroprotection. Brain Res. 1998;780:161–165. [PubMed] [Google Scholar]
- Sari Y, Zhang M, Mechref Y. Differential expression of proteins in fetal brains of alcohol-treated prenatally C57BL/6 mice: a proteomic investigation. Electrophoresis. 2010;31:483–496. doi: 10.1002/elps.200900385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleicher M, Shepherd BR, Suarez Y, Fernandez-Hernando C, Yu J, Pan Y, Acevedo LM, Shadel GS, Sessa WC. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J Cell Biol. 2008;180:101–112. doi: 10.1083/jcb.200706072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler F, Yano T, Di Bernardo S, Yagi T, Yankovskaya V, Singer TP, Casida JE. NADH-quinone oxidoreductase: PSST subunit couples electron transfer from iron-sulfur cluster N2 to quinone. Proc Natl Acad Sci U S A. 1999;96:4149–4153. doi: 10.1073/pnas.96.7.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalla KH, Mikhaylova M, Sahin J, Bernstein HG, Bogerts B, Schmitt A, van der Schors R, Smit AB, Li KW, Gundelfinger ED, Kreutz MR. A comparison of the synaptic proteome in human chronic schizophrenia and rat ketamine psychosis suggest that prohibitin is involved in the synaptic pathology of schizophrenia. Mol Psychiatry. 2008;13:878–896. doi: 10.1038/mp.2008.60. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Chinopoulos C, Fiskum G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium. 2004;36:257–264. doi: 10.1016/j.ceca.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Tatsuta T, Model K, Langer T. Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol Biol Cell. 2005;16:248–259. doi: 10.1091/mbc.E04-09-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauskela JS, Brunette E, Monette R, Comas T, Morley P. Preconditioning of cortical neurons by oxygen-glucose deprivation: tolerance induction through abbreviated neurotoxic signaling. Am J Physiol Cell Physiol. 2003;285:C899–C911. doi: 10.1152/ajpcell.00110.2003. [DOI] [PubMed] [Google Scholar]
- Terashima M, Kim KM, Adachi T, Nielsen PJ, Reth M, Köhler G, Lamers MC. The IgM antigen receptor of B lymphocytes is associated with prohibitin and a prohibitin-related protein. EMBO J. 1994;13:3782–3792. doi: 10.1002/j.1460-2075.1994.tb06689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiss AL, Idell RD, Srinivasan S, Klapproth JM, Jones DP, Merlin D, Sitaraman SV. Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J. 2007;21:197–206. doi: 10.1096/fj.06-6801com. [DOI] [PubMed] [Google Scholar]
- Wang H, Cheng E, Brooke S, Chang P, Sapolsky R. Over-expression of antioxidant enzymes protects cultured hippocampal and cortical neurons from necrotic insults. J Neurochem. 2003;87:1527–1534. doi: 10.1046/j.1471-4159.2003.02123.x. [DOI] [PubMed] [Google Scholar]
- Wang S, Fusaro G, Padmanabhan J, Chellappan SP. Prohibitin co-localizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression. Oncogene. 2002;21:8388–8396. doi: 10.1038/sj.onc.1205944. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Mollnau H, Oelze M, Schulz E, Wickramanayake JM, Müller J, Schuhmacher S, Hortmann M, Baldus S, Gori T, Brandes RP, Münzel T, Daiber A. First evidence for a crosstalk between mitochondrial and NADPH oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid Redox Signal. 2008;10:1435–1447. doi: 10.1089/ars.2007.1969. [DOI] [PubMed] [Google Scholar]
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117–128. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Qian L, Glickstein SB, Golanov EV, Pickel VM, Reis DJ. Electrical stimulation of cerebellar fastigial nucleus protects rat brain, in vitro, from staurosporine-induced apoptosis. J Neurochem. 2001;79:328–338. doi: 10.1046/j.1471-4159.2001.00585.x. [DOI] [PubMed] [Google Scholar]
- Zhou P, Qian L, Iadecola C. Nitric oxide inhibits caspase activation and apoptotic morphology but does not rescue neuronal death. J Cereb Blood Flow Metab. 2005a;25:348–357. doi: 10.1038/sj.jcbfm.9600036. [DOI] [PubMed] [Google Scholar]
- Zhou P, Qian L, Zhou T, Iadecola C. Mitochondria are involved in the neurogenic neuroprotection conferred by stimulation of cerebellar fastigial nucleus. J Neurochem. 2005b;95:221–229. doi: 10.1111/j.1471-4159.2005.03358.x. [DOI] [PubMed] [Google Scholar]