


Abstract
Mammalian genomes encode numerous cis-natural antisense transcripts (cis-NATs). The extent to which these cis-NATs are actively regulated and ultimately functionally relevant, as opposed to transcriptional noise, remains a matter of debate. To address this issue, we analyzed the chromatin environment and RNA Pol II binding properties of human cis-NAT promoters genome-wide. Cap analysis of gene expression data were used to identify thousands of cis-NAT promoters, and profiles of nine histone modifications and RNA Pol II binding for these promoters in ENCODE cell types were analyzed using chromatin immunoprecipitation followed by sequencing (ChIP-seq) data. Active cis-NAT promoters are enriched with activating histone modifications and occupied by RNA Pol II, whereas weak cis-NAT promoters are depleted for both activating modifications and RNA Pol II. The enrichment levels of activating histone modifications and RNA Pol II binding show peaks centered around cis-NAT transcriptional start sites, and the levels of activating histone modifications at cis-NAT promoters are positively correlated with cis-NAT expression levels. Cis-NAT promoters also show highly tissue-specific patterns of expression. These results suggest that human cis-NATs are actively transcribed by the RNA Pol II and that their expression is epigenetically regulated, prerequisites for a functional potential for many of these non-coding RNAs.
INTRODUCTION
In recent years, it has become evident that substantial portions of mammalian genomes are actively transcribed as non-coding RNA, including thousands of cis-natural antisense transcripts (cis-NATs) (1–5). Cis-NATs are transcripts produced from within the protein-coding loci, but from the opposite strand, and are thus complementary to the sense mRNA transcript (Figure 1a). Cis-NATs may play important regulatory roles via transcriptional interference caused by collisions of RNA polymerase complexes moving in opposite directions across the same locus (3,6) or through the formation of double-stranded RNA leading to post-transcriptional silencing through RNA interference (7,8). However, the extent to which non-coding RNAs in general, and cis-NATs in particular are biologically functional remains a matter of debate. Some studies have suggested that the majority of non-coding RNA transcripts are non-functional and simply represent transcriptional noise (9,10), while others have found evidence in support of the function for numerous non-coding RNAs (11–13).
Figure 1.

Delineation and analysis of cis-NAT promoters. Cis-NATs are initiated from protein-coding gene loci and transcribed in the opposite (antisense) direction. (a) Example of a protein-coding gene locus with a genic promoter that drives transcription in the 5′-to-3′ direction along with a cis-NAT promoter that initiates 3′-to-5′ transcription within the locus. (b) cis-NAT TSSs were defined using clusters of overlapping antisense CAGE tags. Specific cis-NAT TSS locations were taken as the base with the highest density of mapped CAGE tags within the cluster. (c) cis-NAT promoter sequences were taken as genomic regions immediately flanking cis-NAT TSS, and the chromatin environment of cis-NAT promoters was analyzed using ChIP-seq data for histone modification and RNA Pol II binding.
Previously, investigators have interrogated the functional potential of novel non-coding RNA transcripts by evaluating the chromatin environment in-and-around their promoters (12,14,15). These studies were motivated by the fact that the promoters of well-characterized human genes have characteristic chromatin properties, including distinct protein binding and histone-modification profiles, and these particular chromatin environments give indications as to the biological mechanisms, both genetic and epigenetic, by which the genes are regulated (12,14,15). For example, chromatin immunoprecipitation (ChIP-seq) studies have revealed that the promoters of actively transcribed genes are occupied by RNA Pol II and marked with a suite of specific histone tail modifications, such as acetylation of the lysine at position 9 of histone H3 (H3K9Ac) (16–18), whereas silent gene promoters are depleted for RNA Pol II and enriched for known repressive modifications such as trimethylation of lysine 27 of histone H3 (H3K27Me3). On the other hand, it has been shown that the promoters of many novel non-coding transcripts that have been characterized by high-throughput sequencing methods, but for which there is no additional supporting information, do not show enrichment for histone modifications or an active chromatin environment (12,14,15). Thus, chromatin can be used to discriminate between the promoters of actively regulated genes versus putative transcription start site (TSS) that probably represents transcriptional noise.
In this study, we evaluated the chromatin environment surrounding hundreds of thousands of human cis-NATs across six different ENCODE cell types for 10 RNA isolation conditions. We sought to establish whether or not cis-NAT promoters show patterns of activity and chromatin modifications that are consistent with epigenetic regulation. We found that active cis-NAT promoters are enriched with active histone modifications and occupied by RNA Pol II, whereas silent cis-NAT promoters are depleted for both active modifications and RNA Pol II and enriched for the repressive modification of H3K27Me3. These data provide evidence for the epigenetic regulation of numerous human cis-NATs, presumably a prerequisite of their potential function as gene regulators.
METHODS
Cap analysis of gene expression data analysis
Human cis-NAT promoters were delineated using Cap analysis of gene expression (CAGE) data from the ENCODE repository on the UCSC Genome Browser (19). CAGE data from six cell types and across 10 RNA isolation conditions were used for this study. The cell types are: GM12878, H1HESC, HepG2, HUVEC, K562 and NHEK. The RNA isolation conditions consist of polyadenylated and non-polyadenylated RNA fractions from whole cells, cytoplasm, nucleus, nucleolus and nucleoplasm. Altogether, a total of 16 different CAGE data sets were analyzed here (Table 1, Supplementary Table S1). CAGE tags from each data set mapped to the reference sequence of the human genome (NCBI build 36.1; UCSC version hg18) (20) were clustered by their genomic locations to identify promoters. CAGE clusters with two or more colocated tags have been previously shown to represent validated TSSs (21,22); accordingly, CAGE clusters containing two or more overlapping tags were used for the promoter analyses reported here. For each CAGE cluster, the actual TSS was characterized by finding the base with the highest density of mapped CAGE 5′-ends (Figure 1b). CAGE clusters that were antisense to a protein-coding locus from the UCSC known genes set were taken to be cis-NAT promoters as previously described (2) (Supplementary Table S2). To reduce contamination of the cis-NAT TSS by the possible degradation products of mRNAs, all CAGE clusters that overlapped an exon of the UCSC gene set were removed from the set of cis-NATs. CAGE clusters within 250 bp of an annotated TSS of a protein-coding loci were taken to be genic promoters and the TSS taken as the base with peak CAGE tag density. As a control, CAGE clusters that overlapped an exon of the UCSC gene set and were in the same orientation as the exon were kept for analysis.
Table 1.
Numbers of cis-NAT promoters identified by CAGE clusters in each cell line, subcellular location and polyadenylation state
| Cell Line | Subcellular location | Poly-A− | Poly-A+ | Total |
|---|---|---|---|---|
| GM12878 | Cytosol | 24 107 | – | – |
| Nucleoplasm | – | – | 165 430 | |
| Nucleus | 62 704 | – | – | |
| H1HESC | Whole cell | 67 216 | – | – |
| HEPG2 | Cytosol | 33 862 | – | – |
| Nucleoplasm | – | – | 214 364 | |
| Nucleus | 265 896 | – | – | |
| HUVEC | Cytosol | 25 309 | – | – |
| K562 | Cytosol | 164 399 | 30 867 | – |
| Nucleoplasm | – | – | 79 677 | |
| Nucleolus | – | – | 112 308 | |
| Nucleus | 313 003 | 148 461 | – | |
| NHEK | Cytosol | 11 650 | – | – |
| Nucleus | 178 016 | – | – |
ChIP-seq data analysis
Histone modification and RNA Pol II occupancy for cis-NAT promoters were evaluated using ChIP-seq data from the ENCODE repository on the UCSC Genome Browser (20). Where available, FASTQ ChIP-seq data for the H3K4Me1, H3K4Me2, H3K4Me3, H3K9Ac, H3K9Me1 H3K27Me3, H3K27Ac, H3K36Me3 and H4K20Me1 modifications in the GM12878, H1HESC, HepG2, HUVEC, K562 and NHEK cell types were taken from the ENCODE repository. A non-specific input ChIP-seq control data set was also analyzed for each of the ENCODE cell types. All ChIP-seq data were mapped to the May 2006 build of the human genome reference sequence (NCBI 36.1; UCSC hg18) using BowTie (23), keeping the best alignments with ties broken by quality scores. Any reads with more than 20 possible mappings were discarded. Remaining reads with multiple, high-quality mappings were resolved using GibbsAM (24) (Supplementary Table S3). Tag counts for a given modification were normalized by dividing the total number of mapped tags for that modification, then multiplying by 10 million. ChIP-seq data were used to characterize the chromatin environment proximal to CAGE-characterized cis-NAT promoters (Figure 1c).
Association mining analysis
For each cell type, we used only the CAGE data from the nucleus (GM12878, HEPG2, K562 and NHEK), cytosol (HUVEC) or whole cell (H1HESC) isolate to classify the activity of sense genic promoters in relation to the sum of cis-NAT activity for the genic promoter, i.e. the sum of downstream cis-NAT promoter activity. For each cell type, genic promoters that had CAGE tags associated were ranked by their CAGE tag counts, and the top 25% were classified as ‘high activity’ in the cell type, while genic promoters that had no CAGE data or were in the bottom 25% were classified as ‘low activity’ in the cell type. The same was done for the cumulative downstream cis-NAT activity of the genic promoters. This resulted in four possible classification combinations for cis-NAT and genic activity levels: (i) high cis-NAT and high gene; (ii) high cis-NAT and low gene; (iii) low cis-NAT and high gene; and (iv) low cis-NAT and low gene. We then used association mining to calculate the value of the ‘Interest (I)’ parameter, as previously described (25), which is the ratio of the observed frequency of co-occurrence of any two classifications divided by their expected co-occurrence based on random association.
Statistical analysis
Student's t-tests were used to compare differences in the average number of normalized ChIP-seq tags ±5 kb of cis-NAT promoters for different cis-NAT-activity levels (Figure 2). We used the statistical software R for calculating the Spearman's rank correlation coefficients for all correlation analyses (Supplementary Figures S16–S31). The statistical significance of Spearman's rank correlation coefficients ρ was determined using the Student's t distribution with df = n−2 with the formula 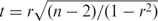 (26).
(26).
Figure 2.

Enrichment of chromatin modifications and RNA Pol II at cis-NAT promoters. Cis-NAT promoters identified in the NHEK cell type were divided into four bins based on their activity (from lowest to highest activity), and the normalized average numbers of ChIP-seq reads from each histone modification ±5 kb of the cis-NAT TSS were calculated for each bin. A ‘+’ or ‘−’ above a bar indicates that the number of ChIP-seq reads for that bin and modification is significantly higher or lower, respectively, than the control for that bin (P < 0.001). A ‘+’ or ‘−’ within the bar indicates that a bin is significantly enriched or depleted, respectively, for the histone modification compared to the next lowest activity bin (P < 0.001). Error bars shown are the SEM.
RESULTS AND DISCUSSION
Large-scale identification of cis-NAT TSS
CAGE is a method for characterizing the 5′-end of RNA transcripts; genomic mapping of CAGE sequence tags identifies TSSs and promoters (27,28). CAGE combined with high-throughput sequencing can identify many thousands of TSS, while at the same time quantifying their promoter activity via the number of reads mapping to each TSS. CAGE data were analyzed as described in the ‘Methods’ section to identify cis-NAT promoters in the human genome for the 16 different combinations of ENCODE cell type and RNA isolation conditions analyzed here. The number of cis-NAT promoters identified in this way ranges from 11 650 to 313 003 across the ENCODE cell types (Table 1, Supplementary Table S2). We evaluated whether the large differences in cis-NAT promoters identified across cell types were due to differences in the numbers of CAGE tags per library or differences in sequencing quality across libraries. Library-specific read-count values and read-quality scores are not significantly correlated with the numbers of cis-NAT promoters identified across cell types, suggesting that the differences observed do not result from the CAGE data abundance or quality.
Enrichment of chromatin modifications and RNA Pol II at cis-NAT promoters
To characterize the relationship between local chromatin modifications and cis-NAT promoter activity, we analyzed the number of ChIP-seq tags from each histone modification and RNA Pol II, proximal (± 5 kb) to each cis-NAT TSS. The analysis of cis-NAT chromatin modifications was conducted for 16 combinations of 6 ENCODE cell types over 10 RNA isolation conditions. Here, we present an example of these results for one cell type and condition (NHEK cis-NATs characterized from nuclear non-polyadenylated RNA); results for all other cell types and conditions are detailed in the Supplementary Data. Cis-NAT promoters were binned into four equal-sized bins based on their promoter activity, from lowest to highest activity, as measured by CAGE tag counts. Histone modifications and RNA Pol II occupancy were then compared for cis-NAT promoters with different levels of activity. Cis-NAT promoters showed significant increases in ChIP-seq tag counts for the activating histone modifications H3K4Me1, H3K4Me2, H3K4Me3, H3K9Ac, H3K27Ac with increasing levels of cis-NAT promoter activity (Figure 2). Furthermore, each of these modifications shows significantly greater average cis-NAT tag counts than seen for the ChIP-seq control (Figure 2). These histone modifications have previously been characterized as activating modifications by virtue of their association with the promoters of actively transcribed genes (16–18). H3K27Me3, on the other hand, is known as a repressive modification that is associated with silent genes, and ChIP-seq tag counts for H3K27Me3 are lower than seen for the control in all cis-NAT promoter activity bins (Figure 2). Similar qualitative patterns are seen for H3K9Me1, H3K36Me3 and H4K20Me1, but the tag counts do not vary as much with cis-NAT promoter activity. This may be due to the fact that these modifications are associated with transcribed regions, where the cis-NAT promoters are located, as opposed to promoter regions per se (16,17). In other words, chromatin signals of promoter activity for these marks may be obscured by the fact that they are enriched within gene bodies where the cis-NATs are located. Overall, the patterns of enrichment seen for histone modifications at cis-NAT promoters suggest that the cis-NATs identified here are epigenetically modified in accordance with their relative expression levels and are thus likely to be specifically regulated, which is a precondition for their functional relevance, as opposed to non-specific artefacts such as RNA degradation products. For all activity levels, the level of Pol II binding is higher than seen for the non-specific input control, suggesting that regions near cis-NAT promoters are bound by Pol II. Qualitatively similar patterns of histone modification and Pol II occupancy across different cis-NAT promoter activity levels were seen for 14 out of the 15 remaining CAGE data sets analyzed here; the only exception was the NHEK cytosol CAGE data set (Supplementary Figures S1–S15).
To further evaluate whether histone modifications were correlated with cis-NAT promoter activity, cis-NAT promoters were divided into 200 bins based on activity as measured by CAGE tag counts. Cis-NAT TSS CAGE tag counts were then compared to ChIP-seq proximal promoter histone modification and RNA Pol II tag counts using the Spearman's rank correlation (Supplementary Figures S16–S31). Cis-NAT promoter activity and histone modifications generally showed positive correlations for the activating H3K4 methylations and H3K9 and H3K27 acetylations and weaker, though still positive correlations for the H3K9Me1, H3K36Me3 and H4K20Me1 modifications. A weaker negative correlation was seen for the repressive H3K27Me3 modification. As would be expected for actively transcribed promoters, there was also a positive and significant correlation between cis-NAT promoter activity and RNA Pol II presence and RNA-seq read density.
Histone modification, RNA Pol II occupancy and transcription near cis-NAT promoters
The enrichment of activating histone modifications and RNA Pol II occupancy near active cis-NAT promoters suggests that cis-NAT expression is epigenetically regulated; however, this enrichment could result from cis-NATs being located in open chromatin regions inside gene bodies, and not from the promoters being specifically modified to regulate their activity. To evaluate this possibility, we analyzed the distribution of histone modifications and RNA Pol II occupancy around cis-NAT TSS. If the enrichment of chromatin modifications observed for cis-NATs is due solely to their location in open chromatin, then we do not expect to see any variability in enrichment along chromosomal regions surrounding cis-NATs. On other hand, actively regulated cis-NATs would be expected to show modification peaks centered around the TSS as has been seen for the promoters of protein-coding loci (14,16–18).
Cis-NAT promoters were broken down by activity level, as described above, and the average numbers of ChIP-seq tags were calculated for 10 base-pair windows ±5 kb from cis-NAT TSS (Figure 3). Methylations of H3K4 (H3K4me1, H3K4me2 and H3K4me3) are known activating marks of promoters (16), and were all found to be enriched near cis-NAT promoters for the NHEK nuclear non-polyadenylated RNA data set (Figure 2). In further accordance with their epigenetic regulation, peaks of ChIP-seq read density from the H3K4Me1, H3K4Me2 and H3K4Me3 modifications were observed on either side of the cis-NAT TSS in this same data set, with more active promoters being more highly modified on average (Figure 3b, c and d). A notable dip can be seen near the cis-NAT TSS for these three modifications, suggesting nucleosome absence, similar to what has been seen at canonical TSS in CD4+ T-cells (18). Similar patterns were seen for the activating acetylations of H3K9 and H3K27 (Figure 3e and g). No discernable difference between bins was seen for the repressive mark H3K27Me3 (Figure 3j). The similarities seen for the genomic distributions of cis-NAT promoter modifications to those of protein-coding loci promoters (14,16–18) provide evidence that cis-NAT expression is not simply transcription resulting from open chromatin, but is specifically regulated. The nucleosome absence seen even at the TSS with the lowest activity suggests that these TSS, which are identified by only a small number of CAGE tags, are bona fide TSS that has been epigenetically silenced by histone deacetylation. Pol II occupancy is seen at the TSS for all activity bins, with the higher activity bins showing a much higher occupancy, in accordance with the activity of the bins (Figure 3k). The H3K9Me1, H3K36Me3 and H4K20Me1 modifications show levels of enrichment similar to the control with no observable enrichment on either side of the TSS (Figure 3g, h and i). This is likely to be due to the fact that these modifications are associated with actively transcribed regions, such as gene bodies, where the cis-NAT TSS in this study is located (16,17). RNA-seq data also peak near the cis-NAT promoters and increase with cis-NAT promoter activity (Figure 3l). Patterns of modification near cis-NAT TSS using CAGE and ChIP-seq data were qualitatively similar for 10 out of the 15 remaining CAGE data sets analyzed here; the HepG2 nucleus, K562 nucleoplasm and both K562 nucleus CAGE sets have greatly distorted patterns of modification (Supplementary Figures S32–S46). Taken together, these data indicated that cis-NAT promoters show genomic distributions of histone modifications and RNA Pol II binding around TSS that are consistent with specific activation of transcription at the TSS as opposed to a simple accumulation of activating marks inside actively transcribed protein-coding gene regions.
Figure 3.

Chromatin modification, RNA Pol II binding, and transcription around cis-NAT promoters. Cis-NAT promoters identified in the NHEK cell type were divided into four bins based on their activity (lowest to highest), and the normalized average numbers of ChIP-seq (a–k) or RNA-seq (l) reads in 10 bp windows ±5kb of the cis-NAT TSS (at position 0) were calculated for each bin.
For comparison, the same chromatin enrichment analyses were done for CAGE clusters associated with genic promoters in the 6 ENCODE cell types. The patterns of local histone modifications for these promoters were largely qualitatively similar to those seen for the cis-NAT promoters (Supplementary Figures S47–S62) (14,16–18). However, histone modification levels and RNA Pol II binding are substantially more enriched around genic promoters. In addition, genic promoters show distinct enrichment patterns for H3K9Me1, H3K36Me3 and H4K20Me1; these differences are likely due to the location of cis-NATs in gene bodies, which differ with respect to the distribution of these particular modifications. Overall, these results further support the functional and regulatory potential of cis-NAT promoters that are actively transcribed, albeit at lower levels than genic promoters.
It is formally possible that the cis-NAT chromatin enrichment patterns observed here can be attributed the fact that the cis-NATs were identified using CAGE, and any CAGE cluster would show such a pattern. To control for this possibility, we performed a similar analysis using CAGE clusters overlapping exons in the sense orientation, which may not be expected to show the same pattern of modification as CAGE clusters associated with genuine promoters. Indeed, sense exonic CAGE clusters have previously been suggested to represent transcriptional degradation products, as opposed to promoters, and were not found to have shown promoter characteristic chromatin profiles (29). Here, we performed the same set of chromatin enrichment analyses done for cis-NATs on exonic CAGE clusters. The patterns of histone modifications near exonic CAGE clusters are markedly different from those seen for cis-NAT promoters and genic promoters (Supplementary Figures S63–S78). These results indicate that the cis-NAT chromatin enrichment profiles observed here are not simply a generic marker for the presence of CAGE clusters.
Differential expression of cis-NAT promoters
Differential expression of cis-NATs was measured by counting the fraction of the six ENCODE cell types in which each cis-NAT promoter was expressed. In order to remove cis-NATs whose expression falls below the limit of CAGE detection, only those cis-NAT promoters that show activity higher than the 90th percentile in some cell type were used. On average, these cis-NAT promoters are expressed in 33% of the ENCODE cell types studied here compared to 43% seen for genic promoters (Figure 4a), this difference is statistically significant (P ≈ 0, Wilcoxon's rank sum) indicating that cis-NAT expression is more cell-type specific than genic expression.
Figure 4.

Differential expression of cis-NAT promoters compared to genic promoters. Cis-NAT promoters that showed activity greater than the 90th percentile of cis-NAT promoters in at least one cell type were considered for analysis. (a) The average fraction of cell types where individual cis-NAT or genic promoters are detected. (b) Rarefaction curve showing the relationship between the number of cis-NAT (black) and genic (grey) promoters found (y-axis) for each possible combination of 1–16 CAGE data sets (x-axis). Error bars shown are the SD.
Rarefaction curve analysis was used to evaluate the extent to which each individual CAGE data set uncovers novel cis-NAT promoters compared to novel genic promoters. For this analysis, the average numbers of cis-NAT or genic promoters detected across all possible CAGE data set combinations, ranging from 1 to 16 data sets, were calculated. Compared to genic promoters, a significantly smaller fraction of cis-NAT promoters is detected when one or only fewer than eight CAGE data sets are considered (P < 0.001, Wilcoxon's rank sum) (Figure 4b). For both genic and cis-NAT promoters, the number of new promoters detected decrease rapidly as more CAGE sets are considered, suggesting that most cis-NAT and genic promoters have been captured. The differences seen for the cis-NAT versus genic curves further underscore the extent to which cis-NATs are specifically regulated.
Association between cis-NAT and genic promoter activity
Previous studies have suggested that the presence of cis-NATs leads to the downregulation of gene expression (6). If cis-NATs are indeed repressive regulatory elements, then one may expect to observe a negative correlation between cis-NAT expression levels and the expression levels of the genes in which they are found. To evaluate this prediction, we regressed the activity levels of genic promoters with those of the corresponding cis-NAT promoters, however, no correlation was apparent (Supplementary Figures S79–S84). Therefore, we used a more sensitive data mining approach to search for possible associations between genic promoter activity and cis-NAT promoter activity. To do this, genic promoters were classified as having high or low activity, and the corresponding genes were classified as having high or low cis-NAT activity in each of the six cell types as described above. Association mining then was used to evaluate the levels of co-occurrence of the four possible gene and cis-NAT activity category combinations: (i) high cis-NAT and high gene; (ii) high cis-NAT and low gene, (iii) low cis-NAT and high gene; and (iv) low cis-NAT & low gene. We found that co-occurrence of high cis-NAT and high genic promoter activity occurs approximately twice as frequently as would be expected by chance (Figure 5, Supplementary Table S4). Similarly, the frequency of high/low associations is much lower than would be expected and the frequency of low/low associations is higher than expected. This association remains when only those cis-NAT promoters distal (>2.5 kb downstream) to the genic promoter or proximal (<2.5 kb downstream) to the genic promoter are considered (Supplementary Figures S85–S86, Supplementary Tables S5 and S6). These results raise the possibility that the majority of cis-NATs are activating rather than repressive regulatory elements.
Figure 5.

Association of cis-NAT promoter activity and genic promoter activity. Cis-NAT and genic promoters were classified into high (H) and low (L) categories based on their activity levels across all cell types analyzed here. The observed versus expected levels of association between the resulting four possible category combinations—(i) high cis-NAT and high gene (H/H); (ii) high cis-NAT and low gene (H/L); (iii) low cis-NAT and high gene (L/H); (iv) low cis-NAT and low gene (L/L)—were then computed using association mining.
CONCLUSIONS
It has been known for some time that there is active antisense transcription in the human genome, though it has only recently become appreciated how pervasive it is. However, the functional significance of human cis-NATs is a matter of debate; it is possible that many of the apparent cis-NATs actually represent transcriptional noise or degraded fragments of sequence processed from larger transcripts. Here, we have attempted to address the potential functional significance of human cis-NATs genome-wide by evaluating the chromatin environment and regulatory properties of their promoters. This approach is based on the rationale that specifically regulated promoters will have distinct chromatin profiles and protein binding properties. Accordingly, the presence and distribution of such chromatin features at the promoters of novel uncharacterized transcripts, when considered together with their relative activity levels, can be used to provide support for their regulation and potential functional significance.
Taking advantage of the methods for characterizing protein binding and histone modifications genome-wide, we demonstrate that active human cis-NAT promoters are in fact enriched for histone modifications and RNA Pol II binding. Furthermore, histone modifications and RNA Pol II binding peak at cis-NAT TSS, and the levels of histone modifications and RNA Pol II binding are correlated with the activity of the cis-NAT promoters. These data suggest that the expression of human cis-NATs is driven by RNA Pol II and at least partially regulated by the modification of histone tails. While the specific function of individual cis-NATs remains an open question, the fact that the cis-NAT promoters are bound by RNA Pol II and epigenetically modified suggests that they are specifically regulated. Indeed, the presence of both cis-NAT promoters with activating marks and cis-NAT promoters with repressive marks is consistent with the high levels of differential expression observed here for cis-NATs and tissue-specific regulation of their function. While the cis-NAT chromatin and expression features uncovered here are consistent with a functional role as regulators, they may also be taken to represent a required precondition of function. Definitive confirmation of the functional role for cis-NATs will await experimental validation of individual cases.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–6, Supplementary Figures 1–86.
FUNDING
Bioinformatics program at the Georgia Institute of Technology (to A.B.C.). Alfred P. Sloan Research Fellowship in Computational and Evolutionary Molecular Biology (BR-4839 to I.K.J.). Funding for open access charge: Alfred P. Sloan Research Fellowship in Computational and Evolutionary Molecular Biology (BR-4839).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Chen J, Sun M, Kent WJ, Huang X, Xie H, Wang W, Zhou G, Shi RZ, Rowley JD. Over 20% of human transcripts might form sense-antisense pairs. Nucleic Acids Res. 2004;32:4812–4820. doi: 10.1093/nar/gkh818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conley AB, Miller WJ, Jordan IK. Human cis natural antisense transcripts initiated by transposable elements. Trends Genet. 2008;24:53–56. doi: 10.1016/j.tig.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Lapidot M, Pilpel Y. Genome-wide natural antisense transcription: coupling its regulation to its different regulatory mechanisms. EMBO Rep. 2006;7:1216–1222. doi: 10.1038/sj.embor.7400857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehner B, Williams G, Campbell RD, Sanderson CM. Antisense transcripts in the human genome. Trends Genet. 2002;18:63–65. doi: 10.1016/s0168-9525(02)02598-2. [DOI] [PubMed] [Google Scholar]
- 5.Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A, Diber A, Biton S, Tamir Y, Khosravi R, et al. Widespread occurrence of antisense transcription in the human genome. Nat. Biotechnol. 2003;21:379–386. doi: 10.1038/nbt808. [DOI] [PubMed] [Google Scholar]
- 6.Osato N, Suzuki Y, Ikeo K, Gojobori T. Transcriptional interferences in cis natural antisense transcripts of humans and mice. Genetics. 2007;176:1299–1306. doi: 10.1534/genetics.106.069484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tam OH, Aravin AA, Stein P, Girard A, Murchison EP, Cheloufi S, Hodges E, Anger M, Sachidanandam R, Schultz RM, et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534–538. doi: 10.1038/nature06904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453:539–543. doi: 10.1038/nature06908. [DOI] [PubMed] [Google Scholar]
- 9.Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat. Struct. Mol. Biol. 2007;14:103–105. doi: 10.1038/nsmb0207-103. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Zhang J, Zheng H, Li J, Liu D, Li H, Samudrala R, Yu J, Wong GK. Mouse transcriptome: neutral evolution of ‘non-coding’ complementary DNAs. Nature. 2004;431 1 p following 757; discussion following 757. [PubMed] [Google Scholar]
- 11.Werner A, Berdal A. Natural antisense transcripts: sound or silence? Physiol. Genomics. 2005;23:125–131. doi: 10.1152/physiolgenomics.00124.2005. [DOI] [PubMed] [Google Scholar]
- 12.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponjavic J, Ponting CP, Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007;17:556–565. doi: 10.1101/gr.6036807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trinklein ND, Karaoz U, Wu J, Halees A, Force Aldred S, Collins PJ, Zheng D, Zhang ZD, Gerstein MB, Snyder M, et al. Integrated analysis of experimental data sets reveals many novel promoters in 1% of the human genome. Genome Res. 2007;17:720–731. doi: 10.1101/gr.5716607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Hon G, Wang W, Ren B. Discovery and annotation of functional chromatin signatures in the human genome. PLoS Comput. Biol. 2009;5:e1000566. doi: 10.1371/journal.pcbi.1000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenbloom KR, Dreszer TR, Pheasant M, Barber GP, Meyer LR, Pohl A, Raney BJ, Wang T, Hinrichs AS, Zweig AS, et al. ENCODE whole-genome data in the UCSC Genome Browser. Nucleic Acids Res. 2010;38: D620–D625. doi: 10.1093/nar/gkp961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ, et al. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010;38:D613–D619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carninci P. Tagging mammalian transcription complexity. Trends Genet. 2006;22:501–510. doi: 10.1016/j.tig.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Faulkner GJ, Kimura Y, Daub CO, Wani S, Plessy C, Irvine KM, Schroder K, Cloonan N, Steptoe AL, Lassmann T, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009;41:563–571. doi: 10.1038/ng.368. [DOI] [PubMed] [Google Scholar]
- 23.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Huda A, Lunyak VV, Jordan IK. A Gibbs sampling strategy applied to the mapping of ambiguous short-sequence tags. Bioinformatics. 2010;26:2501–2508. doi: 10.1093/bioinformatics/btq460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan P-N, Steinbach M, Kumar V. Introduction to Data Mining. Boston: Addison-Wesley; 2005. [Google Scholar]
- 26.Sokal RR, Rohlf JF. Biometry: The Principles and Practice of Statistics in Biological Research. San Francisco: W. H. Freeman; 1981. [Google Scholar]
- 27.Shiraki T, Kondo S, Katayama S, Waki K, Kasukawa T, Kawaji H, Kodzius R, Watahiki A, Nakamura M, Arakawa T, et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl Acad. Sci. USA. 2003;100: 15776–15781. doi: 10.1073/pnas.2136655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kodzius R, Kojima M, Nishiyori H, Nakamura M, Fukuda S, Tagami M, Sasaki D, Imamura K, Kai C, Harbers M, et al. CAGE: cap analysis of gene expression. Nat. Methods. 2006;3:211–222. doi: 10.1038/nmeth0306-211. [DOI] [PubMed] [Google Scholar]
- 29.Mercer TR, Dinger ME, Bracken CP, Kolle G, Szubert JM, Korbie DJ, Askarian-Amiri ME, Gardiner BB, Goodall GJ, Grimmond SM, et al. Regulated post-transcriptional RNA cleavage diversifies the eukaryotic transcriptome. Genome Res. 2010;20:1639–1650. doi: 10.1101/gr.112128.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.