

Abstract
Multiparity is an independent risk factor for obesity in parous females. In addition to being a health issue for the mother, offspring of multiparous females may also be at risk for obesity later in life. The aim of the current study was to establish a mouse model that mimics the human pathology of multiparity and determine the effects of multiparity-induced obesity (MIO) on offspring in adulthood. C57BL/6 mice were mated and studied when primiparous (1st pregnancy) or multiparous (4th pregnancy). Dams became obese with multiparity, an effect that was independent of the age of the dam. Multiparous dams also had increased markers of inflammation (JNK activation, cytokine expression) in adipose tissue and liver that was greater than inflammation in nulliparous females made obese with a high-fat diet. Placental inflammation was prevalent in multiparous vs. primiparous dams as well. Male offspring of the multiparous dams developed increased adiposity by 24 wk of age relative to the progeny of primiparous dams, although food consumption was similar in both groups. Lipid metabolism was altered in liver and fat in that mRNA levels of regulatory genes (PGC-1α) as well as metabolic genes (CPT I) and Akt phosphorylation were decreased in offspring of multiparous dams. Thus, in mice, as in humans, multiparity increases adiposity and is associated with hepatic and placental inflammation and abnormal glucose tolerance. Importantly, MIO leads to increased body fat and metabolic dysfunction in the offspring, suggesting a role in the propagation of obesity.
Keywords: developmental programming, diabetes, peroxisome proliferator-activated receptor-γ coactivator-1α
obesity has become an epidemic in developed countries and is on the rise in underdeveloped countries. Consequently, a significant number of women in their reproductive years, including those that are pregnant, are obese or overweight (30, 31). Obesity during gestation poses a significant clinical problem since it is linked to a spectrum of maternal complications, including gestational diabetes, exaggerated inflammation, hypertension, thromboembolism, preeclamplsia, and delivery complications (26, 31, 55, 62, 69).
Multiparity is a cause of weight gain in women during their reproductive years and is in fact an independent predictor of obesity (12, 20). The increase in multiparity-induced obesity (MIO) has followed overall trends in obesity, since gestational weight gain is additive to prepregnancy weight and the inability to loose weight gained postpartum (51, 53, 54). Moreover, overweight women are more likely than leaner women to retain the weight gained during pregnancy (19). Thus, as the population becomes heavier, the effect of multiparity on body weight is amplified. Similar to obesity, pregnancy has been described as an inflammatory state, with the placenta contributing increased levels of cytokines from both the trophoblasts and resident placental macrophages (reviewed in Ref. 22) and with an increase in the numbers of activated monocytes (39). Thus multiparous women may be subjected to repeated episodes of this placental inflammation with adverse consequences. In fact, it is known that women who have had multiple pregnancies are at an increased risk to develop diabetes, independent of visceral adiposity (1), and diabetes has been associated with chronic inflammation (5, 48, 66). Since obese pregnant women have increased inflammation compared with lean pregnant women (10, 59), obese multiparous women could have an even greater inflammatory state.
Gestational obesity is known to cause acute and chronic consequences in the offspring as well as the female itself. Acute consequences range from shoulder dystocia, low Apgar score, macrosomia, congenital defects, and death (42, 46). Chronic consequences include developmental programming, leading to an increased risk of obesity and glucose intolerance in adulthood (reviewed in Refs. 2, 9, 23, and 44). The fact that obesity during pregnancy increases the incidence of obesity in the offspring raises the worrisome prospect of transgenerational perpetuation of obesity (8, 51). Although the mediators of developmental programming of metabolism are not clear, gestational diabetes, maternal overnutrition, circulating factors associated with maternal obesity, or factors associated with inflammation in the placenta or other tissues of obese pregnant females are all possible routes of regulation (2, 9, 23, 44). Although multiparity is a risk factor for obesity, there is little information about the risks of adverse metabolic outcomes in the offspring of these women.
Adiposity is the result of positive energy balance, and caloric intake greater than caloric expenditure. Hunger and satiety are controlled by a number of factors in specific regions in the brain, and changes in activity of any number of these pathways could affect food consumption and thereby lead to altered adiposity. A reduction in energy expenditure could also lead to accelerated fat deposition. As in the brain, a variety of proteins and pathways are involved in peripheral lipid metabolism and energy expenditure. However, one protein has been suggested to be key in the regulation of energy expenditure, and that protein is peroxisome proliferator-activated receptor (PPAR)γ coactivator-1α (PGC-1α; reviewed in Refs. 15, 21, and 25). PGC-1α is upstream of mitochondrial biogenesis and fatty acid oxidation, as well as oxidative phosphorylation and other mitochondrial functions, and as such plays a pivotal role in energy metabolism. PGC-1α coactivates mitochondrial biogenesis and fatty acid oxidation with other proteins, i.e., the estrogen-related receptor. Although processes are regulated by protein complexes, levels of PGC-1α are thought to be the driving force for gene transcription of the downstream targets (64).
Thus, the present study was designed in two parts. In the first part, we established a mouse model that mimics MIO in humans. Female parous mice became obese by the 4th pregnancy with exaggerated inflammation. In the second part, we demonstrated that male offspring of these multiparous females have altered metabolism, resulting in increased adiposity. The change in body composition was not the result of a change in input of energy intake but was due to a change in energy output (lipid catabolism). Specifically, we found marked decreases in the mRNA levels of carnitine palmitoyltransferase I (CPT I), the rate-limiting step for fatty acid oxidation. The decrease was likely mediated by decreased levels of direct upstream regulators of CPT I, those being PGC-1α and/or PPARα. Thus, multiparous females with marked obesity and inflammation induce developmental programming, leading to increased adiposity in adult male offspring.
RESEARCH DESIGN AND METHODS
Animals
Pregnant dams.
All male and nonpregnant female C57BL/6 mice were purchased from The Jackson Laboratories and fed a pelleted chow (Harlan Laboratories); females were 10 wk old when they arrived. Animals were housed in a temperature- and humidity-controlled room and subjected to 12 h of light and 12 h of darkness. After ≈1 wk acclimation, females were fed a breeder chow that contained 9% fat (wt/wt; PMI Nutrition International) for 1–2 wk prior to mating. To mate females, one male was placed in a cage with three females. The presence of a postcopulatory plug was checked when the lights came on and denoted 0.5 days postconception (dpc). When a plug was detected, female mice were separated into their own cage. After 5 days of mating, males were separated from remaining nonpregnant females. After 2 days, the process was repeated. Females underwent mating until a plug was detected or until three mating cycles were completed. For multiparous females, pups were born and allowed to suckle. Pups were weaned at 21 days postpartum, and females remated within 5 days of the weaning. One group of females was maintained on breeder chow until the same age as the dams mated the fourth time (≈7 mos) and then mated (1st old pregnancy).
At 18.5 dpc of the 1st, 4th, or 1st old pregnancy, either glucose tolerance tests (GTT) were performed on one set of fasted dams or tissues (liver and retroperitoneal, inguinal, and ovarian adipose tissues) were collected from a different set of dams at the beginning of the light phase. At a later time, an additional group of females were fed a high-fat diet starting at 3 mo of age. Diets consisted of 58% fat (calories) primarily as hydrogenated coconut oil plus soybean oil with corn starch (Research Diets); soybean oil was used because this was the primary oil of the breeder chow. These females were never mated, and tissues were collected when nulliparous and multiparous females were the same age.
Male offspring.
In a separate set of studies using different dams, pups were born to dams of their first pregnancy and dams of their fourth pregnancy; all litters used had five to nine pups per litter. At weaning, one male pup from each litter was housed individually and fed chow; thus n equals litter. Each week, animal weights and the amounts of food consumed were measured. At 14 wk of age, GTT were performed, followed by body composition measurements by MRI after animals had time to recover. At 24 wk of age, GTT were performed again. After recovery, body composition was determined by MRI. Approximately 1 wk after the MRI, blood and tissues were collected after a 6-h fast. All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Cincinnati.
GTT
GTT were performed similarly in fasted pregnant females and male offspring (6-h fast). Animals were injected intraperitoneally (ip) with glucose (2 mg/g). Blood was collected from the tail, and glucose was measured in triplicate with a glucometer using glucose strips (Abbott Laboratories) at 0, 15, 30, 60, 120, and 180 min postinjection.
Real-Time PCR
Livers and adipose tissues were collected rapidly in pregnant females (18.5 dpc) or adult offspring (≈5 mos of age) after exsanguination, snap-frozen in liquid nitrogen, and stored at −80°C until use. Tissue RNA was isolated using TRIzol and stored in FORMAzol at −80°C. RNA was treated with RNase-free DNase I and reverse transcribed to cDNA by SuperScript II reverse transcriptase using random hexamers. PCR assays were performed on a Bio-Rad iCycler iQ real-time PCR Detection System, using SYBR green as our fluorophore; primers will be given upon request. A serial dilution of a randomly picked sample was used to generate a standard curve for each gene examined. This standard curve was used to calculate the relative levels of mRNA for the gene of interest compared with the reference/housekeeping gene (cyclophilin).
Hormone Levels and Adiposity
Insulin concentrations were measured by LINCOplex by the Mouse Metabolic Phenotype Center (MMPC) of the University of Cincinnati. Adiposity was measured by MRI (Echo MRI; Echo Medical Systems, Houston, TX).
Plasma and Liver Lipid Concentrations
Plasma lipids were measured enzymatically. Plasma was pooled and separated into lipoproteins by fast-protein liquid chromatography using two Sephadex columns in tandem as described (41), and cholesterol was measured in each fraction enzymatically. Lipids were extracted from livers with Folch, and triglyceride content was measured chemically by the MMPC of the University of Cincinnati (6). Cholesterol was extracted from saponified liver and measured by gas chromatography, using stigmastanol as an internal standard (63).
Western Blots
Expression levels of proteins or phosphorylation of proteins were determined by Western blot. Briefly, tissues were homogenized in a lysis buffer. Proteins were then separated by gel electrophoresis, and the relative amounts of phosphorylated (Cell Signaling Technology) and total JNK (Pharmingen), phosphorylated (Cell Signaling Technology) and total AKT (Cell Signaling Technology), and PGC-1α (Novus Biologicals) were determined by Western blotting using appropriate secondary antibodies and enhanced chemiluminesence plus, as described (68); similar gel loading was verfied as needed after stripping membranes and reprobing with antibodies to tubulin or β-actin (33).
Statistics
Data are presented as means ± SE. Differences between dams of the 1st vs. 4th or offspring of 1st vs. 4th dams were determined by two-tailed Student's t-tests (P < 0.05). When three groups of mice were compared, i.e., 1st, 4th, and 1st old pregnancy, animals were compared by one-way ANOVA, followed by Student-Newman-Keuls test to determine differences between groups (P < 0.05).
RESULTS
Effects of Multiparity on Maternal Adiposity and Inflammation
Previous studies have demonstrated that multiparous women become obese (12, 20) and likely have increased inflammation (1). To establish a mouse model that mimics the human pathology of multiparity, we mated C57BL/6 mice once (primiparous) or four times (multiparous). The body weights for multiparous and primiparous dams were 48.5 ± 2.2 and 33.7 ± 0.9 g, respectively. Both the retroperitoneal (RP) and ovarian (OV) fat depots weighed ≈6.5-fold more, and the inguinal (ING) depot weighed ≈3.5-fold more in the multi- vs primiparous dams (Fig. 1A). In addition, multiparous mice had increased hepatic lipid content and plasma cholesterol concentrations (Table 1); the increase in plasma cholesterol was due to increased LDL as well as HDL cholesterol (data not shown). To assess age as a contributing factor for increased adiposity, dams were mated their first time at a similar age as those of the multiparous dams (1st old pregnancy). There was significantly more fat in the ING (55% more) and OV (87% more) and a trend for more RP fat (45% more; P = 0.09) of the multiparous dams vs. 1st old pregnant dams (Fig. 1A). These data demonstrate that in mice, as in humans, multiparity leads to increased adiposity independent of maternal age.
Fig. 1.

Parameters of multiparous and primiparous dams. A: adipose tissue mass of different fat depots of primi- and multiparous females; primiparous dams were younger (≈3 mo of age; 1st pregnancy) or older and age-matched to multiparous dams (≈7 mo of age; 1st old pregnancy). Retroperitoneal (RP), inguinal (ING), and ovarian (OV) depots were collected from dams at 18.5 days postconception (dpc). Data are presented as means ± SE (n = 9, 14, and 6 for dams of the 4th, 1st, and 1st old pregnancies, respectively). *Differences (P < 0.05) between the amount of fat in depots compared with primiparous younger dams; #difference (P < 0.05) between the amount of fat in 1st old dams compared with dams of 1st and 4th pregnancies. B: glucose tolerance tests of primi- and multiparous dams at 18.5 dpc. Glucose tolerance tests were performed as described. Data are presented as means ± SE (n = 5 for dams in both groups). *Differences (P < 0.05) between the concentrations in the 1st vs. 4th pregnancies.
Table 1.
Metabolite levels in plasma and livers of primi- and multiparous dams
| Parameter | 1st Pregnancy | 4th Pregnancy |
|---|---|---|
| Plasma glucose, mg/dl | 120 ± 11 | 103 ± 7 |
| Plasma cholesterol, mg/dl | 37.0 ± 2.1 | 58.9 ± 4.6* |
| Liver cholesterol, mg/g | 2.55 ± 0.14 | 2.42 ± 0.12 |
| Liver triglyceride, mg/g | 17.6 ± 1.2 | 23.5 ± 2.4* |
Data are means ± SE.
Significant differences between 1st and 4th dams (P < 0.03).
Since increased adiposity is often associated with glucose intolerance and insulin resistance, we examined glucose tolerance in a different set of dams at the same gestational age. Fasting glucose levels were similar among multiparous and primiparous mice (Fig. 1B). However, glucose excursion was greater in dams in their fourth pregnancy compared with those in their first; specific time points post-glucose injection were significantly greater in the multiparous dams, and there was a trend for increased area under the curve of GTT in dams of the fourth compared with the first pregnancy (P = 0.09). Glucose concentrations returned to preinjection levels by 120 min in the primiparous mice, whereas glucose levels were still elevated at 180 min in the multiparous cohort.
Another factor that is associated with obesity and can lead to abnormal glucose tolerance is inflammation (18, 66). Markers of inflammation were measured in the placentas, RP adipose tissues, and livers of the multiparous and primiparous dams. Proinflammatory cytokine (monocyte chemoattractant protein-1, TNFα, IL-6) mRNA levels were increased in the RP adipose tissues of the multiparous females, whereas anti-inflammatory cytokine (arginase) mRNA levels were similar in all dams (Fig. 2A). In the placentas, levels of proinflammatory cytokines (monocyte chemoattractant protein-1 and TNFα) as well as macrophage markers (F4/80 and CD68) were increased, or there was a trend for an increase in the multiparous females (Fig. 2B). Consistent with greater cytokine production and more macrophages, JNK activation, as reflected in increased phosphorylation, was increased in the placentas (Fig. 2C) as well as the livers (Fig. 2D) of the multiparous females. Importantly, the increased inflammation was greater than that which occurs merely with adiposity since expression of proinflammatory cytokines was greater in RP adipose tissues of multiparous females than in nulliparous females fed a high-fat diet to age- and weight-match multiparous females (Fig. 2E). Together, these data indicate that multiparity causes exaggerated inflammation independently of its effect on adiposity.
Fig. 2.

Inflammation in adipose tissues, placentas, and livers of multi- or primiparous females. Adipose tissues (A) are the same as those described in Fig. 1A, and livers and placentas (B–D) were collected from 7 (multiparous) or 4 (primiparous) of the dams described in Fig. 1A. A: relative mRNA levels of proteins associated with inflammation in retroperitoneal adipose tissues of young primi- and multiparous dams compared with cyclophilin are presented. B: relative mRNA levels in the placenta are presented. C: phosphorylated and total JNK were determined in the liver by Western blot using tubulin for gel loading. D: phosphorlated and total JNK were determined in the placenta by Western blot using tubulin for gel loading. E: mRNA levels of cytokines expressed in nulliparous dams fed a high-fat diet for 4 mo (n = 5–6). Animals were age- and weight-matched to the multiparous controls. Dashed lines represent data from multiparous dams presented in B. All PCR results are presented as means ± SE. *Differences (P < 0.05) between tissues of offspring of primi- and multiparous dams fed each diet; §P = 0.09. MCP-1, monocyte chemoattractant protein-1.
Effects of Multiparity on Offspring Glucose Homeostasis and Adiposity
Knowing that diet-induced obese females have offspring with increased adiposity (2, 9, 23, 44), we next set out to determine whether offspring of multiparous obese dams would be at an increased risk of becoming obese. Offspring of multiparous dams weighed slightly more at weaning (12.5 ± 0.6 vs. 10.8 ± 0.3 g, P = 0.001; Fig. 3A), although the differences in neonatal mass disappeared within 1 wk of weaning and were similar during the remainder of the rapid growth phase postweaning. The difference at weaning was not due to litter size, since litters of primiparous dams culled to five pups were the same mass as those not culled (10.3 ± 0.9 g). However, body weight was indeed greater in offspring of multiparous dams than offspring of primiparous dams by 26 wk of age (Fig. 3A). Body composition between offspring of dams at either parity was similar at 14 wk of age (Fig. 3, B–D), but by 24 wk of age the offspring of multiparous dams fed chow had significantly more body fat than the primiparous offspring, without a change in lean mass (Fig. 3, B–D). Food consumption was similar in all offspring, regardless of the parity of the dams (Fig. 4). Together, these data indicate that offspring of multiparous females have increased body weights and elevated adiposity, and these changes are independent of major differences in energy intake.
Fig. 3.
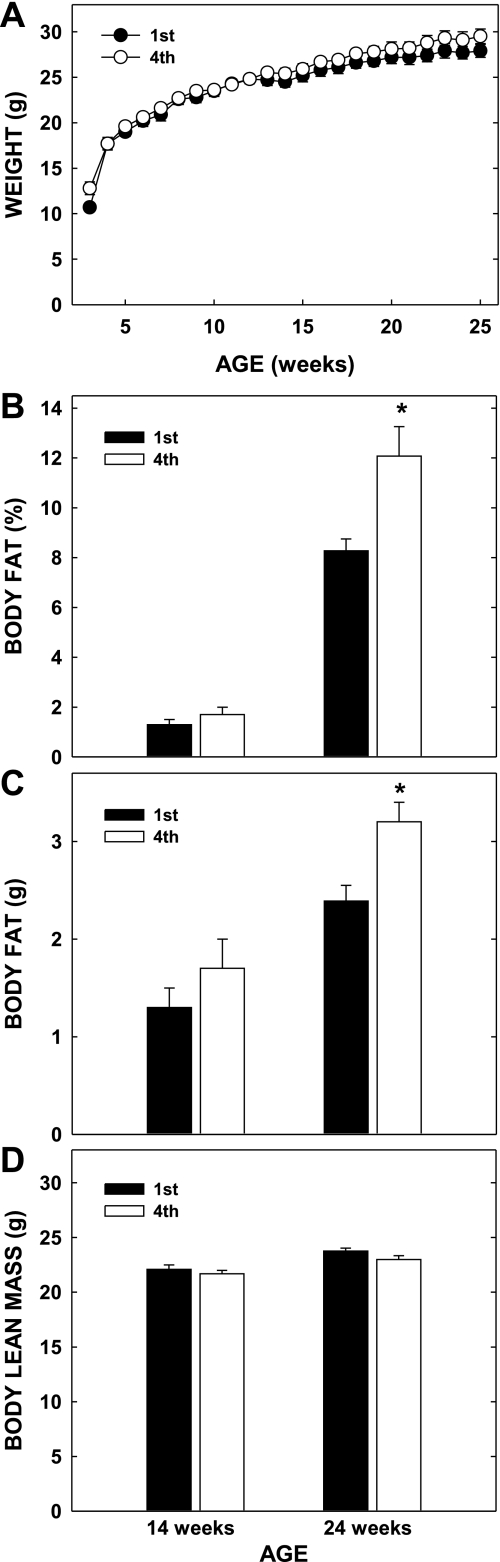
Body weights and composition of male offspring of primi- (1st) and multiparous (4th) dams. Male offspring were weaned at 3 wk of age and housed individually. A: weekly body weights were collected, and composition was measured at 14 and 24 wk of age. B: %fat of offspring at 14 and 24 wk of age. C: mass of fat in offspring at 14 and 24 wk of age. D: lean mass of offspring at 14 and 24 wk of age. Data are presented as means ± SE [n = 11 and 9 offspring (litters) from primi- and multiparous dams, respectively]. *Differences (P < 0.05) between tissues of offspring of primi- and multiparous dams at either age.
Fig. 4.

Food consumption of offspring of multi- and primiparous dams. Consumption was measured weekly from 12 wk of age until the time of the study in the animals described in Fig. 3.
The male offspring of multiparous mothers with increased adiposity also had increased plasma triglyceride levels (Fig. 5, inset). However, there was no difference in plasma cholesterol levels or the distribution of cholesterol between various lipoprotein fractions (Fig. 5). Hepatic lipid content and liver weights were similar in all offspring as well (data not shown).
Fig. 5.

Plasma lipoprotein cholesterol levels in offspring of primi- (1st) or multiparous (4th) dams; animals are the same as those described in Fig. 3. Plasma was collected from offspring at the termination of the study when they were 26 wk of age, and lipoproteins were separated by fast-protein liquid chromatography. Two pooled samples were used, and the results were averaged. Inset: total plasma triglyceride (TG) levels are presented.
Since glucose metabolism and insulin resistance can be affected by adiposity and developmental programming, we next performed GTT. At 25 wk of age, fasting glucose levels were lower in the offspring of primiparous dams (P < 0.05; Fig. 6A), plasma insulin levels were similar (Fig. 6B), and there was evidence of reduced hepatic insulin signaling, as reflected in lesser hepatic Akt phosphorylation (Fig. 6C). During ip GTT, plasma glucose concentrations had higher peaks in the offspring of multiparous dams (P = 0.051), but glucose tolerance was otherwise similar compared with the mice born of primiparous mothers.
Fig. 6.

Glucose tolerance tests and insulin levels and signaling in offspring of multi- and primiparous dams; animals are the same as those described in Fig. 3. A: glucose tolerance tests were performed on offspring of dams of different parity. *Differences (P < 0.05) between plasma glucose levels at zero time; §nearly significant differences (P = 0.051) at 30 min postinjection. B: plasma insulin levels in offspring of dams of different parity. Data are presented as means ± SE. C: phosphorylated (Akt-p) and total Akt in pooled liver samples of offspring of dams with different parity.
Metabolism of Adipose Tissue and Liver of Offspring
We used a targeted gene approach to determine the mechanism(s) responsible for the 40% increased adiposity independent of a change in food consumption. Initially, we studied genes involved in synthesis of fat [fatty acid synthase (FAS)] and oxidation of fat (CPT I) of adipose tissue. There was ≈40% decrease in CPT I and ≈45% decrease in FAS mRNA levels in male offspring of multiparous dams compared with male offspring of primiparous dams (Fig. 7, A and B). We next measured levels of genes responsible for the regulation of these genes (Fig. 7, A and B). PGC-1α mRNA levels were decreased in adipose tissues offspring of multiparous vs primiparous dams, whereas mRNA levels of PGC-1β, PPARγ, liver X receptor (LXR), and sterol regulatory element-binding protein-1 (SREBP-1) were not affected. Compared with expression in adipose tissues, CPT I mRNA levels were decreased almost 40% in liver extracts from offspring of multiparous dams, with only modest changes in FAS mRNA levels (Fig. 7B). PGC-1α mRNA levels were decreased in the livers of offspring of multiparous dams, as occurred in adipose tissue; hepatic PGC-1α protein levels paralleled mRNA levels (Fig. 7C) and were around twofold greater in the primiparous offspring, as noted by ImageJ software. Unlike adipose tissue, PGC-1β mRNA levels were also reduced significantly in the livers of offspring of multiparous dams, and PPARα mRNA levels tended to be less (P = 0.069). LXR and SREBP-1 mRNA levels did not differ from the primiparous group. The expression of phosphoenolpyruvate carboxykinase and glucose-6-phosphatase, key enzymes for hepatic glucose production that are known to be regulated by PGC-1α, was also reduced in the offspring of multiparous dams.
Fig. 7.
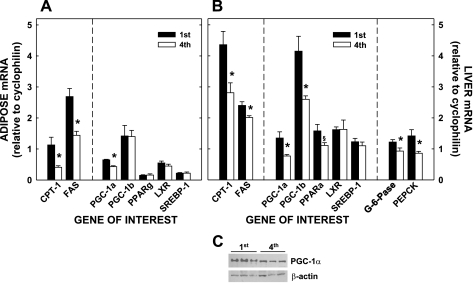
Relative mRNA levels of proteins associated with lipid metabolism in adipose tissues and livers of adult offspring from primi- and multiparous dams; animals are the same as those described in Fig. 3. The relative amounts of mRNA of genes of interest compared with cyclophilin are presented in offspring of dams with different parity in retroperitoneal adipose tissue (A) and liver (B). C: liver homogenates from all offspring from primi- or multiparous dams were pooled into 3 different samples (3–4 different offspring/pooled sample), and the different pooled samples from each group were assayed by Western blot. Data are presented as means ± SE. *Differences (P < 0.05) between tissues of offspring of primi- and multiparous dams. CPT I, carnitine palmitoyltransferase I; FAS, fatty acid synthase; PGC-1α and -β, peroxisome proliferator-activated receptor-γ (PPARγ) coactivator-1α and -β; LXR, liver X receptor; SREBP-1, sterol regulatory element-binding protein-1. G-6-Pase, glucose 6-phosphatase; PEPCK phosphoenolpyruvate carboxykinase.
DISCUSSION
In this set of studies, we have demonstrated that multiparity causes weight gain in mice, independent of age. In addition, multiparous females have increased inflammation in liver, adipose tissue, and placenta, processes that are not accounted for by increased adiposity. Multiparity also has metabolic consequences, since these animals were glucose intolerant compared with primiparous dams; this response could be explained by either the greater adiposity, increased inflammation, or some combination of these. In addition to the detrimental affects of obesity and its comorbidities, exaggerated inflammation is detrimental to the multiparous female and increases its risk for additional age-related diseases. For example, atherosclerosis is known to be a disease associated with chronic inflammation. Initial steps in atherosclerotic plaque formation involve recruitment of monocytes to the vessel walls that are then differentiated into macrophages, thereby initiating lesion formation (36, 50). Inflammation is linked with diabetes as well in that some proinflammatory cytokines can blunt insulin signaling, thereby leading to insulin resistance and diabetes (24). It is likely that obesity and all of its comorbidities and inflammation in the multiparous female could be difficult to overcome if inflammation is prominent in the brain because inflammation in the hypothalamus disrupts satiety signals originating in the brain (13, 73), making weight loss difficult.
The metabolic detriments of multiparity were not limited to the mothers but extended to their male offspring. Importantly, male mice born of multiparous mothers had increased levels of body fatness in adulthood. These findings support a model whereby multiple pregnancies induce fixed metabolic stresses on females that have heritable consequences. Since there are parallels to some of these findings in humans, our results support yet another mechanism of cross-generational transmission of obesity risk. Interestingly, the differences became apparent when the mice were older, after 14 wk of age, suggesting that excess energy was stored as fat only after growth rate slowed down. Since food consumption was not different in offspring of dams with different parity, differences must have occurred in energy expended. Although we did not examine female offspring in these studies, some of the metabolic parameters measured may have been more exaggerated in the female mice based on previous reports by some (4, 34, 61), but not others (45).
We used a targeted gene approach to study metabolism of catabolic processes in two of the tissues where fatty acid oxidation occurs, the liver and adipose tissue (11, 67, 70). The lower CPT I mRNA levels in the offspring of multiparous dams in both tissues suggest that there is less fatty acid oxidation in the whole body, which has been shown to be related to obesity (38, 56). CPT I transcription was regulated differently in the multiparous offspring since expression of key metabolic regulators PGC-1α, PGC-1β, and PPARα was reduced as well. Since PGC-1α levels appear to be most important in mediating transcription compared with other proteins in the transcriptional complexes (64), reduction in its levels is likely enough to manipulate fatty acid oxidation. In addition to a direct effect on CPT I transcription, PGC-1α can manipulate fatty acid oxidation through affects on mitochondria (15, 21, 25). Although we did not measure mitochondrial activity or number in the current studies, we did find that mRNA levels for proteins expressed in the hepatic mitochondria of offspring of multiparous dams were reduced (cyp7a1: 2.72 ± 0.38 vs. 0.84 ± 0.08; citrate synthase: 4.47 ± 0.81 vs. 1.29 ± 0.11 for offspring of primi- vs. multiparous females, respectively), suggesting a change in mitochondrial function.
PGC-1α is one of the key regulatory proteins in mediating cellular metabolism (reviewed in Refs. 15 and 25), including mitochondrial biogenesis and CPT I transcription. PGC-1α is a coregulator that interacts with various DNA-binding factors, including PPARα/γ, estrogen-related receptors, and nuclear receptor factors, to coordinate the regulation of a variety of genes and processes. PGC-1α will form complexes with these various transcription factors and recruit additional proteins that modify histones and remodel nucleosomes (49). PGC-1α levels and phosphorylation (activation)/acetylation (inactivation) appear to be regulated by the energy and/or nutritional status of the cells and are regulated by PPARs, myocyte enhancer factor 2, forkhead box O, cAMP response element-binding protein, estrogen-related receptors, the adiponectin receptors (adipoR1/R2), AMPK, and sirtuin (15, 25, 27, 71). Results from the current studies parallel studies in which pregnant females were overfed in that offspring of overfed mothers had reduced adipoR1/R2 expression and AMPK activity (7, 57), which can lead to less expression and activation of PGC-1α (27, 71).
PGC-1α levels can also be regulated epignetically (3, 58). Previous studies have shown that a variety of conditions can methylate the promoter and lead to reduced mRNA levels. Epigenetic changes in genes have been proposed to be involved in the passing on of traits from one generation to another, and methylation of specific genes at birth can predict adiposity later in life (17). Although most common routes by which the epigenome is affected include DNA methylation and histone modification (72), it is likely that histone modification plays a major role in developmental programming via epigenetic regulation (47). The study of epigenetic regulation is complex, since some epigenetic changes can be silent and their action becomes apparent with different diets, stressors, or stages of life (16, 47, 65).
One surprising observation of these studies was that FAS mRNA levels were decreased in adipose tissues and livers with greater adiposity, and the decreases were independent of changes in SREBP-1 and LXR mRNA levels, the typical route of regulation. Contrary to conventional thinking, recent studies show that PGC-1β and PGC-1α can be coactivators of SREBP-1/LXR and thereby affect their downstream targets independent of a change in SREBP-1/LXR levels (37, 60). Thus, the PGCs can affect the sum of lipid metabolism via both catabolic and anabolic pathways, with the greater impact upon oxidation under the current conditions.
An important outcome of these studies is their implication for mouse research. When efficiently maintaining mouse colonies, a female that is a good breeder is often rebred multiple times. However, this female likely develops multiparity-induced obesity and inflammation. Importantly, the offspring of these multiparous females would accrue more fat independent of any dietary or genetic manipulation. Consequently, a difference in parity of the female could be part of the cause of scatter in data in some studies or lack of effects due to lack of significance. Thus, care should be taken when maintaining mice colonies such that parity of breeding females should be kept equal to minimize scatter and thereby increase significance.
Summary
A question that arises from these studies, as well as others, is, “What is mediating the programming?” The mediator of developmental programming is currently unknown but likely includes circulating maternal factors associated with obesity, inflammation, and/or diabetes and/or the placental inflammatory status (2, 9, 23, 44). Some maternal factors that are associated with these diseases have been proposed as potential mediators because they manipulate nutrient transport across the placenta or placental function, thereby affecting fetal metabolism (14, 23, 28, 29, 32, 33). Another potential mechanism is based on the fact that placentas of obese females contain more inflammation (10, 52, 59, 74), and the inflammatory in utero state that the fetus is exposed to may be mediating the effect. In the current study, the placentas of the multiparous dams were in a more severe proinflammatory state than placentas of the primiparous dams, as depicted by increased JNK activation, and had more macrophages, as do placentas of obese humans, as depicted by increased CD68 and F4/80. Since the studies in mice parallel those in humans and developmental programming did indeed occur, we can use this model to begin to dissect out the role of various maternal factors in mediating programming.
To summarize, these studies support the propagation of the obesity cycle in multiparous females. Since more women are obese at the start of pregnancy, more retain weight during and between pregnancies, thereby developing multiparity-induced obesity. Thus, offspring of the obese multiparous dams with increased inflammation will themselves become obese in adulthood. Interventions targeted to break the obesity cycle could occur at the level of the pregnant female as well as the offspring. One could target inflammation of the pregnant female with various anti-inflammatory treatments, i.e., fish oil or exercise or antiobesity treatments, i.e., food restriction or exercise; it should be noted that the ω-3 fatty acids in fish oil could have a direct impact upon the fetus itself (43). In the offspring, one could target mitochondrial biogenesis or fatty acid oxidation with various factors, such as lipoic acid or carnitine (35, 40). The current studies are important in supporting a healthier, less obese population in that we have defined specific metabolic pathways that are likely involved in the programming of obesity and can be targeted in either the mother or her offspring.
GRANTS
These studies were supported by a grant from the MICROMouse program of the MMPC and Grants HD-34089 and DK-059630 from the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.L.R., T.J., K.T.B., A.J., and L.A.W. performed the experiments; S.L.R., T.J., K.T.B., A.J., P.T., and L.A.W. analyzed the data; S.L.R., T.J., A.J., and L.A.W. prepared the figures; S.L.R., T.J., A.J., P.T., D.A.D., and L.A.W. edited and revised the manuscript; A.J., P.T., D.A.D., and L.A.W. interpreted the results of the experiments; D.A.D. and L.A.W. did the conception and design of the research; L.A.W. drafted the manuscript; L.A.W. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Jodi Caldwell-Lawson for assistance with the various animal studies.
REFERENCES
- 1. Araneta MR, Barrett-Connor E. Grand multiparity is associated with type 2 diabetes in Filipino American women, independent of visceral fat and adiponectin. Diabetes Care 33: 385–389, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Armitage JA, Poston L, Taylor PD. Developmental origins of obesity and the metabolic syndrome: The role of maternal obesity. In: Frontiers of Hormone Research, Obesity and Metabolism, edited by Korbontis M. Basel, Switzerland: Kerger, 2008, p. 73–84 [DOI] [PubMed] [Google Scholar]
- 3. Barres R, Osler ME, Yan J, Rune A, Fritz T, Caldahl K, Krook A, Zierath JR. Non-CpG methylation of the PGC-1α promoter through DNMT3B controls mitochondrial density. Cell Metab 10: 189–198, 2009 [DOI] [PubMed] [Google Scholar]
- 4. Bayol SA, Simbi BH, Bertrand JA, Strickland NC. Offspring from mothers fed a ‘junk food' diet in pregnancy and lactation exhibit exacerbated adiposity that is more pronounced in females. J Physiol 586: 3219–3230, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bertoni AG, Burke GL, Owusu JA, Carnethon MR, Vaidya D, Barr RG, Jenny NS, Ouyang P, Rotter JI. Inflammation and the incidence of type 2 diabetes: the Multi-Ethnic Study of Atherosclerosis (MESA). Diabetes Care 33: 804–810, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Biggs HG, Erikson JM, Moorehead WR. A manual colorimetric assay of triglycerides in serum. Clin Chem 21: 437–441, 1975 [PubMed] [Google Scholar]
- 7. Borengasser SJ, Lau F, Kang P, Blackburn ML, Ronis MJ, Badger TM, Shankar K. Maternal obesity during gestation impairs fatty acid oxidation and mitochondrial sirt3 expression in rat offspring at weaning. PLoS One 6: e24068, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Catalano PM. Obesity and pregnancy—the propagation of a viscous cycle? J Clin Endocrinol Metab 88: 3505–3506, 2003 [DOI] [PubMed] [Google Scholar]
- 9. Catalano PM, Ehrenberg HM. The short- and long-term implications of maternal obesity on the mother and her offspring. BJOG 113: 1126–1133, 2006 [DOI] [PubMed] [Google Scholar]
- 10. Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Hauguel-de Mouzon S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 29: 274–281, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Choo HJ, Kim JH, Kwon OB, Lee CS, Mun JY, Han SS, Yoon YS, Yoon G, Choi KM, Ko YG. Mitochondria are imparied in the adipocytes of type 2 diabetic mice. Diabetologia 49: 784–791, 2006 [DOI] [PubMed] [Google Scholar]
- 12. Davis EM, Zyzanski SJ, Olson CM, Stange KC, Horwitz RI. Racial, ethnic, and socioeconomic differences in the incidence of obesity related to childbirth. Am J Public Health 99: 294–299, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Denis RG, Arruda AP, Romanatoo T, Milanski M, Coope A, Solon C, Razolli DS, Velloso LA. TNF-α transiently induces endoplasmic reticulum stress and an incomplete unfolded protein response in the hypothalamus. Neuroscience 170: 1035–1044, 2010 [DOI] [PubMed] [Google Scholar]
- 14. Farley DM, Choi J, Dudley DJ, Li C, Jenkins SL, Myatt L, Nathanielsz PS. Placental amino acid transport and placental leptin resistance in pregnancies complicated by maternal obesity. Placenta 31: 713–724, 2010 [DOI] [PubMed] [Google Scholar]
- 15. Fernandez-Marcos PJ, Auxerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93: 884S–890S, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gluckman PD, Lillycrop KA, Vickers MH, Pleasants AB, Phillips ES, Beedle AS, Burdge GC, Hanson MA. Metabolic plasticity during mammalian development is directionally dependent on early nutritional status. Proc Natl Acad Sci USA 104: 12796–12800, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, Rodford J, Slater-Jefferies JL, Garratt E, Crozier SR, Emerald BS, Gale CR, Inskip HM, Cooper C, Hanson MA. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes 60: 1528–1534, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greenberg AS, Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr 83: 461S–465S, 2006 [DOI] [PubMed] [Google Scholar]
- 19. Gunderson EP, Abrams B, Selvin S. Does the pattern of postpartum weight change differ according to pregravid body size? Int J Obes Relat Metab Disord 25: 853–862, 2001 [DOI] [PubMed] [Google Scholar]
- 20. Gunderson EP, Jacobs DR, Jr, Chiang V, Lewis CE, Tsai A, Quesenberry CP, Jr, Sidney S. Childbearing is associated with higher incidence of the metabolic syndrome among women of reproductive age controlling for measurements before pregnancy: the CARDIA study. Am J Obstet Gynecol 201: 177.e1–177.e9, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Handschin C, Speigelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 27: 728–735, 2006 [DOI] [PubMed] [Google Scholar]
- 22. Hauguel-de Mouzon S, Guerre-Millo M. The placenta cytokine network and inflammatory signals. Placenta 27: 794–798, 2006 [DOI] [PubMed] [Google Scholar]
- 23. Heerwagen MJ, Miller MR, Barbour LA, Friedman JE. Maternal obesity and fetal metabolic programming: a fertile epigenetic soil. Am J Physiol Regul Integr Comp Physiol 299: R711–R722, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature 420: 333–336, 2002 [DOI] [PubMed] [Google Scholar]
- 25. Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol 71: 177–203, 2009 [DOI] [PubMed] [Google Scholar]
- 26. Huda SS, Brodie LE, Sattar N. Obesity in pregnancy: prevalence and metabolic consequences. Semin Fetal Neonatal Med 15: 70–76, 2010 [DOI] [PubMed] [Google Scholar]
- 27. Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, Ogata H, Kubota N, Takamoto I, Hayashi YK, Yamauchi N, Waki H, Fukayama M, Nishino I, Tokuyama K, Ueki K, Oike Y, Ishii S, Hirose K, Shimizu T, Touhara K, Kadowaki T. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 464: 1313–1319, 2010 [DOI] [PubMed] [Google Scholar]
- 28. Jansson N, Nilsfelt A, Gellerstedt M, Wennergran M, Rossander-Hulthen L, Powell TL, Jansson T. Maternal hormones linking maternal body mass index and dietary intake to birth weight. Am J Clin Nutr 87: 1743–1749, 2008 [DOI] [PubMed] [Google Scholar]
- 29. Jansson T, Powell TL. Role of the placenta in fetal programming: underlying mechanisms and potential interventional approaches. Clin Sci (Lond) 113: 1–13, 2007 [DOI] [PubMed] [Google Scholar]
- 30. Jarvie E, Hauguel-de-Mouzon S, Nelson SM, Sattar N, Catalano PM, Freeman DJ. Lipotoxicity in obese pregnancy and its potential role in adverse pregnancy outcome and obesity in the offspring. Clin Sci (Lond) 119: 123–129, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jarvie E, Ramsay JE. Obstetric management of obesity in pregnancy. Semin Fetal Neonatal Med 15: 83–88, 2010 [DOI] [PubMed] [Google Scholar]
- 32. Jones HN, Jansson T, Powell TL. IL-6 stimulates system A amino acid transporter activity in trophoblast cells through STAT3 and increased expression of SNAT2. Am J Physiol Cell Physiol 297: C1228–C1235, 2009 [DOI] [PubMed] [Google Scholar]
- 33. Jones HN, Woollett LA, Barbour N, Prasad PD, Powell TL, Jansson T. High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice. FASEB J 23: 271–278, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khan IY, Taylor PD, Dekou V, Seed PT, Lakasing L, Grahan D, Dominiczak AF, Hanson MA, Poston L. Gender-linked hypertension in offspring of lard-fed pregnant rats. Hypertension 41: 168–175, 2003 [DOI] [PubMed] [Google Scholar]
- 35. Koh EH, Lee WJ, Lee SA, Kim EH, Cho EH, Jeong E, Kim DW, Kim MS, Park JY, Park KG, Lee HJ, Lee IK, Lim S, Jang HC, Lee KH, Lee KU. Effects of alpha-lipoic acid on body weight in obese subjects. Am J Med 124: 85.e1–85.e8, 2011 [DOI] [PubMed] [Google Scholar]
- 36. Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J 74: 213–220, 2010 [DOI] [PubMed] [Google Scholar]
- 37. Lin J, Yang R, Tarr PT, Wu PH, Handschin C, Li S, Yang W, Pei L, Uldry M, Tontonoz P, Newgard CB, Spiegelman BM. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell 120: 261–273, 2005 [DOI] [PubMed] [Google Scholar]
- 38. Liu C, Lin JD. PGC-1 coactivators in the control of energy metabolism. Acta Biochim Biophys Sin (Shanghai) 43: 248–257, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luppi P, Haluszczak C, Betters D, Richard CA, Trucco M, DeLoia JA. Monocytes are progressively activated in the circulation of pregnant women. J Leukoc Biol 72: 874–884, 2002 [PubMed] [Google Scholar]
- 40. Makowski L, Noland RC, Koves TR, Xing W, Ilkayeva OR, Muehlbauer MJ, Stevens RD, Muoio DM. Metabolic profiling of PPARalpha−/− mice reveals defects in carnitine and amino acid homeostasis that are partially reversed by oral carnitine supplementation. FASEB J 23: 586–604, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McConihay JA, Honkomp AM, Granholm NA, Woollett LA. Maternal high density lipoproteins affect fetal mass and extra-embryonic fetal tissue sterol metabolism in the mouse. J Lipid Res 41: 424–432, 2000 [PubMed] [Google Scholar]
- 42. Mills JL, Troendle J, Conley MR, Carter T, Druschel CM. Maternal obesity and congenital heart defects: a population-based study. Am J Clin Nutr 91: 1543–1549, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Muhlhausler BS, Miljkovic D, Fong L, Xian CJ, Duthoit E, Gibson RA. Maternal omega-3 supplementation increases fat mass in male and female rat offspring. Front Gene 2: 48, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nijland MJ, Ford SP, Nathanielsz PW. Prenatal origins of adult disease. Curr Opin Obstet Gynecol 20: 132–138, 2008 [DOI] [PubMed] [Google Scholar]
- 45. Nivoit P, Morens C, Van Assche FA, Jansen E, Poston L, Remacle C, Reusens B. Established diet-induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia 52: 1133–1142, 2009 [DOI] [PubMed] [Google Scholar]
- 46. Ovesen P, Rasmussen S, Kesmodel U. Effect of prepregnancy maternal overweight and obesity on pregnancy outcome. Obstet Gynecol 118: 305–312, 2011 [DOI] [PubMed] [Google Scholar]
- 47. Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest 118: 2316–2324, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286: 327–334, 2001 [DOI] [PubMed] [Google Scholar]
- 49. Puigserver P, Adelmant G, Wu Z, Fan M, Xu J, O'Malley B, Spiegelman BM. Activation of PPARgamma coactivator-1 through transcription factor docking. Science 286: 1368–1371, 1999 [DOI] [PubMed] [Google Scholar]
- 50. Rader DJ, Daugherty A. Translating molecular discoveries into new therapies for atherosclerosis. Nature 451: 904–913, 2008 [DOI] [PubMed] [Google Scholar]
- 51. Rasmussen KM, Abrams B, Bodnar LM, Butte NF, Catalano PM, Maria Siega-Riz A. Recommendations for weight gain during pregnancy in the context of the obesity epidemic. Obstet Gynecol 116: 1191–1195, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roberts KA, Riley SC, Reynolds RM, Barr S, Evans M, Statham A, Hor K, Jabbour HN, Norman JE, Denison FC. Placental structure and inflammation in pregnancies associated with obesity. Placenta 32: 247–254, 2011 [DOI] [PubMed] [Google Scholar]
- 53. Rooney BL, Schauberger CW. Excess pregnancy weight gain and long-term obesity: one decade later. Obstet Gynecol 100: 245–252, 2002 [DOI] [PubMed] [Google Scholar]
- 54. Rooney BL, Schauberger CW, Mathiason MA. Impact of perinatal weight change on long-term obesity and obesity-related illnesses. Obstet Gynecol 106: 1349–1356, 2005 [DOI] [PubMed] [Google Scholar]
- 55. Schmatz M, Madan J, Marino T, Davis J. Maternal obesity: the interplay between inflammation, mother and fetus. J Perinatol 30: 441–446, 2009 [DOI] [PubMed] [Google Scholar]
- 56. Schreurs M, Kupers F, van der Leij FR. Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes Res 11: 380–388, 2010 [DOI] [PubMed] [Google Scholar]
- 57. Shankar K, Kang P, Harrell A, Zhong Y, Marecki JC, Ronis MJ, Badger TM. Maternal overweight programs insulin and adiponectin signaling in the offspring. Endocrinology 151: 2577–2589, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sookoian S, Rosselli MS, Gemma C, Burgueño AL, Fernández Gianotti T, Castaño GO, Pirola CJ. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor γ coactivator 1α promoter. Hepatology 52: 1992–2000, 2010 [DOI] [PubMed] [Google Scholar]
- 59. Stewart FM, Freeman DJ, Ramsay JE, Greer IA, Caslake M, Ferrell WR. Longitudinal assessment of maternal endothelial function and markers of inflammation and placental function throughout pregnancy in lean and obese mothers. J Clin Endocrinol Metab 92: 969–975, 2007 [DOI] [PubMed] [Google Scholar]
- 60. Summermatter S, Baum O, Santos G, Hoppeler H, Handschin C. Peroxisome proliferator-activated receptor {gamma} coactivator 1 {alpha} (PGC-1{alpha}) promotes skeletal muscle lipid refueling in vivo by activating de novo lipogenesis and the pentose phosphate pathway. J Biol Chem 285: 32793–32800, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Taylor PD, McConnell JM, Khan IY, Holemans K, Lawrence KM, Asare-Anane H, Persaud SJ, Jones PM, Petrie L, Hanson MA, Poston L. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am J Physiol Regul Integr Comp Physiol 288: R234–R239, 2005 [DOI] [PubMed] [Google Scholar]
- 62. Tsoi E, Shaikh H, Rbonison S, Teoh TG. Obesity in pregnancy: a major healthcare issue. Postgrad Med J 86: 617–623, 2010 [DOI] [PubMed] [Google Scholar]
- 63. Turley SD, Herndon MW, Dietschy JM. Reevaluation and application of the dual-isotope plasma ratio method for the measurement of intestinal cholesterol absorption in the hamster. J Lipid Res 35: 328–339, 1994 [PubMed] [Google Scholar]
- 64. Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res 79: 208–217, 2008 [DOI] [PubMed] [Google Scholar]
- 65. Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr 27: 363–388, 2007 [DOI] [PubMed] [Google Scholar]
- 66. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest 115: 1111–1119, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wilson-Fritch L, Nicoloro S, Chouinard M, Lazar MA, Chui PC, Leszyk J, Straubhaar J, Czech MP, Corvera S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest 114: 1281–1289, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yao L, Jenkins K, Horn PS, Lichtenberg MH, Woollett LA. Inability to fully suppress sterol synthesis rates with exogenous sterol in embryonic and extraembyronic fetal tissues. Biochim Biophys Acta 1171: 1372–1379, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yogev Y, Catalano PM. Pregnancy and obesity. Obstet Gynecol Clin North Am 36: 285–300, 2009 [DOI] [PubMed] [Google Scholar]
- 70. Yoon M. The role of PPARalpha in lipid metabolism and obesity: focusing on the effects of estrogen on PPARalpha actions. Pharmacol Res 60: 151–159, 2009 [DOI] [PubMed] [Google Scholar]
- 71. You M, Considine RV, Leone TC, Kelly DP, Crabb DW. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology 42: 568–577, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang X, Ho SM. Epigenetics meets endocrinology. J Mol Endocrinol 46: R11–R32, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135: 61–73, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhu MJ, Du M, Nathanielsz PW, Ford SP. Maternal obesity up-regulates inflammatory signaling pathways and enhances cytokine expression in the mid-gestation sheep placenta. Placenta 31: 387–391, 2010 [DOI] [PubMed] [Google Scholar]