

Abstract
Insulin receptors in podocytes are essential for normal kidney function. Here, we show that insulin evokes a rapid increase in the surface expression of canonical transient receptor potential-6 channel (TRPC6) channels in cultured podocytes, but caused a decrease in surface expression of TRPC5. These effects are accompanied by a marked increase in outwardly rectifying cationic currents that can be blocked by 10 μM SKF96365 or 100 μM La3+. Application of oleoyl-2-acetyl-sn-glycerol (OAG) also increased SKF96365- and La3+-sensitive cationic currents in podocytes. Importantly, current responses to a combination of OAG and insulin were the same amplitude as those evoked by either agent applied alone. This occlusion effect suggests that OAG and insulin are targeting the same population of channels. In addition, shRNA knockdown of TRPC6 markedly reduced cationic currents stimulated by insulin. The effects of insulin on TRPC6 were mimicked by treating podocytes with H2O2. Insulin treatment rapidly increased the generation of H2O2 in podocytes, and it increased the surface expression of the NADPH oxidase NOX4 in cultured podocytes. Basal and insulin-stimulated surface expression of TRPC6 were reduced by pretreatment with diphenylene iodonium, an inhibitor of NADPH oxidases and other flavin-dependent enzymes, by siRNA knockdown of NOX4, and by manganese (III) tetrakis (4-benzoic acid) porphyrin chloride, a membrane-permeable mimetic of superoxide dismutase and catalase. These observations suggest that insulin increases generation of ROS in part through activation of NADPH oxidases, and that this step contributes to modulation of podocyte TRPC6 channels.
Keywords: focal and segmental glomerulosclerosis, diabetes
trpc6 channels are widely expressed Ca2+-permeable cation channels that are activated in the course of G protein and growth factor signaling cascades (12). In 2005 a pair of landmark studies showed that TRPC6 channels are expressed in podocytes and that gain-of-function mutations in TRPC6 channels lead to progressive forms of focal and segmental glomerulosclerosis (FSGS) (44, 55). Additional TRPC6 mutations associated with proteinuric kidney disease have been identified more recently (18), and it was subsequently shown that wild-type TRPC6 channels are expressed at significantly higher levels in acquired proteinuric diseases, including membranous glomerulonephritis and minimal change disease (38). Moreover, selective overexpression of wild-type or mutant TRPC6 channels in mouse podocytes in vivo leads to proteinuria and glomerulosclerosis that resembles human FSGS (31).
TRPC6 channels typically function as downstream plasma membrane components of phospholipase C signaling cascades that liberate diacylglycerol (DAG) and cause local depletion of plasma membrane phosphatidylinositol 4,5-bisphosphate (12). In cultured podocytes, TRPC6 activation can be evoked by angiotensin II (39, 48, 57) and probably by other signals (40). In podocytes, TRPC6 signaling leads to activation of transcription factors such as NFAT (46, 52), and sustained TRPC6 activation in cultured podocytes can induce apoptosis (57). TRPC6 forms part of a larger signaling complex at the glomerular slit diaphragm (12). For example, TRPC6 interacts with the slit diaphragm protein nephrin and its total expression is increased in nephrin knockout mice (44). TRPC6 also interacts with podocin, which may regulate its gating (21), as well as with large-conductance Ca2+-activated K+ (BKCa) channels (26), which may play a role to maintain a driving force for Ca2+ influx in the course of TRPC6 activation (12, 14).
An important recent study showed that selective deletion of insulin receptors in mouse podocytes in vivo causes albuminuria and glomerulosclerosis (53). These knockout mice appeared normal at 3 wk of age, but proteinuria, glomerulosclerosis, foot process effacement, and thickening of the glomerular basement membrane were present by 8 wk of age, as is seen in several mouse models of diabetic nephropathy (5). Moreover, the podocyte-specific insulin receptor knockout mice were normoglycemic (53), indicating that insulin signaling to podocytes is crucial for the normal function of the entire glomerulus. Relatively little is known about the cellular actions of insulin on podocytes. Recent studies showed that insulin stimulates glucose uptake by increasing the cell surface expression of glucose transporters (9, 10). More recently, we showed that insulin also increases surface expression of BKCa channels in podocytes (29).
In this study, we present evidence that insulin modulates TRPC6 channels in cultured podocytes. We observed that treatment of cultured podocytes with insulin causes an increase in the steady-state surface expression of TRPC6 channels that is apparent within minutes, and which is accompanied by an increase in the mean amplitude of outwardly rectifying cationic currents with pharmacological properties that implicate TRPC6 (14, 23). We also show that the effects of insulin on surface expression of TRPC6 are at least partly dependent on generation reactive oxygen species (ROS) such as H2O2, that are generated by NADPH oxidases that include NOX4. Finally, we show that insulin evokes essentially the opposite effect on TRPC5 channels, as we observed a decrease in the steady-state surface expression of those channels after insulin treatment.
MATERIALS AND METHODS
Cell culture protocols, transfection, and drugs.
Cell culture protocols have been described previously (26–30). Briefly, MPC-5 mouse podocyte cell lines (obtained from Dr. Peter Mundel) were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum and 100 U/ml penicillin-streptomycin, with recombinant mouse γ-interferon, in humidified 5% CO2 incubators and propagated at 33°C. Podocyte differentiation and expression of podocyte markers were induced by removal of γ-interferon and temperature switch to 37°C for 14 days. A subclone of the MPC-5 podocyte cell line stably expressing shRNA against TRPC6 described previously (39) was obtained from Dr. Jochen Reiser. Differentiated podocytes were transferred to serum-free media 24 h before experiments with insulin. Cells were then treated with 100 nM recombinant human insulin for various lengths of time (1 min to 72 h) before analyses. A panel of siRNAs directed against NOX4, as well as nontargeted siRNAs for use in control experiments, was obtained from Santa Cruz Biotechnology and transfected into podocytes using Oligofectamine (Invitrogen) in serum-reduced medium according to the manufacturer's directions and as described previously (26–28). Biochemical analyses were carried out 24–48 h after transfection of NOX4 or control siRNA. Recombinant human insulin, 1-oleoyl-2-acetyl-sn-glycerol (OAG), and diphenylene iodonium (DPI) were from Sigma, SKF96365 was from Calbiochem, and manganese (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP) was from Percipio Biosciences.
Immunoblot analysis and cell surface biotinylation assays.
These were carried out as described in detail previously (26–30). Briefly, for immunoblot analyses, proteins in podocyte lysates were separated by SDS-PAGE on 10% gels and transferred to filters. Blots were blocked, incubated with a primary antibody overnight at 4°C, washed again, and the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature. Proteins were visualized using a chemiluminescent substrate. The primary antibodies were rabbit anti-TRPC6 (Alomone), mouse monoclonal anti-TRPC5 clone N67/15 (University of California Davis/NIH NeuroMab Facility), rabbit anti-NOX4, and rabbit anti-β-actin (Sigma). For cell surface biotinylation assays, intact podocytes were treated with a membrane-impermeable biotinylation reagent, sulfo-N-hydroxy-succinimidobiotin (Pierce Biotechnology, Rockford, IL; 1 mg/ml) for 1 h. The reaction was stopped, cells were lysed, and biotinylated proteins from the cell surface were recovered by incubation with immobilized streptavidin-agarose beads. A sample of the initial cell lysate was retained for analysis of total proteins. These samples were quantified by immunoblot analysis followed by densitometric analysis using Image J software. Bar graphs describing these data were constructed from three to five repetitions of each experiment.
Assay of ROS generation.
Generation of H2O2 by podocytes was measured using the OxiSelect fluorometric assay (Cell Biolabs). In this assay, H2O2 in the presence of HRP causes oxidation of 10-acetyl-3,7-dihydroxyphenoxazine (ADHP) to resorufin, a fluorescent product with a high extinction coefficient (60). Briefly, ADHP/HRP working solution was added to 50-μl samples of serum-free culture supernatant from control or experimental cells according to the manufacturer's directions. Media were then incubated with rocking for 30 min in the dark, and fluorescence was measured with a microplate reader in standard 96-well fluorescence black microtiter plates using excitation at 530 nm and detection at 590 nm. Background fluorescence, measured from culture media that did not contain ADHP, was subtracted before statistical analysis of data.
Electrophysiology.
Whole cell recordings were made using a modification of methods described previously (26–30). Briefly, the bath solution contained 150 mM NaCl, 5.4 mM CsCl, 0.8 mM MgCl2, 5.4 mM CaCl2, and 10 mM HEPES, pH 7.4. Pipette solutions contained 10 mM NaCl, 125 mM CsCl, 6.2 mM MgCl2, 10 mM HEPES, and 10 mM EGTA, pH 7.2. The recording electrodes had resistances of 3–4 MΩ and it was possible to compensate up to 85% of this without introducing oscillations into the current output of the patch-clamp amplifier. Outwardly rectifying currents were evoked by a ramp voltage commands (−80 to + 80 mV over 2.5 s) from a holding potential of −40 mV. Note that there is no K+ in either the bath or pipette solutions, and the dominant cation in the pipette is Cs+, which is designed to prevent contamination from K+ currents. The concentration of Ca2+ in the bath solution (5.4 mM) in all experiments is threefold higher than the normal physiological concentration, by design. After experimenting with many conditions, we found that this helps maintain the stability of whole cell recordings from podocytes, which are flat and relatively fragile. Note, however, that 5.4 mM external Ca2+ markedly reduces inward currents through TRPC6 channels (14, 23) and increases the outward rectification that is a normal feature of these channels under any conditions. For this reason, whole cell currents were quantified at +80 mV. Voltage ramps were applied before and after bath superfusion of 10 μM SKF96365, which produces a rapid and reversible blockade of cation channels in the TRPC family (23, 37). SKF96365-sensitive currents were then obtained by digital subtraction and currents at +80 mV were quantified for statistical analyses. We used a similar procedure in experiments using 100 μM La3+ as an inhibitor of TRPC6 (48). It was not possible to maintain stable recordings long enough to measure currents in a single cell before and after insulin treatments. Therefore, in these experimental designs, statistical analyses were carried out on currents measured from groups of cells (control or insulin-treated). For consistency, we used the same statistical design for experiments with OAG.
Statistics.
Summaries of quantitative data are presented as means ± SE. Electrophysiological data and results of assays for generation of ROS were analyzed by one-way or two-way ANOVA, as indicated, followed by Tukey's post hoc test when multiple groups were compared. We used Student's unpaired t-test in experiments when only two groups were compared. Throughout, P < 0.05 is regarded as significant. For densitometric analyses, immunochemical experiments were run in triplicate, as per standard practice. Means ± SE are provided to illustrate variability of data.
RESULTS
Insulin modulates podocyte TRPC6 channels.
These experiments were performed on differentiated cells of a conditionally immortalized mouse podocyte cell line (MPC-5 cells). After differentiation for 2 wk at 37°C in our laboratory, these cells express the podocyte markers nephrin, Neph1, and synaptopodin (27, 28, 30) and they show physiological responses to insulin (29). We initially examined the effect of insulin on the steady-state surface expression of TRPC6 by means of cell surface biotinylation assays. Differentiated podocytes were maintained in serum-free media for 24 h before insulin treatment. We observed that 100 nM insulin evoked a robust increase in the steady-state surface expression of TRPC6 (Fig. 1). In every experiment, this effect could be seen after 15 min of insulin treatment, and in several experiments this effect was discernible with as little as 1 min of exposure to insulin. However, based on densitometric quantification of these experiments, the largest effects were seen with 24-h or longer insulin treatments, and these effects were typically more than twofold greater than those evoked by 15-min exposures to insulin.
Fig. 1.
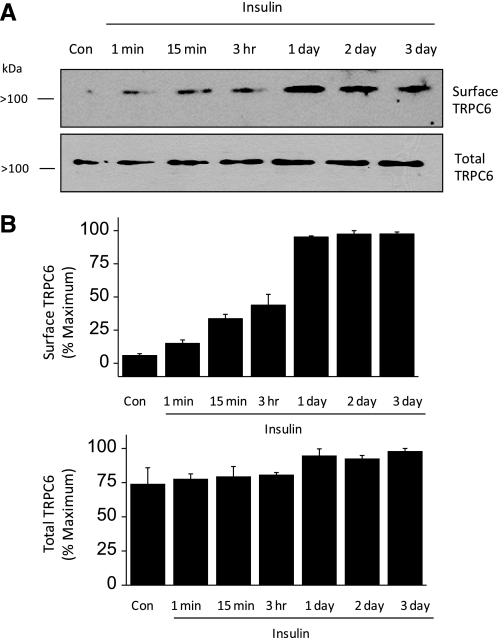
Insulin increases the steady-state surface expression of TRPC6 in differentiated cells of an immortalized podocyte cell line. A: representative cell surface biotinylation assay showing surface and total expression of TRPC6 as indicated after exposing podocytes to insulin for the durations indicated. Biotinylated (surface) and total cellular proteins were quantified by immunoblot analysis. In this experiment, note increase in surface expression of TRPC6 with insulin exposures of 1 and 15 min, with larger effects seen after 1 day of insulin treatment. In this and subsequent figures, molecular weights are shown to the left of each blot. B: bar graphs showing densitometric analysis of 3 repetitions of this experiment. Bars denote mean signal intensity from immunoblot for surface TRPC6 (top) and total TRPC6 (bottom), and error bars denote SE.
Previous studies on TRPC6 channels in heterologous expression systems showed that their activation gives rise to outwardly rectifying macroscopic cationic currents that are much larger in the presence of DAG analogs (20) and that are reduced in the presence of millimolar external Ca2+ (14, 23). We observed macroscopic currents with these characteristics by means of conventional whole cell recordings from differentiated podocytes (Fig. 2). In these experiments, K+ ions that would normally be present in physiological solutions were replaced by Cs+ in both the bath and pipette solutions. In addition, external Ca2+ was raised to 5.4 mM to improve the stability of whole cell recordings. Macroscopic currents were evoked by 2.5-s voltage ramps from −80 to +80 mV, and currents at +80 mV were quantified for statistical analysis. We observed that cationic currents evoked by voltage ramps under these conditions had an average reversal potential of −4 mV under the recording conditions used in these experiments (N = 18 cells). These currents were substantially larger in cells exposed for 5 min to 100 μM OAG, a membrane-permeable analog of DAG that can activate TRPC6, TRPC3, and TRPC7 channels (14, 20). Moreover, these cationic currents were markedly reduced after superfusion of 10 μM SKF96365, an inhibitor of nonselective cation channels of the TRPC family (37) (Fig. 2, A and B). They were also blocked by superfusion of 100 μM La3+, which blocks TRPC6 channels (48), but increases activation of TRPC5 channels (22, 48, 56) (Fig. 2, C and D). Using a similar design, we observed that treating podocytes with 100 nM insulin could also evoke a significant increase in cationic currents in podocytes that could be instantly blocked by bath superfusion of either 10 μM SKF96365 (Fig. 3, A and B) or 100 μM La3+ (Fig. 3, C and D).
Fig. 2.
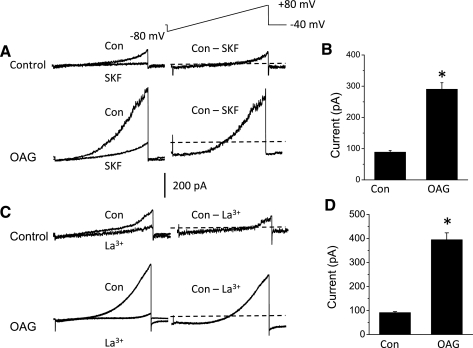
Properties of podocyte cationic currents activated by the diacylglycerol analog oleoyl-2-acetyl-sn-glycerol (OAG). A: examples of currents recorded during 2.5-s ramp voltage commands as indicated above the traces in cells treated with vehicle (top traces) or after 5-min exposure to 100 μM OAG (bottom traces) for 5 min. Superimposed traces to the left are from single cells before and after bath application of 10 μM SKF96365. The traces on the right show SKF96365-sensitive currents obtained by digital subtraction. The dotted lines running through the subtracted traces denote the zero-current level. Note that SKF96365-sensitive currents are much larger after OAG treatment. B: summary of results of several repetitions of the experiment shown in A. Ordinate is the amount of mean SKF96365-sensitive current measured at +80 mV. *Significant (P < 0.0001, Student's unpaired t-test) increase in current after OAG treatment (n = 10 cells in each group). Note that these are group means. The cells exposed to vehicle were not exposed to OAG. C: as in A except that currents in vehicle- or OAG-treated cells were blocked by bath superfusion of 100 μM La3+. D: summary of several repetitions of the experiment shown in C, showing that La3+-sensitive currents are significantly larger in OAG-treated cells (P < 0.001, Student's unpaired t-test, n = 5 cells in each group).
Fig. 3.
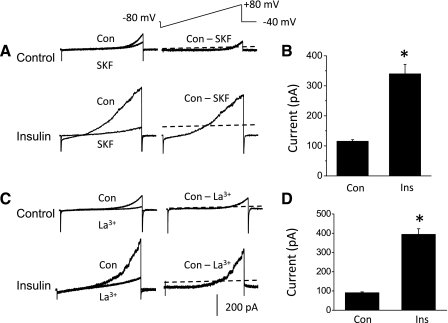
Insulin increases cationic currents in podocytes. A: currents evoked during voltage ramps in control podocytes (top traces) and in podocytes treated with 100 nM insulin for 12 h (bottom traces), as indicated. Superimposed traces to the left are from the same cell before and after bath application of 10 μM SKF96365. The traces on the right show SKF96365-sensitive current calculated by digital subtraction. The dotted lines running through the subtracted traces denote the zero-current level. Note that SKF96365-sensitive currents are much larger in cells treated with insulin. B: summary of results of several repetitions of the experiment shown in A. Ordinate is the mean SKF96365-sensitive current measured at +80 mV. *Significant (P < 0.0001, Student's unpaired t-test) increase in current after insulin treatment (n = 10 cells in each group). C: as in A except that currents in control or insulin-treated cells were blocked by bath superfusion of 100 μM La3+. D: summary of several repetitions of the experiment shown in C, showing that La3+-sensitive currents are significantly larger in insulin-treated cells (P < 0.001, Student's unpaired t-test, n = 5 cells in each group).
More than one functional member of the TRPC family is expressed in podocytes (12, 48). Because of the lack of selective inhibitors, there is no single experimental design that would allow for completely unambiguous isolation of whole cell currents–or, for that matter, any functional responses–that are mediated by endogenously expressed TRPC6 channels. However, we performed two other experiments that suggest that most or all of the insulin-stimulated current is due to modulation of TRPC6 channels. In one set of experiments, we examined the effects of OAG in control cells and in podocytes previously treated with insulin for 24 h, and we analyzed the results using a two-way ANOVA (Fig. 4A). Currents were recorded from 10 cells in each group (for a total of 40 cells in this experiment). There was a significant effect of both insulin (F1 = 199.38, P < 0.0001) and OAG (F2 = 26.35, P < 0.0005) in the overall data set. Importantly, there was also a statistically significant interaction between these two treatments (F1,2 = 37.74, P < 0.0002). This last result indicates that the response to OAG depends on whether cells were previously treated with insulin. Post hoc analysis indicated that both OAG and insulin were effective when applied by themselves, but that applying OAG to cells immediately after 24-h exposure to insulin treatment did not produce any additional effect. In other words, insulin appears to completely occlude the response to OAG, an observation that is consistent with the hypothesis that OAG and insulin are modulating the same population of channels in podocytes.
Fig. 4.

Insulin activates OAG-sensitive currents in podocytes that are eliminated by TRPC6 knockdown. A: insulin treatment occludes electrophysiological responses to OAG in podocytes. Control podocytes, or podocytes treated with 100 nM insulin for 12 h, were exposed to vehicle or 100 μM OAG for 5 min, and whole cell quantification of currents at +80 mV was carried out as in Figs. 2 and 3. Bars represent means ± SE for 10 cells in each group. Data were analyzed by 2-way ANOVA and Tukey's post hoc test. The effect of a combination of insulin and OAG was not significantly different from those evoked by either OAG or insulin alone. B: representative immunoblot analysis (left) and densitometric quantification of 3 repetitions of that experiment showing reduction in total TRPC6 expression in a cell line stably expressing an shRNA targeting TRPC6 compared (39) to a control cell line. C: mean currents at +80 mV recorded cells treated with vehicle, with 5 min of OAG, or 24 h of insulin in wild-type podocytes or in TRPC6 knockdown podocytes, as indicated. Data were analyzed by 2-way ANOVA, which showed that TRPC6 knockdown had a significant (P < 0.005) interaction effect on responses to OAG and insulin. Post hoc analysis indicated that responses to OAG and insulin were eliminated in TRPC6 knockdown cells.
Previous studies showed that the set of channels that can be activated by OAG is comprised of TRPC3, TRPC6, and TRPC7 (20). Therefore, in another set of experiments, we examined the effects of OAG and insulin in wild-type podocytes and in a subclone of the same cell line stably expressing an shRNA targeting TRPC6 channels (39). With immunoblot analysis, we observed a ∼75% reduction in total TRPC6 expression in these cells compared with controls (Fig. 4B). We also observed a nearly complete loss of both OAG-evoked cation currents and insulin-stimulated cation currents in the TRPC6 knockdown cells compared with controls (Fig. 4C), even though the knockdown is less than 100%. Our cell surface biotinylation assays indicate that only a fraction of TRPC6 signal, less than 25%, normally reaches the cell surface, even after insulin treatment, and we suspect that the amount of TRPC6 that remains after siRNA knockdown, and which then moves to the cell surface, is not sufficient to give a signal reliably discernible above the normal variance seen in our electrophysiological experiments. Currents were recorded from 10 cells in each group (for a total of 60 cells in this experiment). Analysis of these data by two-way ANOVA revealed significant effects of the TRPC6 knockdown (F1 = 17.18, P < 0.005), as well as significant effects of insulin or OAG treatment (F2 = 66.21, P < 0.0001). Most importantly, there was a highly statistically significant interaction between these two classes of independent variables (F1,2 = 55.47, P < 0.0001). Post hoc analysis indicated that TRPC6 knockdown abolished the ability of podocytes to increase cationic currents in response to either insulin or OAG. Thus, several lines of biochemical and electrophysiological evidence converge to indicate that insulin causes a rapid and functionally significant modulation of TRPC6 channels that is associated with increases in their steady-state expression on the cell surface. In addition, we also examined whether insulin could affect podocyte TRPC5 channels, since this cation channel is also expressed in podocyte cell lines (48). We observed using cell surface biotinylation assays that in marked contrast to its effects on TRPC6, insulin treatment caused a modest decrease in the steady-state surface expression of endogenous TRPC5 channels in podocyte cell lines (data not shown).
Role of ROS and NADPH oxidases.
ROS are now known to play a role in the regulation of a wide range of reversible regulatory processes and in cell signaling (3), including responses to insulin (32, 35). Recent studies also showed that ROS such as H2O2 can increase the amplitude of currents through TRPC6 channels expressed in heterologous expression systems, at least in part owing to rapid increases in TRPC6 expression on the cell surface (16, 52a). We observed that increasing H2O2 can modulate endogenously expressed TRPC6 channels in podocytes (Fig. 5). Thus, simply applying 250 μM or 1 mM H2O2 to podocytes for 30 min caused a robust increase in the steady-state surface expression of TRPC6 as assessed by cell surface biotinylation assays (Fig. 5, A and B). Moreover, insulin treatment increased production of H2O2 by podocytes, with significant effects seen after 15 min of treatment and larger effects observed at 24 h (Fig. 6A). The insulin-evoked increases in ROS generation were blocked by pretreating podocytes with 100 μM MnTBAP, a membrane-permeable mimetic of superoxide dismutase (2) and catalase (13) (Fig. 6B). Importantly, pretreatment with MnTBAP attenuated the increases in surface expression of TRPC6 evoked by 12 h of treatment with 100 nM insulin (Fig. 6, C and D), although we also observed that MnTBAP by itself evoked some increase in surface TRPC6. These data suggest that generation of ROS is necessary for normal insulin-evoked mobilization of TRPC6 channels.
Fig. 5.
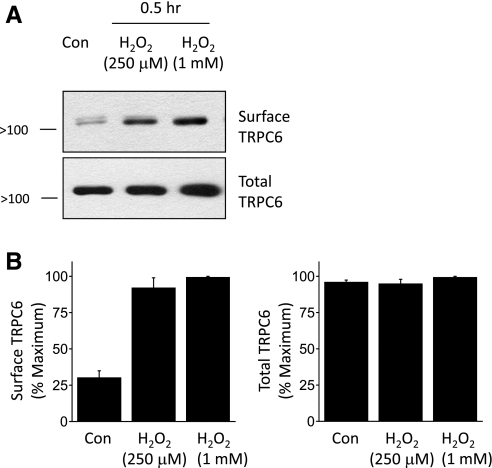
Increase in surface expression of TRPC6 induced by exogenous H2O2. A: representative cell surface biotinylation assay showing increase in steady-state surface expression of TRPC6 after treating podocytes with H2O2 for 30 min at the concentrations shown. B: summary of densitometric analyses of 3 repetitions of the experiment in A.
Fig. 6.

Insulin increases generation of reactive oxygen species (ROS) that are required for normal insulin mobilization of podocyte TRPC6 channels. A: fluorometric assay showing increase in H2O2 generation in podocytes after insulin treatment for the indicated times. Data were analyzed by 1-way ANOVA followed by Tukey's post hoc test with n = 22 wells in each group. Single * denotes mean value significantly greater than control. Double ** indicates that insulin effect at 24 h is significantly greater than effects at 15 min or 6 h or control. B: generation of H2O2 evoked by 24 h of insulin treatment is inhibited by pretreating podocytes with the ROS scavenger manganese (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP; 100 μM). C: representative cell surface biotinylation assay showing reduced insulin mobilization of TRPC6 after pretreatment with MbTBAP. D: summary of densitometric analyses of 3 repetitions of the experiment shown in C.
How does insulin evoke generation of ROS in podocytes? In other cells, insulin has been reported to increase ROS by activation of NAPH oxidases (13). There are seven known members of the NOX family (NOX1–5 and DUOX1–2), which are subjected to distinct regulatory cascades (3). Among these, NOX4 is reported to be most heavily expressed in the kidney (15, 47). We detected two molecular weight variants of NOX4 in podocytes; a 66-kDa protein and a larger 75-kDa protein thought to be a glycosylated form (Fig. 7A) (19, 47). Importantly, we observed that insulin treatment caused a strong increase in the surface expression of the larger molecular weight form of NOX4 (Fig. 7, A and B), which is the only form that we detect on the surface. Using siRNA directed against NOX4, we were able to achieve a substantial knockdown of NOX4 (to ∼25% of control levels) as assessed by immunoblot analysis (Fig. 7, C and D). Insulin-evoked modulation of NADPH oxidase appears to be functionally significant, as we observed that basal and insulin-stimulated TRPC6 mobilization was reduced after NOX4 knockdown (Fig. 8, A and B). Similarly, pretreatment with 10 μM DPI, an inhibitor of ROS-generating flavoenzymes such as NOX4 and NOX2 (47, 54), also reduced insulin-evoked mobilization of TRPC6 (Fig. 8, C and D). Collectively, these data suggest that insulin modulation of NADPH oxidases, including NOX4 and possibly other isoforms, is a component of the pathway underlying insulin regulation of TRPC6.
Fig. 7.
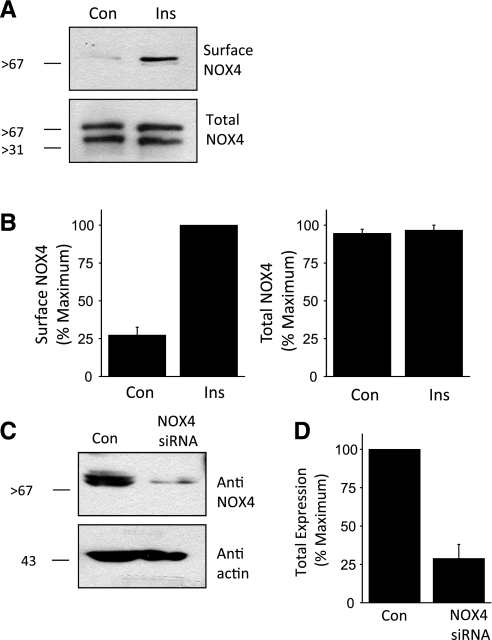
Insulin evokes mobilization of an NADPH oxidase in podocytes. A: cell surface biotinylation assay showing that insulin treatment for 24 h markedly increased surface expression of the higher molecular weight form of NOX4 in podocytes. B: summary of densitometric quantification of 3 repetitions of the experiment shown in A. C: immunoblot analysis showing effective knockdown of total NOX4 expression 24 h after transient transfection with an appropriate siRNA. Control cells were transfected with a nontargeted siRNA. D: summary of 3 repetitions of the experiment shown in C.
Fig. 8.

Normal insulin-evoked mobilization of TRPC6 channels in podocytes requires active NOX4. A: representative cell surface biotinylation assay showing reduced basal and insulin-stimulated surface expression of TRPC6 after siRNA knockdown of NOX4. Control cells were transfected with nontargeted siRNA. B: densitometric analysis of 3 repetitions of the experiment shown in A. C: insulin-evoked mobilization of TRPC6 is blocked by pretreatment with 10 μM diphenylene iodonium (DPI), an inhibitor of ROS-generating flavoenzymes such as NOX4. D: densitometric analysis of 3 repetitions of the experiment shown in C.
DISCUSSION
The most important observation in this study is that insulin evokes a marked and rapid increase in the steady-state surface expression of TRPC6 channels in differentiated cells of a podocyte cell line. This effect is at least partially dependent on increased generation of ROS through insulin mobilization of NADPH oxidases, and it is accompanied by an increase in outwardly rectifying cationic currents that can be activated by OAG and blocked by a pan-TRPC channel inhibitor, by micromolar concentrations of La3+, or by knockdown of TRPC6 channels. Finally, we observed that insulin produced a decrease in the steady-state surface expression of endogenous TRPC5 channels, an observation consistent with previous suggestions of a distinct and partially antagonistic role for TRPC5 and TRPC6 channels in the cellular physiology of podocytes (48).
The effect of insulin on podocyte TRPC6 proteins was observed within minutes after application of insulin to serum-starved cells by cell surface biotinylation assays–in some cases in as little as 1 min. A previous study showed that recombinant TRPC6 channels in heterologous expression systems can be modulated by ROS, especially H2O2, which increases TRPC6 channels on the cell surface (16, 52a) and increases their mean open-time (16). We observed similar effects on endogenous TRPC6 channels after treating podocytes with H2O2. There is growing recognition that ROS such as H2O2 and O2·− can function as physiological intracellular messengers, especially in growth factor signaling cascades. ROS can be generated in part through regulated stimulation of NADPH oxidases (3) and generation of these species is a feature of the responses to insulin in several cell types (13, 32, 35). This also occurs after chronic exposure to high glucose (42). The NADPH oxidases comprise a family of homologous flavin-binding transmembrane proteins (NOX1–5 and DUOX1–2) that catalyze transfer of electrons from NADPH to molecular oxygen. These reactions lead typically to production of O2·− (3), which can then be transformed to other ROS, especially the highly lipid-soluble and more persistent H2O2 (41). NOX4 is unusual among the NOX enzymes in that its primary product appears to be H2O2 rather than O2·− (36). NOX4 is also the most abundantly expressed NADPH oxidase in the kidney (15, 47), and we were able to readily detect NOX4 in podocyte cell lines. In vascular endothelial cells, NOX4 is largely localized in intracellular compartments such as endoplasmic reticulum (19). Therefore, it is interesting that we observed a marked increase in the steady-state expression of NOX4 on the surface of podocytes after insulin treatment. To our knowledge, regulated movement of NOX4 to the surface has not been reported in other systems, although newly synthesized NOX4 can be detected by total internal reflectance fluorescence microscopy at the surface of HEK293 cells following tetracycline-induced induction (58). NOX2 is also expressed in podocytes, but we did not observe a change in its location in response to insulin (data not shown), although it was present on the cell surface and could still contribute to insulin-evoked ROS generation in these cells. In this regard, the effects of insulin on TRPC6 were also reduced by the membrane-permeable ROS scavenger MnTBAP and by NOX4 knockdown. They were also reduced by pretreating cells with DPI, which should inhibit catalytic activity of all known NADPH oxidases (54). In summary, these data strongly suggest that insulin activation leads to generation of ROS close to the cell surface, which in turn contributes to modulation of TRPC6 channels of podocytes.
An important recent study showed that insulin signaling in podocytes is essential for normal kidney function (53). This same group also showed that insulin can stimulate insulin uptake into podocytes owing in part to translocation of glucose transporters to the podocyte cell surface (9, 10). The question arises as to what role insulin modulation of podocyte TRPC6 and glucose uptake may play in the integrative physiology of glomerular filtration? One possibility is that an increase in glomerular filtration rate (GFR) is observed in response to intravenous infusions of glucose (6) or after feeding (8, 21a, 49, 51). These stimuli also trigger insulin secretion. This increase in GFR is due in part to increased glomerular transmural pressure gradients driven by proximal tubule glucose reabsorption by the SLGT2 transporter (25, 51). SGLT2 is a Na+-dependent uptake system, and transport of Na+ and glucose by this system will passively drive H2O uptake by proximal tubules. Moreover, the resulting fall in distal tubule Na+ causes dilation of the afferent arteriole owing to inhibition of tubuloglomerular feedback (25, 41, 50, 51). In an intact nephron, one or both of these processes should increase transmural pressure gradients whether the elevated circulating glucose is transient (as in normal subjects following a glucose load) or sustained (as in the early stages of diabetic nephropathy).
Changes in transmural pressure gradients are thought to require mechanical compensation by glomerular cells to maintain the integrity of the filtration barrier (33, 34). We propose that insulin signaling, by increasing surface expression of TRPC6 channels as reported here, and BKCa channels as we reported previously (29), may prepare podocytes to carry out this compensation by allowing for increased Ca2+ influx. Podocytes contract in response to increased Ca2+ influx (45), and this could contribute necessary stability to the glomerular capillary wall–either by helping to maintain the integrity of the slit diaphragm or by preventing podocytes from detaching from the glomerular basement membrane. One point worth noting is that if this model is correct and mobilization of TRPC6 is normally part of a compensatory response to mechanical or metabolic stress of podocytes, then inhibiting these channels may actually be counterproductive in glomerular diseases.
We should note that a recent study examined regulation of TRPC6 channels by H2O2 in cultured human mesangial cells (17). Interestingly, those workers found that application of H2O2 caused a concentration-dependent decrease in total expression of TRPC6–at least after 2–6 h of continuous exposure. They noted that sustained overexpression of NOX4 also reduced total amounts of TRPC6 that they could detect in mesangial cells, whereas knockdown of NOX4 increased total TRPC6 (17). Assuming that measurements of total channel expression reflect expression at the mesangial cell surface, these changes are in the opposite direction of what we observed here for podocytes and what the same group reported earlier for TRPC6 channels in heterologous expression systems (16). One difference is that the mesangial cells were subjected to substantially longer exposures to H2O2 (17). It is possible that ROS has multiple effects that are a function of the concentration and duration of exposure to these species, such that brief exposure causes initial stimulation of TRPC6 channels, but eventually causes a fall in their total expression. However, it is also quite possible that the effects of ROS on TRPC6 are cell-type specific and that a single diffusible mediator could have opposite effects on TRPC6 channels of mesangial cells and podocytes at all times. For example, it is possible that relaxation of mesangial cells (as might be expected from a ROS-induced fall in TRPC6) could increase compliance and allow for some dilation of the capillary tuft, thereby releasing some pressure, while simultaneous ROS-induced activation of TRPC6 in podocytes, as seen here, and after treatment with puromycin aminonucleoside (52a) could promote stability of the slit diaphragm or adhesion of podocytes to the glomerular basement membrane.
In summary, we observed that insulin evokes an increase in the steady-state surface expression of TRPC6 channels in podocytes that depends on NOX-dependent generation of ROS. This response may be part of a mechanism to maintain the stability of the glomerular filtration in the face of stimuli, such as feeding, that are normally associated with changes in renal hemodynamics.
GRANTS
The authors gratefully acknowledge support from National Institutes of Health Grant RO1 DK-82529 and an M. James Scherbenske Grant from the American Society of Nephrology to S. E. Dryer.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: E.Y.K. and M.A. performed experiments; E.Y.K. and M.A. analyzed data; E.Y.K. and S.E.D. prepared figures; S.E.D. conception and design of research; S.E.D. interpreted results of experiments; S.E.D. drafted manuscript; S.E.D. edited and revised manuscript; S.E.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We are grateful to Drs. Peter Mundel and Jochen Reiser for supplying podocyte cell lines and to Dr. Anna Greka for essential advice on TRPC5 assays.
REFERENCES
- 1. Bank N, Aynedjian HS. Progressive increases in luminal glucose stimulate proximal sodium absorption in normal and diabetic rats. J Clin Invest 86: 309–316, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Batinicć-Haberle I. Manganese porphyrins and related compounds as mimics of superoxide dismutase. Methods Enzymol 349: 223–233, 2002 [DOI] [PubMed] [Google Scholar]
- 3. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007 [DOI] [PubMed] [Google Scholar]
- 4. Brenner BM, Troy JL, Daugharty TM. The dynamics of glomerular ultrafiltration in the rat. J Clin Invest 50: 1776–1780, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brosius FC, 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T. Mouse models of diabetic nephropathy. J Am Soc Nephrol 20: 2503–2512, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Christiansen JS, Frandsen M, Parving HH. Effect of intravenous glucose infusion on renal function in normal man and in insulin-dependent diabetics. Diabetologia 21: 368–373, 1981 [DOI] [PubMed] [Google Scholar]
- 7. Christiansen JS, Gammelgaard J, Frandsen M, Parving HH. Increased kidney size, glomerular filtration rate and renal plasma flow in short-term insulin-dependent diabetics. Diabetologia 20: 451–456, 1981 [DOI] [PubMed] [Google Scholar]
- 8. Corman B, Chami-Khazraji S, Schaeverbeke J, Michel JB. Effect of feeding on glomerular filtration rate and proteinuria in conscious aging rats. Am J Physiol Renal Fluid Electrolyte Physiol 255: F250–F256, 1988 [DOI] [PubMed] [Google Scholar]
- 9. Coward RJ, Welsh GI, Koziell A, Hussain S, Lennon R, Ni L, Tavaré JM, Mathieson PW, Saleem MA. Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes 56: 1127–1135, 2007 [DOI] [PubMed] [Google Scholar]
- 10. Coward RJ, Welsh GI, Yang J, Tasman C, Lennon R, Koziell A, Satchell S, Holman GD, Kerjaschki D, Tavaré JM, Mathieson PW, Saleem MA. The human glomerular podocyte is a novel target for insulin action. Diabetes 54: 3095–3102, 2005 [DOI] [PubMed] [Google Scholar]
- 11. Day BJ, Fridovich I, Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide-mediated injury. Arch Biochem Biophys 347: 256–262, 1997 [DOI] [PubMed] [Google Scholar]
- 12. Dryer SE, Reiser J. TRPC6 channels and their binding partners in podocytes: role in glomerular filtration and pathophysiology. Am J Physiol Renal Physiol 299: F689–F701, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Espinosa A, García A, Härtel S, Hidalgo C, Jaimovich E. NADPH oxidase and hydrogen peroxide mediate insulin-induced calcium increase in skeletal muscle cells. J Biol Chem 284: 2568–2575, 2009 [DOI] [PubMed] [Google Scholar]
- 14. Estacion M, Sinkins WG, Jones SW, Applegate MA, Schilling WP. Human TRPC6 expressed in HEK 293 cells forms nonselective cation channels with limited Ca2+ permeability. J Physiol 572: 359–377, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Geiszt M, Kopp JB, Várnai P, Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci USA 97: 8010–8014, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Graham S, Ding M, Ding Y, Sours-Brothers S, Luchowski R, Gryczynski Z, Yorio T, Ma H, Ma R. Canonical transient receptor potential 6 (TRPC6), a redox-regulated cation channel. J Biol Chem 285: 23466–23476, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Graham S, Gorin YC, Abboud HE, Ding M, Lee DY, Shi H, Ding Y, Ma R. Abundance of TRPC6 protein in glomerular mesangial cells is decreased by ROS and PKC in diabetes. Am J Physiol Cell Physiol 301: C304–C315, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heeringa SF, Möller CC, Du J, Yue L, Hinkes B, Chernin G, Vlangos CN, Hoyer PF, Reiser J, Hildebrandt F. A novel TRPC6 mutation that causes childhood FSGS. PLos One 4: e7771, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 24: 677–683, 2004 [DOI] [PubMed] [Google Scholar]
- 20. Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259–263, 1999 [DOI] [PubMed] [Google Scholar]
- 21. Huber TB, Schermer B, Müller RU, Höhne M, Bartram M, Calixto A, Hagmann H, Reinhardt C, Koos F, Kunzelmann K, Shirokova E, Krautwurst D, Harteneck C, Simons M, Pavenstädt H, Kerjaschki D, Thiele C, Walz G, Chalfie M, Benzing T. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc Natl Acad Sci USA 103: 17079–17086, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21a. Hummel CS, Lu C, Loo DD, Hirayama BA, Voss AA, Wright EM. Glucose transport by human renal Na+/D-glucose cotransporters SGLT1 and SGLT2. Am J Physiol Cell Physiol 300: C14–C21, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jung S, Muhle A, Schaefer M, Strotmann R, Schultz G, Plant TD. Lanthanides potentiate TRPC5 currents by an action at extracellular sites close to the pore mouth. J Biol Chem 278: 3562–3571, 2003 [DOI] [PubMed] [Google Scholar]
- 23. Jung S, Strotmann R, Schultz G, Plant TD. TRPC6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am J Physiol Cell Physiol 282: C347–C359, 2002 [DOI] [PubMed] [Google Scholar]
- 25. Karlsen FM, Holstein-Rathlou NH, Leyssac PP. A re-evaluation of the determinants of glomerular filtration rate. Acta Physiol Scand 155: 335–350, 1995 [DOI] [PubMed] [Google Scholar]
- 26. Kim EY, Alvarez-Baron CP, Dryer SE. Canonical transient receptor potential channel (TRPC)3 and TRPC6 associate with large-conductance Ca2+-activated K+ (BKCa) channels: role in BKCa trafficking to the surface of cultured podocytes. Mol Pharmacol 75: 466–477, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim EY, Chiu YH, Dryer SE. Neph1 regulates steady-state surface expression of Slo1 Ca2+-activated K+ channels: different effects in embryonic neurons and podocytes. Am J Physiol Cell Physiol 297: C1379–C1388, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim EY, Choi KJ, Dryer SE. Nephrin binds to the COOH terminus of a large-conductance Ca2+-activated K+ channel isoform and regulates its expression on the cell surface. Am J Physiol Renal Physiol 295: F235–F246, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim EY, Dryer SE. Effects of insulin and high glucose on mobilization of Slo1 BKCa channels in podocytes. J Cell Physiol 226: 2307–2315, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim EY, Suh JM, Chiu YH, Dryer SE. Regulation of podocyte BKCa channels by synaptopodin, Rho, and actin microfilaments. Am J Physiol Renal Physiol 299: F594–F604, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krall P, Canales CP, Kairath P, Carmona-Mora P, Molina J, Carpio JD, Ruiz P, Mezzano SA, Li J, Wei C, Reiser J, Young JI, Walz K. Podocyte-specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLos One 5: e12859, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krieger-Brauer HI, Kather H. Human fat cells possess a plasma membrane-bound H2O2-generating system that is activated by insulin via a mechanism bypassing the receptor kinase. J Clin Invest 89: 1006–1013, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kriz W, Hackenthal E, Nobiling R, Sakai T, Elger M, Hähnel B. A role for podocytes to counteract capillary wall distension. Kidney Int 45: 369–376, 1994 [DOI] [PubMed] [Google Scholar]
- 34. Kriz W, Kretzler M, Provoost AP, Shirato I. Stability and leakiness: opposing challenges to the glomerulus. Kidney Int 49: 1570–1574, 1996 [DOI] [PubMed] [Google Scholar]
- 35. Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JT, Goldstein BJ. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J Biol Chem 276: 48662–48669, 2001 [DOI] [PubMed] [Google Scholar]
- 36. Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 18: 69–82, 2006 [DOI] [PubMed] [Google Scholar]
- 37. Merritt JE, Armstrong WP, Benham CD, Hallam TJ, Jacob R, Jaxa-Chamiec A, Leigh BK, McCarthy SA, Moores KE, Rink TJ. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem J 271: 515–522, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Möller CC, Wei C, Altintas MM, Li J, Greka A, Ohse T, Pippin JW, Rastaldi MP, Wawersik S, Schiavi S, Henger A, Kretzler M, Shankland SJ, Reiser J. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol 18: 29–36, 2007 [DOI] [PubMed] [Google Scholar]
- 39. Nijenhuis T, Sloan AJ, Hoenderop JGJ, Flesche J, van Goor H, Kistler AD, Bakker M, Nindels RJM, de Boer RA, Moller C, Hamming I, Navis G, Wetzels JFM, Berden JHM, Reiser J, Faul C, van der Vlag J. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pavenstädt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev 83: 253–307, 2003 [DOI] [PubMed] [Google Scholar]
- 41. Persson P, Hansell P, Palm F. Tubular reabsorption and diabetes-induced glomerular hyperfiltration. Acta Physiol (Oxf) 200: 3–10, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Piwkowska A, Rogacka D, Audzeyenka I, Jankowski M, Angielski S. High glucose concentration affects the oxidant-antioxidant balance in cultured mouse podocytes. J Cell Biochem 112: 1661–1672, 2011 [DOI] [PubMed] [Google Scholar]
- 43. Pryor WA. Oxy-radicals and related species: their formation, lifetimes, and reactions. Annu Rev Physiol 48: 657–667, 1986 [DOI] [PubMed] [Google Scholar]
- 44. Reiser J, Polu KR, Möller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 37: 739–744, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saleem MA, Zavadil J, Bailly M, McGee K, Witherden IR, Pavenstadt H, Hsu H, Sanday J, Satchell SC, Lennon R, Ni L, Bottinger EP, Mundel P, Mathieson PW. The molecular and functional phenotype of glomerular podocytes reveals key features of contractile smooth muscle cells. Am J Physiol Renal Physiol 295: F959–F970, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schlöndorff J, Del Camino D, Carrasquillo R, Lacey V, Pollak MR. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am J Physiol Cell Physiol 296: C558–C569, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, Hattori M, Sakaki Y, Sumimoto H. A novel superoxide-producing NAD(P)H oxidase in kidney. J Biol Chem 276: 1417–1423, 2001 [DOI] [PubMed] [Google Scholar]
- 48. Tian D, Jacobo SM, Billing D, Rozkalne A, Gage SD, Anagnostou T, Pavenstädt H, Hsu HH, Schlondorff J, Ramos A, Greka A. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci Signal 3: ra77, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Uemasu J, Hori T, Uemasu Y, Kawasaki H. Effects of a rice meal on renal hemodynamics and excretory functions in normal subjects. Nephron 57: 187–191, 1991 [DOI] [PubMed] [Google Scholar]
- 50. Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 300: R1009–R1022, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, Koepsell H, Rieg T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22: 104–112, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang Y, Jarad G, Tripathi P, Pan M, Cunningham J, Martin DR, Liapis H, Miner JH, Chen F. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol 21: 1657–1666, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52a. Wang Z, Wei X, Zhang Y, Ma X, Li B, Zhang S, Du P, Zhang X, Yi F. NADPH oxidase-derived ROS contributes to upregulation of TRPC6 expression in puromycin aminonucleoside-induced podocyte injury. Cell Physiol Biochem 24: 619–626, 2009 [DOI] [PubMed] [Google Scholar]
- 53. Welsh GI, Hale LJ, Eremina V, Jeansson M, Maezawa Y, Lennon R, Pons DA, Owen RJ, Satchell SC, Miles MJ, Caunt CJ, McArdle CA, Pavenstädt H, Tavaré JM, Herzenberg AM, Kahn CR, Mathieson PW, Quaggin SE, Saleem MA, Coward RJ. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab 12: 329–340, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wind S, Beuerlein K, Eucker T, Müller H, Scheurer P, Armitage ME, Ho H, Schmidt HH, Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol 161: 885–898, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308: 1801–1804, 2005 [DOI] [PubMed] [Google Scholar]
- 56. Wong CO, Huang Y, Yao X. Genistein potentiates activity of the cation channel TRPC5 independently of tyrosine kinases. Br J Pharmacol 159: 1486–1496, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang H, Ding J, Fan Q, Liu S. TRPC6 upregulation in Ang II-induced podocyte apoptosis might result from ERK activation and NF-kappaB translocation. Exp Biol Med (Maywood) 234: 1029–1036, 2009 [DOI] [PubMed] [Google Scholar]
- 58. Zhang L, Nguyen MV, Lardy B, Jesaitis AJ, Grichine A, Rousset F, Talbot M, Paclet MH, Qian G, Morel F. New insight into the Nox4 subcellular localization in HEK293 cells: first monoclonal antibodies against Nox4. Biochimie 93: 457–468, 2011 [DOI] [PubMed] [Google Scholar]
- 60. Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem 253: 162–168, 1997 [DOI] [PubMed] [Google Scholar]