

Abstract
Homozygous ataxic mice (axJ) express reduced levels of the deubiquitinating enzyme Usp14. They develop severe tremors by 2–3 wk of age, followed by hindlimb paralysis, and death by 6–8 wk. While changes in the ubiquitin proteasome system often result in the accumulation of ubiquitin protein aggregates and neuronal loss, these pathological markers are not observed in the axJ mice. Instead, defects in neurotransmission were observed in both the central and peripheral nervous systems of axJ mice. We have now identified several new alterations in peripheral neurotransmission in the axJ mice. Using the two-microelectrode voltage clamp technique on diaphragm muscles of axJ mice, we observed that under normal neurotransmitter release conditions axJ mice lacked paired-pulse facilitation and exhibited a frequency-dependent increase in rundown of the end plate current at high-frequency stimulation (HFS). Combined electrophysiology and styryl dye staining revealed a significant reduction in quantal content during the initial and plateau portions of the HFS train. In addition, uptake of styryl dyes (FM dye) during HFS demonstrated that the size of the readily releasable vesicle pool was significantly reduced. Destaining rates for styryl dyes suggested that axJ neuromuscular junctions are unable to mobilize a sufficient number of vesicles during times of intense activity. These results imply that axJ nerve terminals are unable to recruit a sufficient number of vesicles to keep pace with physiological rates of transmitter release. Therefore, ubiquitination of synaptic proteins appears to play an important role in the normal operation of the neurotransmitter release machinery and in regulating the size of pools of synaptic vesicles.
Keywords: neuromuscular transmission, vesicle cycling, ubiquitin-proteasome system, ataxic mice
the ubiquitin-proteasome system (UPS) is best known as a pathway for the clearance of damaged and misfolded cellular proteins. Several studies (5, 13, 14, 21, 30, 31, 43) have also demonstrated that ubiquitin-mediated protein degradation plays a critical role in controlling protein abundance in all spatially restricted domains within the cell. Proteins are identified for degradation by the covalent attachment of the 76 amino acid protein ubiquitin. This modification of protein substrates with ubiquitin can occur through attachment of a mono- or polyubiquitin chain. While the covalent attachment of a polyubiquitin chain is critical for proteasomal degradation, a monoubiquitin tag can act as a signal for receptor endocytosis and trafficking of vesicles. In addition, different types of polyubiquitin chains can be formed and are thought to signal a variety of different cellular processes. These varied ubiquitin signals have been shown to be important in the regulation of many cellular processes including cell cycle control, DNA repair, transcription, cell signaling, and regulation of protein trafficking (1, 66).
In the nervous system, ubiquitination plays important roles in synapse development and function and in the genesis of different neurodegenerative diseases. The accumulation of ubiquitin conjugates and/or proteasome-associated inclusion bodies has been reported in a broad array of chronic neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, Huntington's disease, and spinocerebellar ataxias (7, 19, 33, 42). Most importantly, the observation that mutations in the components of the UPS (i.e., Parkin and UBE3A) result in neurological dysfunction and neurodegeneration clearly demonstrates the importance of this pathway in nervous system development and function.
We previously identified the mutated gene in axJ mutant mice as the deubiquitinating enzyme Usp14 (68). These mice exhibit severe tremors by 2–3 wk of age, followed by increased muscle wasting from 3 wk onwards resulting in hindlimb paralysis and death by 6–8 wk of age. We also demonstrated that axJ mice exhibit defects in synaptic transmission in the central (CNS) and peripheral nervous systems (PNS; Ref. 68). In the axJ CNS, short-term synaptic plasticity was altered at the Schaeffer collateral synapse in the hippocampus. In the axJ PNS, a significant reduction in the frequency of spontaneous release and quantal content was observed. Subsequent studies (16) have demonstrated that loss of Usp14 destabilizes ubiquitin pools and causes a developmental block in the structural and functional aspects of the neuromuscular junction. It is not clear if the observed block in neuromuscular junction (NMJ) development in axJ mice is caused by reduced protein degradation secondary to ubiquitin deficiency or if other ubiquitin signaling pathways, such as vesicular transport and receptor-mediated endocytosis, contribute critically to motor end plate function in the axJ mice. These phenotypes could be rescued by expressing Usp14 exclusively in the nervous system, indicating that the neuromuscular defects were caused by nervous system dysfunction (18) and also indicating that synaptic ubiquitin pools are particularly vulnerable to fluctuations in ubiquitin stability due to their remote location away from the site of ubiquitin synthesis (16). In adult mice, lack of Usp14 causes reduced size of the free ubiquitin pool and abnormal neuronal activity, indicating that USP14 is required for maintaining ubiquitin levels in the adult nervous system (4, 39, 64, 68). A consistent dysregulation of synaptic transmission in response to UPS malfunction is reported to contribute to several neurodegenerative diseases (20, 60). Recently, restoration of neuronal ubiquitin by transgenic complementation prevented the neuromuscular disease resulting from the lack of Usp14 and restored synaptic transmission at the NMJ, showing that ubiquitin homeostasis is an important component of synaptic function (17). These data indicate a critical role for ubiquitin homeostasis in synaptic development and function, which may contribute to diseases characterized by synaptic dysfunction.
Ubiquitination of inhibitory GABAA receptors (GABAARs) has recently been shown to be activity dependent and to regulate synaptic GABAAR accumulation (58) and suggested that Usp14 directly participates in the regulation of synaptic GABAAR turnover (39) in CNS. These results suggest the UPS may be necessary for normal neurotransmission to occur in the adult CNS and PNS.
Thus it is important to further explore the mechanisms underlying the abnormal neurotransmission and extensive muscle wasting observed in axJ mice; we now report multiple functional defects at the neuromuscular junction resulting from loss of Usp14 expression. In this study, we found that repetitive stimulations resulted in profound synaptic depression. Combined electrophysiological and FM dye labeling studies suggested that there was an inability to mobilize a sufficient number of vesicles at high but physiological rates of transmitter release. These studies further define the role of ubiquitin signaling in the regulation of neurotransmitter release and synaptic transmission.
MATERIALS AND METHODS
Animals.
Homozygous ataxic mice (axJ) display reduced levels of the deubiquitinating enzyme ubiquitin-specific protease Usp14 as a result of the insertion of an intracisternal-A particle into intron 5 of Usp14 (68). Wild-type littermates and axJ mutant mice of 5–6 wk of age were used throughout this study. All handling of animals and subsequent experiments followed the animal care and use protocols established and approved by the Institutional Animal Care and Use Committee (Northwestern University, Chicago, IL).
Electrophysiology.
All experiments were carried out in vitro at room temperature on phrenic nerve hemidiaphragm muscle preparations of 5- to 6-wk-old axJ mice and their wild-type littermate controls. The diaphragm muscles were chosen because these muscles are functional in axJ until death (>8 wk). Mice were deeply anesthetized with isoflurane before dissection of the diaphragm muscle. The muscle was perfused with Tyrode's solution (137 mM NaCl, 2.8 mM KCl, 1.8 mM CaCl2, 1.1 mM MgCl2, 11.9 mM NaHCO3, 0.33 mM NaH2PO4, and 11.2 mM dextrose pH 7.4 when bubbled with a mixture of 95% O2-5% CO2) at 30°C. Intracellular potentials and currents were measured using glass microelectrodes filled with 3M KCl of 10–15 mΩ resistance and with an Axoclamp-2A amplifier system. With the use of a cut diaphragm preparation and two-microelectrode voltage-clamp system (11, 22, 68), synaptic currents, including miniature end plate currents (MEPCs) and end plate currents (EPCs), were obtained at −70 mV holding potential. The currents were digitized at 50 μs per point and stored, captured, and analyzed using pClamp9 software (Axon Instruments).
EPCs were elicited by stimulating the corresponding phrenic nerve with rectangular pulses of a 0.05-ms duration. During single shock stimulation, the nerve was stimulated at 0.2 Hz, and during tetanic stimulation, it was stimulated at 20, 50, and 100 Hz for 0.5 to 2 s. The amplitude decay and time-to-peak of the EPCs and MEPCs were captured and subsequently analyzed with pClamp9.0 and MiniAnalyses program (Synaptosoft). The “rundown” of EPC trains was defined as the fractional decrease in EPC amplitude from the peak response to the plateau (mean of last 10 responses). The quantal content in a train was determined by dividing the first EPC amplitude or plateau EPC amplitude of an high-frequency stimulation (HFS; 50 Hz) train by the MEPC amplitude recorded just prior to the train (11, 29, 68). Facilitation of transmitter release [paired-pulse facilitation (PPF)] was measured by paired responses of EPCs elicited by supramaximal twin-pulse nerve stimuli at different time intervals of 6, 8, 10, 20, and 50 ms ( 63, 68 ). PPF index (P2/P1) was obtained by comparing second (P2) and first EPCs (P1) averaged over 10 trials.
Nerve action potential studies.
Conduction properties of myelinated axons were examined in acutely isolated sciatic nerves from axJ mice and their littermate controls. The proximal end of the nerve was stimulated, and the distal end was used to record the compound action potential (CAP). Microelectrodes were back filled with 2–3 M KCl. The preparation was super fused with Tyrode's solution, and voltage outputs were monitored using an oscilloscope. Response to low-frequency stimulation was assessed at 0.2 Hz, while response to high-frequency train stimulation was assessed at 30 Hz. Automated analyses of peak and latency were performed using pClamp9 software.
Immunostaining studies.
For the immunohistochemical studies of diaphragm acetylcholine receptors, mice were anesthetized with ketamine and xylazine. Mice were then perfused with PBS, followed with 2% paraformaldehyde. Hemidiaphragms were dissected out and further fixed with 2% paraformaldehyd and then washed with wash buffer (PBS containing 1% Triton X-100) three times at room temperature for 15 min. Hemidiaphragms were then blocked with 2% (wt/vol) BSA and 4% goat serum in wash buffer for 1 h. To label the acetylcholine receptors (AChRs), samples were incubated with 1 μg/ml α-bungarotoxin-conjugated with tetramethyl rhodamine for 1 h. After being washed three times at room temperature for 15 min with wash buffer, samples from Thy1-Yfp mice were mounted in PBS containing 50% glycerol and imaged by confocal microscopy. Images were digitally acquired using an upright Olympus BX51W (Tokyo, Japan) microscope equipped with a ×40 objective digital camera. Images were analyzed with the Metamorph imaging system (Universal Imaging, West Chester, PA).
Immunostaining for sodium and potassium channels and the contactin-associated protein 2 (Caspr2) and contactin proteins in the paranodal and juxtaparanodal regions was carried out with teased sciatic nerve preparations (10). Sciatic nerves were removed from axJ mice and their wild-type littermate controls and fixed in PBS with 1–4% paraformaldehyde for 1.5 h. The nerves were then stored in PBS until teased. The sciatic nerves were placed onto a coverslip, and individual fibers were teased apart with a fine needle. The preparation was then dried overnight and treated with acetone at −20°C for 10 min. The coverslips were washed several times with PBS before being treated with antibodies against sodium channels, potassium channels, contactin, and Caspr2. Preparations were washed for 1 h with 0.1% Triton X-100 and then treated overnight with the following antibodies affinity-purified rabbit anti-mCNTN-Ig1-6 (1:500–1:1,000; Ref. 8); rabbit polyclonal antibodies to Caspr2 (1:500; Ref. 46); mouse monoclonal anti Kv1.2 (1:100); and rabbit anti-sodium channel (1:75; Refs. 6, 50); all were purchased from Chemicon. Following treatment with the primary antibodies, the tissue was incubated with the fluorochrome-conjugated secondary antibody Alexa-488 (Molecular Probes). Nerves were then washed in PBS and mounted in Slow Fade light (Molecular Probes).
Optical imaging with FM1-43 and FM2-10 for vesicular loading and unloading.
Synaptic vesicle dynamics in axJ mice and littermate controls were studied using the phrenic nerve diaphragm preparations described above. The nerve was taken up into a suction electrode, and synaptic vesicles were labeled by nerve stimulation in the presence of fluorescent styryl dye FM1-43 (6–8 μM) and FM2-10 (30–50 μM) solutions (Molecular Probes). FM1-43 reversibly binds to presynaptic membrane and becomes trapped within recycled synaptic vesicles during endocytosis- FM1-43 loading.
The protocol used was based on the differential staining properties of FM1-43 and FM2-10 to potentially identify two vesicle recycling pathways that selectively refill two functionally distinct vesicle pools (53, 54). When stimulation and imaging took place, the simultaneously immobilized diaphragm (FM dye experiments) was used after μ-conotoxin GIII B (2 μM) treatment, which blocks Na+ channels in mouse muscle but does not affect the quantal release of acetylcholine (45). A modified protocol was followed using the dye FM1-43 (54) to label both the “readily releasable pool” (RRP) and the “reserve pool” (RP),which together constitute the “total pool.” Muscles were exposed to FM1-43 shortly before and during a 1-min, 30-Hz stimulation. Synaptic vesicles were allowed to take up the dye for 15 min before being washed by high Mg2+ solution, resulting in bright fluorescence at the nerve terminal. A “quick wash” loading protocol (54) was used to label the RRP alone with FM2-10, which was applied just before tetanic stimulation (30 Hz) for 1 min and immediately washed off (9, 54).
In both experiments, the washing (35–45 min) was carried out with a low Ca 2+(0.05 mM) and high Mg 2+ (12 mM) Tyrode's solution. The images were captured (100 ms exposition) using a ×40 water immersion objective on an Olympus BX51W microscope equipped with an intensified Penta-MAX CCD camera (Princeton Instruments, Trenton, NJ) and controlled by MetaMorph software (Universal Imaging, Downingtown, PA; Version 5.0). The following filter sets were used for FM1-43 and FM2-10: (500 ± 10 nm bandpass excitation filter and 515 nm long-pass beam splitter). In both cases, vesicular exocytosis experiments were conducted by washing the preparation for 5 min in normal saline (1.8 mM Ca 2+ and 1 mM Mg 2+) followed by trains of stimuli at 30 Hz, resulting in a loss of fluorescence. To image vesicle release, first we collected several control images. Next, we stimulated phrenic nerve for synaptic vesicle exocytosis while imaging. As vesicles containing dye exocytose, dye releases into extracellular space and quickly washes away. Any loss in fluorescence measured during stimulation is indicative of the rate and amount of synaptic vesicle exocytosis. For quantification of the fluorescence intensity of the staining, presynaptic terminals that are superficially located were selected from each hemidiaphragm preparations. The fluorescence of terminal was calculated in arbitrary units as average brightness of all pixels of the part (selected spots) of terminal after subtraction of background fluorescence (71). The background is attributed to nonterminal FM dye, auto fluorescence, or image detector settings (26). After this value was subtracted from the terminal fluorescence for each time point, fluorescence images were aligned and the outline of each selected terminal's area of interest was marked. Afterwards, the average fluorescence intensity of all pixels inside the outline was calculated. The same outline was measured after destaining, and the resulting data were converted into a percentage of dye loss. Time-lapse sequences for destaining were captured using the Metamorph program. Graphs and exponential curve best-fits were calculated using SigmaPlot 8.0 software (Jandel Scientific).
Statistical analyses.
All data were analyzed by ANOVA or independent Student's t-test using either Excel or Sigma plot programs. All experimental data are presented as means ± SE unless otherwise specified.
RESULTS
Properties of synaptic transmission at axJ terminals and the morphology of axJ NMJs.
To determine whether the absence Usp14 affects overall synaptic morphology, we examined wild-type and axJ diaphragms. As visualized by fluorescence using a Thy1-YFP transgenic mouse (green) >60–70% of the nerve terminals in the diaphragms of the axJ mice were branched and showed no signs of NMJ pathology, as it was previously reported (16) (Fig. 1A). The postsynaptic acetylcholine receptors from wild-type (n = 13 animals) and axJ mice (n = 8 animals) containing the Thy1-YFP transgene were identified by staining with α-bungarotoxin-conjugated with tetramethyl rhodamine (Fig. 1A, red), and these acetylcholine receptor-labeled clusters in the diaphragms of axJ mice (Fig. 1A, right) and ranged from 97.5 ± 5.6 μm2 in wild type (n = 221) to 110.44 ± 12.48 μm2 in axJ (n = 173).
Fig. 1.
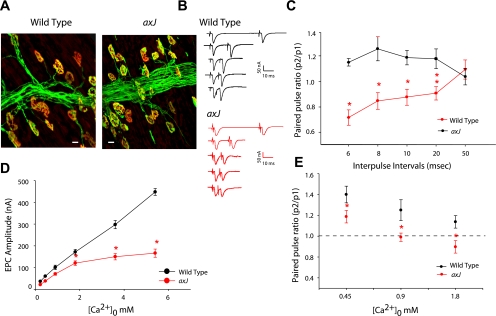
Morphology and paired-pulse stimulation in wild-type and axJ neuromuscular junction. A: Motor end plates from wild-type and axJ diaphragms. Diaphragms were dissected from wild-type and axJ mice containing the Thy1-Yfp transgene and were fixed and stained with α-bungarotoxin-conjugated with tetramethyl rhodamine (red) to label the postsynaptic acetylcholine receptors. Presynaptic axons and nerve terminals were visualized by fluorescence from the Yfp transgene (green). Scale = 20 μm. B and C: end plate currents (EPCs) recording under voltage clamp conditions (−70 mV) illustrating that wild-type neuromuscular junctions (NMJs) exhibit paired-pulse facilitation at physiological levels of Ca2+ and Mg2+ and with stimulus intervals of 6, 8, 10, 20, and 50 ms (bottom to top), whereas axJ NMJs do not . Paired-pulse ratios (P2/P1) indicate a significant (*P < 0.001; **P < 0.02) decrease in PPF in axJ mice compared with the wild type (C). D: Ca2+ dependence of EPC amplitude. Relationship between extracellular Ca2+ levels and the amplitude of evoked EPCs is shown for a wild type and an axJ NMJ voltage clamped at −70 mV. EPC amplitude continually increased as Ca2+ levels rose; in contrast at axJ NMJs the EPC amplitude rose rapidly to reach a near maximal value at the in vivo level of ∼2 mM and increasing Ca2+ beyond this concentration did not result in appreciably more increase in EPC amplitude (*P < 0.001). E: ratios of paired-pulse facilitation from wild-type and axJ NMJ at various Ca 2+ concentrations. At in vivo Ca2+ levels, lack of facilitation is significant (*P < 0.001), compared with the wild type NMJ.
To study synaptic transmission at in vivo levels of transmitter release, recordings from NMJs in normal Ca2+ and Mg2+ were taken using a cut diaphragm preparation to reduce transmission to a level below threshold for muscle action potential generation, thereby preventing contraction. In our previous study (68), it was reported that axJ mice have defects in CNS synaptic transmission causing changes in short-term synaptic plasticity, represented by a lack of PPF in the hippocampus. As in the CNS, axJ NMJ synapses failed to show PPF using short interpulse intervals (6 to 20 ms; n = 9 animals and 20 end plates) compared with wild-type synapses (n = 6 animals and 20 end plates), which instead continued to show PPF at normal levels of transmitter release. Example traces at 6- to 50-ms intervals are shown in Fig. 1B (top, wild type, and bottom, axJ), and pooled PPF ratio data showed a significant difference between wild-type and axJ NMJs (Fig. 1C). A decrease in PPF is observed if the intracellular Ca2+ level is elevated or if the release machinery is more sensitive to Ca2+. This results in a high probability of release in response to the first stimulus (59). It is also possible that the size of the vesicle pool is decreased causing the entire RRP of vesicles to be released by a single stimulus, leaving no additional vesicles or docking sites for enhanced release by the second stimulus (72).
To examine these possibilities, the Ca2+ dependence of the release process in the axJ terminals was examined by determining mean EPC amplitudes at different extracellular Ca2+ concentrations. For wild-type NMJs (n = 5 animals and 14 end plates), it was found that the EPC amplitude continually increased as the Ca2+ concentration rose (Fig. 1D), as previously described by Dodge and Rahamimoff (23). In contrast, at axJ NMJs (n = 4 animals and 12 end plates) the EPC amplitude increased rapidly to reach a near maximal value at 2 mM Ca2+, a level consistent with in vivo signaling, yet increasing Ca2+ beyond this concentration did not result in further increases in EPC amplitude (Fig. 1D). Wild-type junctions (n = 6 animals 12 end plates) exhibited PPF (20-ms intervals) at Ca2+ levels ranging from 0.45–2 mM with lesser facilitation as Ca2+ concentrations rose further (Fig. 1E). However, axJ junctions (n = 14 animals 26 end plates) only exhibited PPF at low Ca2+ levels and were shifted down at different Ca2+concentrations (Fig. 1E). PPF is generally explained as an increase of release probability (Pr) during the second stimulus, arising from prior accumulation of residual Ca2+ near active zones or a lingering effect of Ca2+ on a Ca2+ sensor (44, 72), whereas a decrease in PPF may be caused by decreases in both release probability and quantal size (15). A Ca2+ dependence of the paired-pulse ratio and near-saturating EPCs amplitude at 2 mM Ca2+ suggests that the Pr might be close to maximum levels at axJ junctions in vivo Ca2+ levels (Fig. 1E), and a decreased transmitter output could be associated with observed decrease in PPF in axJ NMJ.
The axJ terminals are incapable of sustaining normal rates of neurotransmitter release during HFS.
Following high stimulation rates (20, 50, and 100 Hz), NMJs from wild-type mice elicited trains of EPCs that increased in amplitude during the first two to four EPCs (cells voltage clamped at −70 mV). This increase was normally followed by a reduction in amplitude over the subsequent 5–10 EPCs until a plateau level was reached. The amplitude of the plateau was always less than the maximum EPC amplitude in the train (Fig. 2A). The average amplitude of the plateau compared with that of the first EPC of the train provided an estimate of the degree of rundown observed during the EPC train. An example of this EPC train rundown (20, 50, and 100 Hz) in wild-type and axJ terminals is presented in Fig. 2, A-C. In Fig. 2B, the amplitude of the EPC evoked at 50 Hz in wild-type terminals ran down by an average of 19 ± 2% (n = 10 animals and 41 end plates). In contrast, examination of axJ terminals demonstrated that the EPC rundown was increased to 49 ± 2% (n = 16 animals and 49 end plates; P < 0.001; Fig. 2D; Table 1). At 100 Hz, this “tetanic fade” or tetanic rundown was more prominent in axJ than wild-type mice at the plateau portion of the train (Fig. 2C). A similar rapid decline of successive end plate currents during repetitive nerve stimulation was previously reported by several investigators as a characteristic effect of neuromuscular blocking agents (27, 28, 40, 41), neurotransmission in a transgenic mouse model of slow channel syndrome (11), in NCAM-deficient mice, and in CD24 mutant mice (32, 47, 48). Each of these cases results in either blockade of the postsynaptic acetylcholine receptor-associated ion channel and/or alteration in presynaptic function.
Fig. 2.
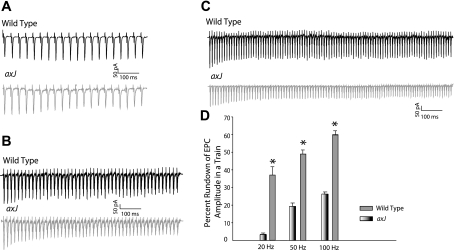
Neuromuscular transmission defects in axJ animals in response to high-frequency stimulation (HFS). A–C: data for EPC (voltage clamped at −70 mV) responses were obtained from axJ and wild type mice after HFS at 20 (A), 50 (B), and 100 Hz (C). Wild-type end plates showed an initial increase of EPC amplitude during the first 2–4 EPCs, followed by a reduction in amplitude over approximately the next 10 EPCs until a plateau level was reached. In all trains, the amplitude at the plateau was lower than the amplitude of the maximum EPC in the train. At axJ end plates, initial depression of EPC amplitudes was observed compared with the wild type and then followed by increased EPC rundown. (D). A comparison of percent rundown of EPC trains from axJ and wild type end plates at 3 different stimulation frequencies, (20, 50, and 100 Hz). In axJ NMJs, percent rundown of EPC amplitude in a train is significantly higher (*P < 0.001) at each stimulation frequency compared with the wild type.
Table 1.
Effects of membrane potential on MEPC and EPC amplitudes during HFS, quantum content, and mobilization rate in cut diaphragm muscle preparations of axJ mice and littermate controls
| 1st EPC in Train |
Plateau EPC in Train |
|||||||
|---|---|---|---|---|---|---|---|---|
| Membrane Potentials Holding | Mean MEPC Amplitude, nA (Before HFS) | Mean amplitude, nA | Mean amplitude, nA† | Mean MEPC amplitude, nA (After HFS) | 1st EPC Quantal Content | Plateau EPC Quantal Content | Mobilization Rate, quanta/s | |
| Wild type | ||||||||
| −70 mV | 2.14 ± 0.11 | 173.45 ± 11 | 144.32 ± 9.7 | 2.03 ± 0.26 | 80.85 ± 5.35 | 70.44 ± 4.75 | 3,543.91 ± 238.25 | |
| −90 mV | 2.37 ± 0.23 | 185.22 ± 14 | 152.02 ± 11 | 2.11 ± 0.5 | ||||
| axJ | ||||||||
| −70 mV | 4.1 ± 0.35 | 126.17 ± 10 | 63.11 ± 4† | 2.7 ± 0.3 | 41.37 ± 3.8* | 22.97 ± 1.65* | 1,148.51 ± 83.5* | |
| −90 mV | 4.9 ± 0.30 | 140.03 ± 3 | 65.01 ± 3† | 3.9 ± 0.2 | ||||
Values are means ± SE. EPC, end plate current; MEPC, miniature end plate current; HFS, high-frequency stimulation (50 Hz).
P < 00.001, significantly different from the corresponding control.
P < 0.001, significant difference from the 1st EPC of the train.
To assess any possible contribution of AChR-associated ion channel block to the increased rundown of EPC trains observed in axJ terminals, the EPC decay time constants (τEPC) were measured throughout the train (50 Hz) at two different holding membrane potentials (−90 and −70 mV). In axJ terminals, there was no significant change in τEPC during the train at any holding potential (−70 mV, 2.2 ± 0.12 ms; −90 mV, 2.53 ± 0.23 ms). Additionally in axJ terminals, the percent rundown of the EPC (50 Hz) amplitude remained independent of the membrane potential (voltage clamped at −70 mV, 50 ± 2%; n = 16 animals and 49 end plates; and voltage clamped at −90 mV, 53 ± 1.6%; n = 4 animals and 10 end plates; Table 1), suggesting that there was no contribution of acetylcholine receptor-associated ion channel blockade to the EPC rundown. This is in contrast to several nicotinic antagonists that have shown voltage-dependent rundown with changes in the EPC decay time constant during HFS (27). To compare the effect of nerve stimulation on the amplitude of miniature end plate currents, MEPCs were recorded before and after the train of stimulation. The MEPC amplitude in axJ and wild-type mice measured before and after the train remained unaltered (Table 1). Thus axJ end plates exhibited voltage-independent tetanic rundown and no change in posttetanic MEPC amplitude or shortening of EPC decay time (τEPC). These results suggested that the “tetanic fade” or tetanic rundown observed after Usp14 mutation was not use dependent and that preactivation of AChR was not necessary (3). Thus it appeared that the observed increase in tetanic rundown at axJ NMJs was associated with an alteration of presynaptic function.
AxJ muscle transmission is interrupted by failures.
In addition to a significant depression of EPC amplitudes, 27% of axJ end plates also showed an inability to sustain synaptic transmission resulting from HFS (20, 50, and 100 Hz). This transmission failure is shown in Fig. 3, A–C. The random transmission failures observed at axJ terminals were frequency dependent (Fig. 3D) and more prominent at the end of trains of stimulations (Fig. 3, A–C). For example, during 50-Hz stimulation a 44 ± 4% transmission failure was observed at the end of the stimulus train compared with the start of the train (32 ± 3.7%; n = 15 animals and 12 end plates).
Fig. 3.
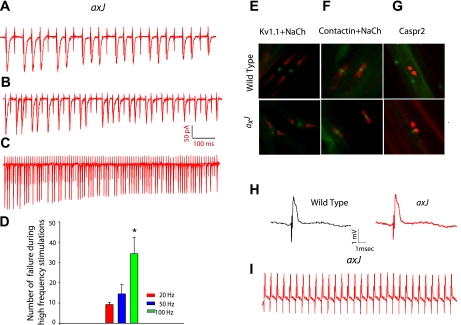
HFS causes transmission failures in axJ NMJs with unaltered compound action potentials during HFS and voltage-gated channel distribution. A–C: HFS causes occasional complete transmission failure as well as EPC rundown at axJ NMJs stimulated at 20 (A)-, 50 (B)-, and 100 (C)-Hz stimulations. D: at the axJ NMJ, the occurrence of failure increased with increasing stimulation frequencies, *P < 0.01, significantly different from 20-Hz stimulation. E and G: teased sciatic nerves from axJ and wild-type mice were stained with antibodies to ion channels responsible for saltatory conduction. Top: wild type. Bottom: axJ. E and F: labeling for Na+ channels (green) and K+ channels (red) were unaltered in wild type and axJ. F: labeling for contactin (red)/Na+ channels (green) remained same in axJ and wild type. G: contactin-associated protein 2 (Caspr2; red) shows no alteration in juxtaparanodal localization in wild-type and axJ mice. H: low-frequency (0.2 Hz) compound action potential generated from sciatic nerves of wild-type and axJ mice showed no differences in peak-to-peak amplitude and time of onset. I: HFS of sciatic nerve of axJ at 30 Hz showed no failure.
A similar type of transmitter output failure has been reported during HFS in CD24 mutant mice (32) and NCAM null NMJs (48). Further, these mice exhibited profound depression along with periods of transmission failures. These studies have suggested that transmission failures may be the result of defects in neurotransmitter release machinery or an inability to propagate nerve conduction, which would then result in failed depolarization of presynaptic terminals. For example, in NCAM-deficient animals and CD24 mutants, these defects were found to be presynaptic in origin (32, 47, 48) and were speculated to result from deregulation of vesicle mobilization/cycling. To determine whether presynaptic release failure and/or conduction failure in motor axons was playing a role in the high-frequency rundown observed in axJ junctions, the researchers performed experiments to isolate these potential defects.
Peripheral nerve conduction and ion channel distribution remain unaltered in axJ mice.
Myelinated axons invading muscles are organized into distinct domains that are necessary for saltatory conduction. These domains include the nodes of Ranvier, the internodes, the paranodes, and the juxtaparanodal regions (10, 12). Voltage-gated sodium (Na+) channels are concentrated at the nodes of Ranvier, whereas delayed rectifier potassium (K+) channels are localized in the juxtaparanodal regions. These junctions also contain a Neurexin IV-related protein, Caspr/Paranodin (NCP1). Mice that lack NCP1 exhibit tremors and ataxia (10), which were reportedly due to K+ channel displacement from the juxtaparanodal and paranodal domains. To determine whether loss of Usp14 in axJ mice can affect the distribution of ion channels and contribute to abnormal electrophysiological properties in peripheral nerves, juxtaparanodal and paranodal domains were fluorescently stained and field potential recordings were taken from sciatic nerves.
In axJ mice, the pattern of K+ and Na+ channel distribution appeared identical to the wild type and largely confined to the nodes of Ranvier (Fig. 3, E and F). Immunostaining of the paranodal markers contactin-associated protein 2 (Caspr2) and contactin showed no obvious change in distribution in axJ mice compared with wild-type mice (Fig. 3, F and G). Electrophysiological recording demonstrated that no difference was observed in the characteristics of the CAP after stimulation of sciatic nerves from wild-type and axJ mice. The population conduction velocities evoked at low frequency stimulation (0.2 Hz) obtained from the measured CAP at maximal intensities were the same in wild-type (22.3 ± 2 m/s; n = 12 animals and 20 sciatic nerves) and axJ mice (21.5 ± 1.6 m/s; n = 10 animals and 18 sciatic nerves; Fig. 3H). The latency and CAP peak-to-peak amplitude also remained the same. When the sciatic nerve was stimulated at high frequency (30 Hz), the axJ nerves were able to effectively conduct (Fig. 3I). These results indicated that axJ axons could successfully conduct action potentials during HFS, so they cannot explain the observed rundown and failures in neuro-transmission during HFS in axJ mice.
HFS in axJ mice produces a decrease in quantum content and transmitter mobilization.
It was previously reported that a combination of low frequency, larger amplitude miniature end plate potentials (a mixtures of both small and giant miniature end plate potentials) and lower quantal content are the characteristic defects at the axJ NMJ (68). In addition to our previous study, one of the most striking defects in the axJ junction is a failure to sustain optimal transmission over time. This would most likely represent a presynaptic defect in the ability either to release synaptic vesicles or to mobilize vesicles from the RP, after those in the RRP (38, 62, 65) have been exhausted. To confirm that the reduction in quantal content in axJ NMJs is due to a decrease in the acetylcholine mobilization, the quantal content was measured at the start and at the end of high-frequency trains of stimulation (50 Hz), by dividing the EPC amplitude at the start of the HFS train and during the resulting plateau, by the MEPC amplitude recorded just before the EPC train. Results obtained from this series of experiments are presented in Table 1 demonstrating that at the end plates of axJ mice (n = 18 animals and 35 end plates) compared with wild type (n = 16 animals and 30 end plates), quantal content was significantly lowered both at the start (P < 0.001) and the plateau portion of the train (P < 0.001). According to Harborne et al. (29), transmitter mobilization can also be calculated from the plateau portion of the train (mean quantal content multiplied by stimulation frequency). With the use of this method (11, 29), the transmitter mobilization observed (Table 1) at axJ end plates (n = 35) was significantly lower (P < 0.001) than that observed with the corresponding controls (n = 30). Thus it appears that transmission failures in axJ NMJs may result from defects in mobilizing synaptic vesicles from the RP to presynaptic release sites.
Synaptic vesicle dynamics are altered in axJ mice.
Efficient vesicle membrane recycling, i.e., loading, mobilizing, and release of neurotransmitter, is necessary to sustain normal neurotransmission during HFS at the NMJ. Most synapses exhibit a characteristic rundown of synaptic signals during repetitive stimulation, which has been interpreted as a sequential depletion of readily releasable and reserve vesicle pools (38, 69). Optical studies using fluorescent dyes in frog (54) and the Drosophila mutant, Shibire (35, 36, 38), have provided additional support for two recycling pools, the RP and the RRP. Together these constitute the total pool of synaptic vesicles (53, 54). To understand the vesicular dynamics in axJ mice, two FM dyes, FM1-43 and FM2-10, were selected for their different staining properties(53, 54 ). The kinetics of FM-dye release after different loading conditions provided further information about release from these two pools. Selective loading of the two pools was achieved by using the fluorescent styryl dye FM1-43 to label the total pool and by using FM2-10 to label the RRP, as described previously (53, 54) (see methods).
In Fig. 4A, the muscle/nerve preparation was stimulated for 1 min at 30 Hz in the presence of FM1-43 and the dye was left for 15 min before washing with a high Mg 2+ solution. The protocol used (Fig. 4A, top) produced maximally stained terminals with FM1-43. Spots of FM1-43 staining appeared similar (Fig. 4, A and B) in wild-type and in axJ terminals, indicating both terminals could participate in exo-/endocytotic vesicle cycling and activity-dependent uptake of styryl dye. Comparison of activity-dependent dye uptake of FM1-43 into the nerve terminals of wild-type and axJ mice is illustrated in Fig. 4C. The average fluorescence intensity of the terminals was significantly reduced in axJ mice (n = 11 animals and 551 terminals) compared with their littermate controls (n = 18 animals and 772 terminals). A 21% decrease in total dye uptake in axJ terminals was noted compared with their corresponding controls (285 ± 5.35 and axJ 226.95 ± 5.61 arbitrary units; P < 0.001; Fig. 4C), indicating differential loading of dye in axJ terminals. Next, after FM1-43 administration, the ability of terminals to release the dye taken up during 1 min, 30 Hz tetanus was examined. Stimulation of labeled terminals caused dye loss as vesicle exocytosis took place. In Fig. 4, D and E, both wild-type and axJ nerve terminals displayed significant dye loss (FM1-43) after 30-Hz stimulations. The normalized destaining rates obtained from both types of terminals are presented in Fig. 4 F. At time zero, tetanic stimulation began and continued for the duration of the experiment, causing a net decrease in terminal fluorescence as the dye was released. The time course of activity-dependent nerve terminal FM1-43 destaining in wild-type and axJ mice could be well fitted (R2 > 0.98) by the sum of two exponentials, suggesting the presence of two vesicle pools, as previously described for rat and mouse NMJs (51, 52). An initial, accelerated dye loss followed by a slower decline suggests that the two pools may be released in a strictly sequential fashion during a tetanus (53). This may be due to the fact that the dye first labels an “immediately releasable” store of vesicles and with time, a fraction of this becomes consolidated in a store with a lower probability of release that is mobilized only during HFS (52). Results obtained from these experiments demonstrated a shorter time constant in axJ NMJs (∼24 s) compared with the wild type (∼30 sec). Interestingly, the slower component of the destaining kinetics of FM1-43 in axJ was more prolonged (∼600 s) compared with the wild type (∼400 s; P < 0.01; Fig. 4F). It appears that in axJ terminals a slower rate of dye loss after HFS could be due to reduced mobilization of the vesicles stored in the RP with a lower probability of release.
Fig. 4.
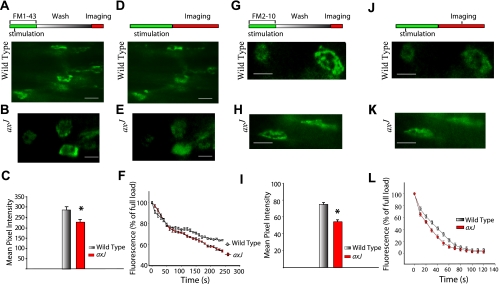
Characteristics of synaptic vesicular pool sizes and release dynamics in wild-type and axJ muscle. A and B: photographs of wild-type (A) and axJ end plates (B) after complete loading with FM1-43 (6–8 μM) staining during and after a 1-min, 30-Hz stimulation and a 15-min incubation following the wash (top). C: average fluorescence of the terminals demonstrates a decrease in overall loading of FM1-43 in the axJ (*P < 0.01). D and E: photographs of FM1-43 unloading after 1 min stimulation at 50 Hz (top) and reduction of fluorescence in wild-type (D) and axJ (E) muscle. F: time course of nerve terminal destaining in wild-type and axJ NMJs is shown. Nerve stimulation (50 Hz) started at time point zero. Inset: average data from destaining spots in wild-type and axJ terminals together with the best nonlinear, least squares, double exponential curve fit using the Marquardt-Levenberg algorithm. Time constant for faster component was ∼24 s in axJ NMJs compared with the wild type ∼30 s. Slower component of the destaining kinetics of FM1-43 in axJ was more prolonged, ∼600 s, compared with the wild type (∼400 s; P < 0.01). G and H : photographs of wild-type (G) and axJ terminals (H) after loading with FM2-10 dye. Preparations were stimulated in the presence of dye (30 Hz for 1 min) and then washed immediately (top). I: mean pixel intensity after complete loading of FM2-10 (30 μM) during tetanic stimulations shows significant reduction (*P < 0.001) of FM2-10 uptake into the nerve terminals of axJ mice. J and K: photographs of destaining of FM2-10 from the nerve terminals of the wild type (J) and axJ (K) after 50-Hz stimulation and imaging (protocol, top). L: destaining kinetics of FM2-10 after 50-Hz stimulation. Destaining rates from both groups were well fitted by a single exponential (R2 > 0.99). Time to destain was ∼19 s in axJ terminals and ∼28 s in wild-type terminals. Scale bar = 20 μM.
The difference in the FM1-43 uptake and destaining pattern evident between axJ and wild-type terminals prompted a closer examination using FM2-10, employing a tetanus, and “quick wash” paradigm (Fig. 4, G–L). With the use of this protocol (Fig. 4G, top), only the RRPs were labeled (53). The protocol consisted of stimulating the preparation for 1 min at 30 Hz with dye present in the bath followed by immediate washing and imaging. A typical dye uptake experiment using wild-type and axJ terminal is shown in Fig. 4, G and H. A comparison of the mean pixel intensity (Fig. 4I) revealed a significant reduction (27%; n = 27 animals and 1,151 terminals) in dye uptake in axJ compared with the wild type (P < 0.01; n = 35 animals and 1,790 terminals). After a quick wash, these terminals were destained by application of 50-Hz stimulations (Fig. 4, J and K). The destaining rates from both groups (Fig. 4L) were well fitted by a single exponential (R2 > 0.99) indicating the participation of single pool (53). These observations indicate that, like CD24 mutant mice which also showed transmission failures, a reduced mobilization rate together with a smaller RRP suggests fewer active zones for transmitter release (32). These alterations could contribute to the reduction in quantal content and observed transmission failure in axJ mice. Overall, in axJ terminals, a decrease in dye uptake and a prolonged decay after FM1-43 destaining suggests that, in addition to a smaller RRP, there may be a defect in transmitter mobilization from the reserve to the RRP during high stimulus rates.
DISCUSSION
The ataxia (axJ) mutation is one of the few genetic lesions in the UPS signaling pathway that leads to a neurological phenotype. Loss of Usp14 in mice results in a 30% decrease in the free pool of ubiquitin indicating that Usp14 functions to stabilize ubiquitin by preventing its proteasomal degradation (4). These findings suggest that proteasomal recycling of ubiquitin by Usp14 is required to maintain the ubiquitin levels necessary for normal synaptic function. Recent reports (16) indicate that Usp14 is indispensable for synaptic development and suggest a requirement for local ubiquitin recycling by proteasome to control the development as well as function of NMJs . The transgenic rescue of the axJ mouse with neuronal-specific expression of Usp14 also demonstrated that the full-length form of Usp14 was sufficient to restore viability and motor system function to the axJ mice and indicates that changes in proteasome function may contribute to neurological dysfunction in the axJ mice (18). Many neurological disorders display changes in protein turnover and synaptic function, indicating that therapies targeted to the UPS may be beneficial for treatment of neurological diseases associated with synaptic function. Recently, transgenic complementation studies (17) to restore ubiquitin levels to the axJ mice showed that neurally expressed ubiquitin corrected abnormal synaptic transmission at the NMJ, indicating that ubiquitin homeostasis is essential for synaptic activity. These findings demonstrate a unique role for Usp14 in regulating synaptic ubiquitin pools and indicate that fluctuations in ubiquitin may contribute to other neuronal diseases that exhibit alterations in protein turnover (17). Our present study further confirms the fact that ubiquitination of synaptic proteins is important for the normal operation of the neurotransmitter release machinery and in regulating the size of synaptic vesicle pools.
In axJ mice, together with our previous study (16), 60–70% of diaphragm muscles of mixed fiber type composition were morphologically less abnormal compared with the distal muscles, probably because it is an active muscle till death. Heterogeneity in FM-dye uptake was reported across NMJs at different fiber types in rat diaphragm muscles The fiber-type differences reflect differences in quantal size and safety factor for neuromuscular transmission (24). Although similar findings have not been studied for the mouse, heterogeneity in synaptic vesicle pool size and fiber type differences is reported in NMJ ultrastructural studies (25). A detailed ultrastructural study is needed to understand the important role of ubiquitin proteasome system on vesicular dynamics at type-identified diaphragm muscle fibers in axJ.
Our previous study using axJ mice demonstrated defects in synaptic transmission in the central as well as PNS. These included decreased quantal content at peripheral synapses and reduced short-term synaptic plasticity at central synapses (68). The present result provide additional evidence suggesting that loss of Usp14 is associated with crucial defects in synaptic transmission which may, in turn, be caused by defects in Ca2+ sensitive quantal release and mobilization of neurotransmitter vesicles. Like the CNS, axJ NMJs showed no PPF and transmitter release had an altered sensitivity to extracellular Ca2+ with output saturating at physiological levels. Two factors govern the overall efficiency of transmission: the unitary probability of release (Pr) of individual release sites and the number of functional release sites (N). Ca2+ dependence of the paired-pulse ratio and near-saturating EPC amplitudes at 2 mM Ca2+ suggest that the Pr might be close to maximum levels at axJ junction's in vivo Ca2+ levels. A significant alteration of paired-pulse ratios are discussed as the possibility of Pr compensation at physiological Ca2+ in mice lacking NCAM or P/Q-type Ca2+ channels (47, 49, 63). Alternatively, it was also suggested that the absence of PPF may be associated with changes in release machinery, such as the recruitment of synaptic vesicles. Therefore, a significant decrease in vesicle pool size may cause the depletion of the entire RRP by each stimulus resulting in a decrease in PPF. Similar defects in PPF have been reported in mice lacking the presynaptic component, synapsin (55). Together with the finding that Usp14 is present in the synaptoneurosome fraction, normally enriched in synaptic vesicles, these data are consistent with a role for Usp14 in synaptic function (68).
The rundown of the EPC amplitude during HFS in axJ mice is also quite a characteristic phenomenon. In previous reports (2, 3, 11, 27–29, 41, 56), the increased EPC rundown during HFS induced by several nicotinic antagonists (e.g., d-tubocurarine, local anesthetics, or barbiturates) or mutations of the acetylcholine receptor were attributed to either pre- or postsynaptic defects. In axJ muscles, the EPC rundown observed during HFS appeared to be presynaptic in origin since axJ end plates exhibited a voltage-independent tetanic rundown, no change in posttetanic MEPC amplitude, or shortening of EPC decay time (τEPC). The greater reduction of quantal content observed at the plateau phase of the train compared with littermate controls was also associated with a concomitant reduction in transmitter mobilization. Neurotransmitter mobilization serves as a basic measure of exocytosis; thus the data indicated a defect in neurotransmitter release at axJ nerve terminals.
The observed rundown accompanied with transmission failures seen at axJ NMJs after repetitive stimulation under normal Ca2+ concentrations appears similar to the transmission failures reported in CD24 mutants (32) and NCAM 180 (48)- and NCAM-deficient mice. In these mice, transmission failures result from defects in vesicle mobilization/cycling (47, 48). The results presented here indicate that these failures are due to the novel combination of abnormally low quantal content, significant depression during repetitive activity, and/or defects in vesicle cycling (32, 47, 48). Like NCAM 180- and NCAM-deficient junctions, total transmission failures were not due to nerve conduction failure (47, 48), because direct recording of compound action potentials in axJ mice suggests that axonal conduction block did not contribute to neuromuscular transmission failures during high stimulation rates. However, the branch point failure could still remain a possibility for the observed transmission failure at axJ junctions (47, 61). The two-paranodal markers Caspr2 and contactin as well as labeling of Na+ and K+ channels in sciatic nerves revealed no change in distribution in axJ mice; characteristics that were frequently observed in mice with ablated contactin gene expression, ataxia, and extreme hind leg weakness (8, 12). All these findings suggest that events downstream of Ca2+ influx, such as an inability to deliver vesicles at rates needed to maintain physiological transmission level, may be responsible for the transmission failure at the axJ NMJ. This could be due to alterations in the size of the pool of releasable vesicles (as supported by the FM1-43 data), defects in mobilizing vesicles, or subsequent steps in docking and fusion of vesicles in high rates. The conclusions drawn from these electrophysiological studies were further supported by results of activity-dependent FM-dye uptake into synaptic vesicles at axJ terminals and subsequent destaining during tetanic stimulation. In this study, a significant decrease in FM-dye loading with FM1-43 can be correlated to the electrophysiological findings in axJ terminals that showed that initial depression in the EPC and reduced quantal content observed after HFS might be attributed to a reduction in the total vesicle pool size, specifically the RRP. The rate of FM1-43 loss nerve terminal destaining in wild-type NMJs can be well fitted by the sum of two exponentials (51, 52). An initial faster dye loss is followed by a slower decline indicative of two releasable stores of vesicles. These two pools may be released in a strictly sequential fashion during a tetanus (53). In axJ mice, the prolonged slower component of destaining kinetics could be interpreted as an optical readout of the decrease in vesicle mobilization. The alterations in the size of the RRP, due to reduced mobilization from the RP, may be responsible for the inability to deliver vesicles at rates needed to maintain transmission at high but physiological rates. Mobilization of vesicles from the reserve to the immediately releasable pool helps sustain transmitter release during high-frequency repetitive stimulation (37, 57), which was affected in axJ terminals. Therefore, a smaller cycling vesicle pool, due to reduced mobilization from the RP, may be responsible for significant reduction of quantal content, thus accounting for the observed transmission defects in axJ NMJ.
Although there are very few reports on the molecular mechanisms involved in ubiquitin protease-dependent regulation of synaptic vesicle mobilization, docking, or release, it has been reported that the activity-dependent alteration in the size of the recycling vesicle pool may be regulated by cAMP/PKA-dependent pathway. This appears to be the case, for example, in Drosophila mutants dunce and rutabaga that have deficiencies in the cAMP pathway (34). Generally speaking it will be of interest to determine the level at which ubiquitin and cAMP signaling interact in the regulation of vesicle dynamics in the nerve terminal. To this end, animals with mutations in the ubiquitin pathway as we have described here will be important tools in such investigations. Our studies also indicate that the UPS is intimately associated with synaptic function and ubiquitin conjugation is critical for efficient protein turnover at the synaptic terminals. Several E3 ligases have been implicated in the turnover of presynaptic proteins such as RIM1 and synaptophysin (67, 70). Although the importance of the proteasome-associated DUB Usp14 in maintaining ubiquitin levels required for both synaptic development and function is clear, further study is needed to understand whether the block in NMJ development in axJ mice is due to reduced protein degradation that is secondary to the ubiquitin deficiency or if other ubiquitin signaling pathways like vesicular transport, kinase activation, and receptor-mediated endocytosis contribute to the motor end plate disease.
In summary, Usp14 is a component of the proteasome and the axJ mice represent a valuable mouse model to examine how changes in proteasome activity affect synaptic function. Although the role the UPS system plays either for regulation of synaptic function in adult neuromuscular junctions and/or normal development is yet to be resolved, this report demonstrates evidence that ubiquitination of synaptic proteins is important for recruitment and the proper regulation of vesicle pool dynamics and also points out the role of ubiquitin protease in this process. This is a very important observation since there is only neuronal culture supporting a role for the proteasome in regulating synaptic vesicle pools. Here we have characterized the phenotype of this synapse, revealing several new and unexpected properties including vesicular dynamics and UPS malfunction. This study is also important because steady dysregulation of synaptic transmission in response to UPS malfunction is reported to contribute to several neurodegenerative diseases (20, 60).
GRANTS
This work was supported by National Institutes of Health Grants RO1-NS-043095, RO1-DAO-13141, and R37-MH-040165 (to R. J. Miller) and RO1-NS-047533-01 (to S. M. Wilson).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
B.J.B., S.M.W., and H.J. conception and design of research; B.J.B., S.M.W., and H.J. performed experiments; B.J.B., S.M.W., and H.J. analyzed data; B.J.B., S.M.W., and H.J. interpreted results of experiments; B.J.B. and S.M.W. prepared figures; B.J.B. drafted manuscript; B.J.B., S.M.W., and R.J.M. edited and revised manuscript; B.J.B., S.M.W., and R.J.M. approved final version of manuscript.
ACKNOWLEDGMENTS
Thanks to Caroline Freitag for helping with the manuscript.
REFERENCES
- 1. Adams J. The proteasome: structure, function, and role in the cell. Cancer Treat Rev 29 Suppl 1: 3–9, 2003 [DOI] [PubMed] [Google Scholar]
- 2. Adams PR. Acetylcholine receptor kinetics. J Membr Biol 58: 161–174, 1981 [DOI] [PubMed] [Google Scholar]
- 3. Adams PR. Drug blockade of open end-plate channels. J Physiol 260: 531–552, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anderson C, Crimmins S, Wilson JA, Korbel GA, Ploegh HL, Wilson SM. Loss of Usp14 results in reduced levels of ubiquitin in ataxia mice. J Neurochem 95: 724–731, 2005 [DOI] [PubMed] [Google Scholar]
- 5. Aravamudan B, Broadie K. Synaptic Drosophila UNC-13 is regulated by antagonistic G-protein pathways via a proteasome-dependent degradation mechanism. J Neurobiol 54: 417–438, 2003 [DOI] [PubMed] [Google Scholar]
- 6. Bekele-Arcuri Z, Matos MF, Manganas L, Strassle BW, Monaghan MM, Rhodes KJ, Trimmer JS. Generation and characterization of subtype-specific monoclonal antibodies to K+ channel alpha- and beta-subunit polypeptides. Neuropharmacology 35: 851–865, 1996 [DOI] [PubMed] [Google Scholar]
- 7. Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292: 1552–1555, 2001 [DOI] [PubMed] [Google Scholar]
- 8. Berglund EO, Murai KK, Fredette B, Sekerkova G, Marturano B, Weber L, Mugnaini E, Ranscht B. Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron 24: 739–750, 1999 [DOI] [PubMed] [Google Scholar]
- 9. Betz WJ, Bewick GS. Optical monitoring of transmitter release and synaptic vesicle recycling at the frog neuromuscular junction. J Physiol 460: 287–309, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, St Ching W, Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 30: 369–383, 2001 [DOI] [PubMed] [Google Scholar]
- 11. Bhattacharyya BJ, Day JW, Gundeck JE, Leonard S, Wollmann RL, Gomez CM. Desensitization of mutant acetylcholine receptors in transgenic mice reduces the amplitude of neuromuscular synaptic currents. Synapse 27: 367–377, 1997 [DOI] [PubMed] [Google Scholar]
- 12. Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 30: 385–397, 2001 [DOI] [PubMed] [Google Scholar]
- 13. Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM. Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron 35: 107–120, 2002 [DOI] [PubMed] [Google Scholar]
- 14. Chain DG, Casadio A, Schacher S, Hegde AN, Valbrun M, Yamamoto N, Goldberg AL, Bartsch D, Kandel ER, Schwartz JH. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron 22: 147–156, 1999 [DOI] [PubMed] [Google Scholar]
- 15. Chen G, Harata NC, Tsien RW. Paired-pulse depression of unitary quantal amplitude at single hippocampal synapses. Proc Natl Acad Sci USA 101: 1063–1068, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen PC, Qin LN, Li XM, Walters BJ, Wilson JA, Mei L, Wilson SM. The proteasome-associated deubiquitinating enzyme Usp14 is essential for the maintenance of synaptic ubiquitin levels and the development of neuromuscular junctions. J Neurosci 29: 10909–10919, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen PC, Bhattacharyya BJ, Hanna J, Minkel H, Wilson JA, Finley D, Miller RJ, Wilson SM. Ubiquitin homeostasis is critical for synaptic development and function. J Neurosci 31: 17505–17513, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crimmins S, Jin Y, Wheeler C, Huffman AK, Chapman C, Dobrunz LE, Levey A, Roth KA, Wilson JA, Wilson SM. Transgenic rescue of ataxia mice with neuronal-specific expression of ubiquitin-specific protease 14. J Neurosci 26: 11423–11431, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL, Zoghbi HY. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 24: 879–892, 1999 [DOI] [PubMed] [Google Scholar]
- 20. Detera-Wadleigh SD, Badner JA, Berrettini WH, Yoshikawa T, Goldin LR, Turner G, Rollins DY, Moses T, Sanders AR, Karkera JD, Esterling LE, Zeng J, Ferraro TN, Guroff JJ, Kazuba D, Maxwell ME, Nurnberger JI, Jr, Gershon ES. A high-density genome scan detects evidence for a bipolar-disorder susceptibility locus on 13q32 and other potential loci on 1q32 and 18p112. Proc Natl Acad Sci USA 96: 5604–5609, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS. Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 412: 449–452, 2001 [DOI] [PubMed] [Google Scholar]
- 22. Dionne VE, Stevens CF. Voltage dependence of agonist effectiveness at the frog neuromuscular junction: resolution of a paradox. J Physiol 251: 245–270, 1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol 193: 419–432, 1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ermilov LG, Mantilla CB, Rowley KL, Sieck GC. Safety factor for neuromuscular transmission at type-identified diaphragm fibers. Muscle Nerve 35: 800–803, 2007 [DOI] [PubMed] [Google Scholar]
- 25. Fahim MA, Holley JA, Robbins N. Topographic comparison of neuromuscular junctions in mouse slow and fast twitch muscles. Neuroscience 13: 227–235, 1984 [DOI] [PubMed] [Google Scholar]
- 26. Gaffield MA, Betz WJ. Imaging synaptic vesicle exocytosis and endocytosis with FM dyes. Nat Protoc 1: 2916–2921, 2006 [DOI] [PubMed] [Google Scholar]
- 27. Gibb AJ, Marshall IG. Pre-and post-junctional effects of tubocurarine and other nicotinic antagonists during repetitive stimulation in the rat. J Physiol 351: 275–297, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Giniatullin RA, Khamitov G, Khazipov R, Magazanik LG, Nikolsky EE, Snetkov VA, Vyskocil F. Development of desensitization during repetitive end-plate activity and single end-plate currents in frog muscle. J Physiol 412: 113–122, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harborne AJ, Bowman WC, Marshall IG. Effects of tubocurarine on end-plate current rundown and quantal content during rapid nerve stimulation in the snake. Clin Exp Pharmacol Physiol 15: 479–490, 1988 [DOI] [PubMed] [Google Scholar]
- 30. Hegde AN, DiAntonio A. Ubiquitin and the synapse. Nat Rev Neurosci 3: 854–861, 2002 [DOI] [PubMed] [Google Scholar]
- 31. Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH. Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in aplysia. Cell 89: 115–126, 1997 [DOI] [PubMed] [Google Scholar]
- 32. Jevsek M, Jaworski A, Polo-Parada L, Kim N, Fan J, Landmesser LT, Burden SJ. CD24 is expressed by myofiber synaptic nuclei and regulates synaptic transmission. Proc Natl Acad Sci USA 103: 6374–6379, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem 75: 436–439, 2000 [DOI] [PubMed] [Google Scholar]
- 34. Kidokoro Y, Kuromi H, Delgado R, Maureira C, Oliva C, Labarca P. Synaptic vesicle pools and plasticity of synaptic transmission at the Drosophila synapse. Brain Res Brain Res Rev 47: 18–32, 2004 [DOI] [PubMed] [Google Scholar]
- 35. Kuromi H, Kidokoro Y. The optically determined size of exo/endo cycling vesicle pool correlates with the quantal content at the neuromuscular junction of Drosophila larvae. J Neurosci 19: 1557–1565, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuromi H, Kidokoro Y. Selective replenishment of two vesicle pools depends on the source of Ca2+ at the Drosophila synapse. Neuron 35: 333–343, 2002 [DOI] [PubMed] [Google Scholar]
- 37. Kuromi H, Kidokoro Y. Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron 27: 133–143, 2000 [DOI] [PubMed] [Google Scholar]
- 38. Kuromi H, Kidokoro Y. Two distinct pools of synaptic vesicles in single presynaptic boutons in a temperature-sensitive Drosophila mutant, shibire. Neuron 20: 917–925, 1998 [DOI] [PubMed] [Google Scholar]
- 39. Lappe-Siefke C, Loebrich S, Hevers W, Waidmann OB, Schweizer M, Fehr S, Fritschy JM, Dikic I, Eilers J, Wilson SM, Kneussel M. The ataxia (axJ) mutation causes abnormal GABAA receptor turnover in mice. PLoS Genet 5: e1000631, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Magleby KL, Pallotta BS. A study of desensitization of acetylcholine receptors using nerve-released transmitter in the frog. J Physiol 316: 225–250, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Magleby KL, Pallotta BS, Terrar DA. The effect of (+)-tubocurarine on neuromuscular transmission during repetitive stimulation in the rat, mouse, and frog. J Physiol 312: 97–113, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin-proteasome system in Parkinson's disease. Nat Rev Neurosci 2: 589–594, 2001 [DOI] [PubMed] [Google Scholar]
- 43. Miller RJ, Wilson SM. Neurological disease: UPS stops delivering. Trends Pharmacol Sci 24: 18–23, 2003 [DOI] [PubMed] [Google Scholar]
- 44. Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20: 389–399, 1998 [DOI] [PubMed] [Google Scholar]
- 45. Plomp JJ, Vergouwe MN, Van den Maagdenberg AM, Ferrari MD, Frants RR, Molenaar PC. Abnormal transmitter release at neuromuscular junctions of mice carrying the tottering alpha(1A) Ca(2+) channel mutation. Brain 123: 463–471, 2000 [DOI] [PubMed] [Google Scholar]
- 46. Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, Trimmer JS, Shrager P, Peles E. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 24: 1037–1047, 1999 [DOI] [PubMed] [Google Scholar]
- 47. Polo-Parada L, Bose CM, Landmesser LT. Alterations in transmission, vesicle dynamics, and transmitter release machinery at NCAM-deficient neuromuscular junctions. Neuron 32: 815–828, 2001 [DOI] [PubMed] [Google Scholar]
- 48. Polo-Parada L, Plattner F, Bose C, Landmesser LT. NCAM 180 acting via a conserved C-terminal domain and MLCK is essential for effective transmission with repetitive stimulation. Neuron 46: 917–931, 2005 [DOI] [PubMed] [Google Scholar]
- 49. Rafuse VF, Polo-Parada L, Landmesser LT. Structural and functional alterations of neuromuscular junctions in NCAM-deficient mice. J Neurosci 20: 6529–6539, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci 19: 7516–7528, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reid B, Slater CR, Bewick GS. Synaptic vesicle dynamics in rat fast and slow motor nerve terminals. J Neurosci 19: 2511–2521, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ribchester RR, Mao F, Betz WJ. Optical measurements of activity-dependent membrane recycling in motor nerve terminals of mammalian skeletal muscle. Proc Biol Sci 255: 61–66, 1994 [DOI] [PubMed] [Google Scholar]
- 53. Richards DA, Guatimosim C, Betz WJ. Two endocytic recycling routes selectively fill two vesicle pools in frog motor nerve terminals. Neuron 27: 551–559, 2000 [DOI] [PubMed] [Google Scholar]
- 54. Richards DA, Guatimosim C, Rizzoli SO, Betz WJ. Synaptic vesicle pools at the frog neuromuscular junction. Neuron 39: 529–541, 2003 [DOI] [PubMed] [Google Scholar]
- 55. Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Sudhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375: 488–493, 1995 [DOI] [PubMed] [Google Scholar]
- 56. Ruff RL. The kinetics of local anesthetic blockade of end-plate channels. Biophys J 37: 625–631, 1982 [PMC free article] [PubMed] [Google Scholar]
- 57. Ryan TA. Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J Neurosci 19: 1317–1323, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Saliba RS, Michels G, Jacob TC, Pangalos MN, Moss SJ. Activity-dependent ubiquitination of GABA(A) receptors regulates their accumulation at synaptic sites. J Neurosci 27: 13341–13351, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23: 399–409, 1999 [DOI] [PubMed] [Google Scholar]
- 60. Schwab SG, Hallmayer J, Lerer B, Albus M, Borrmann M, Honig S, Strauss M, Segman R, Lichtermann D, Knapp M, Trixler M, Maier W, Wildenauer DB. Support for a chromosome 18p locus conferring susceptibility to functional psychoses in families with schizophrenia, by association and linkage analysis. Am J Hum Genet 63: 1139–1152, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sieck GC, Prakash YS. Fatigue at the neuromuscular junction. Branch point vs presynaptic vs postsynaptic mechanisms. Adv Exp Med Biol 384: 83–100, 1995 [PubMed] [Google Scholar]
- 62. Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron 21: 885–893, 1998 [DOI] [PubMed] [Google Scholar]
- 63. Urbano FJ, Piedras-Renteria ES, Jun K, Shin HS, Uchitel OD, Tsien RW. Altered properties of quantal neurotransmitter release at end plates of mice lacking P/Q-type Ca2+ channels. Proc Natl Acad Sci USA 100: 3491–3496, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Walters BJ, Campbell SL, Chen PC, Taylor AP, Schroeder DG, Dobrunz LE, Artavanis-Tsakonas K, Ploegh HL, Wilson JA, Cox GA, Wilson SM. Differential effects of Usp14 and Uch-L1 on the ubiquitin proteasome system and synaptic activity. Mol Cell Neurosci 39: 539–548, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394: 384–388, 1998 [DOI] [PubMed] [Google Scholar]
- 66. Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol 2: 169–178, 2001 [DOI] [PubMed] [Google Scholar]
- 67. Wheeler TC, Chin LS, Li Y, Roudabush FL, Li L. Regulation of synaptophysin degradation by mammalian homologues of seven in absentia. J Biol Chem 277: 10273–10282, 2002 [DOI] [PubMed] [Google Scholar]
- 68. Wilson SM, Bhattacharyya B, Rachel RA, Coppola V, Tessarollo L, Householder DB, Fletcher CF, Miller RJ, Copeland NG, Jenkins NA. Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat Genet 32: 420–425, 2002 [DOI] [PubMed] [Google Scholar]
- 69. Wu LG, Betz WJ. Kinetics of synaptic depression and vesicle recycling after tetanic stimulation of frog motor nerve terminals. Biophys J 74: 3003–3009, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, Fukuda Y, Chiba T, Morone N, Yuasa S, Inokuchi K, Ohtsuka T, Macgregor GR, Tanaka K, Setou M. SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell 130: 943–957, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zefirov AL, Abdrakhmanov MM, Mukhamedyarov MA, Grigoryev PN. The role of extracellular calcium in exo- and endocytosis of synaptic vesicles at the frog motor nerve terminals. Neuroscience 143: 905–910, 2006 [DOI] [PubMed] [Google Scholar]
- 72. Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol 9: 305–313, 1999 [DOI] [PubMed] [Google Scholar]