

Abstract
Steatotic livers are sensitive to ischemic events and associated ATP depletion. Hepatocellular necrosis following these events may result from mitochondrial uncoupling protein-2 (UCP2) expression. To test this hypothesis, we developed a model of in vitro steatosis using primary hepatocytes from wild-type (WT) and UCP2 knockout (KO) mice and subjected them to hypoxia/reoxygenation (H/R). Using cultured hepatocytes treated with emulsified fatty acids for 24 h, generating a steatotic phenotype (i.e., microvesicular and broad-spectrum fatty acid accumulation), we found that the phenotype of the WT and UCP2 KO were the same; however, cellular viability was increased in the steatotic KO hepatocytes following 4 h of hypoxia and 24 h of reoxygenation; Hepatocellular ATP levels decreased during hypoxia and recovered after reoxygenation in the control and UCP2 KO steatotic hepatocytes but not in the WT steatotic hepatocytes; mitochondrial membrane potential in WT and UCP2 KO steatotic groups was less than control groups but higher than UCP2 KO hepatocytes. Following reoxygenation, lipid peroxidation, as measured by thiobarbituric acid reactive substances, increased in all groups but to a greater extent in the steatotic hepatocytes, regardless of UCP2 expression. These results demonstrate that UCP2 sensitizes steatotic hepatocytes to H/R through mitochondrial depolarization and ATP depletion but not lipid peroxidation.
Keywords: liver, mitochondria, steatosis
the biochemical mechanisms responsible for steatotic liver sensitivity to ischemic stressors are numerous; however, the primary cause of hepatocellular death in these livers is necrosis and is oncotic in nature (26). Mitochondrial dysfunction, namely depolarization and/or membrane permeability transition, impairs ATP formation and shifts cell death away from apoptosis (26, 33). As the incidence of liver steatosis increases in the United States, this becomes a greater concern for liver surgeries that involve periods of ischemia, including transplantation and resection (2, 4, 11).
The condition of liver steatosis includes presence of increased metabolic substrates, increased oxidative potential, and increased reactive oxygen species (ROS) production in the mitochondria (26). Under these conditions, a compensatory decrease in mitochondrial membrane potential (MMP) would be an appropriate response, both as an antioxidant response and as a control of the metabolic state. To do this, steatotic livers upregulate mitochondrial uncoupling protein-2 (UCP2), which is thought to combat these cellular disorders by acting as an inducible protonophore in the mitochondrial inner membrane (6, 30, 31). In normal livers, UCP2 expression is specific to Kupffer cells; however, parenchymal UCP2 levels increase in response to steatosis (1, 6, 29). UCP2 proton conductance is activated in the mitochondria by superoxide and possibly by free fatty acids (FAs) (15, 16, 20). Once activated, mitochondrial proton leak is increased, and the efficiency of ATP synthesis is diminished (7, 27).
The capacity of UCP2 to reduce MMP and circumvent ATP production raises the possibility that the protein increases the risk of hepatocyte cell death following ischemia. We have shown that, in steatotic livers subjected to ischemia/reperfusion (I/R), UCP2 deficiency resulted in increased ATP levels, decreased necrosis, and increased survival (17). Whether UCP2 expression in hepatocytes is responsible for these effects is unknown. To test this, we developed and characterized a model of in vitro steatosis using primary isolated hepatocytes and studied the effects of UCP2 deficiency on MMP, ATP, lipid peroxidation, and cell death following hypoxia and reoxygenation (H/R).
MATERIALS AND METHODS
Animal subjects.
Founders of UCP2 heterozygous mice were a gift from Dr. Bradford Lowell (Beth Israel Deaconess, Harvard, Boston, MA). Genotypes of animals were determined as previously described (36). Leptin knockout (KO) (Ob/Ob) mice were purchased from Jackson Laboratories. Breeding, housing, and surgery all complied with the protocols approved by the Medical University of South Carolina Institutional Animal Care and Use Committee.
Hepatocyte isolation and purification.
Wild-type (WT) and UCP2 KO (male, 9–11 wk old) mice were anesthetized with pentobarbital sodium (50 mg/kg) and placed on a temperature-controlled (37°C) pad (Gaymar). A transverse incision was made, accessing the entire abdomen, and the portal vein was exposed. An 18-gauge cannula was inserted into the portal vein, an extension tube was threaded, and the vena cava was cut to allow liver blood outflow. Three milliliters of heparin (50 IU/ml in saline) were infused into the liver, immediately followed by two 25-ml perfusion stages, the second including collagenase (37°C, 1.5 ml/min, Harvard Apparatus) as described previously (3). Perfused livers were physically manipulated, and the suspension was filtered through a 70-μm cell strainer (Falcon). Hepatocyte purification was performed using density gradient separation (Redigrad, Amersham Bioscience) as previously described (3). The layered isodensity gradient suspension was centrifuged at 4°C at 50 g for 10 min. The pellet was washed, resuspended, counted, and then plated in supplemented William's E medium at 1 × 106 cells/35-mm dish. Viability was confirmed by Trypan blue staining and was >90%.
Hepatocyte culture.
Isolated hepatocytes (1 × 106 cells/35-mm dish) were plated on collagen-coated dishes (BD BioCoat) in William's E medium (with Pen/Strep, 200 μM l-glutamine, 12.5 U/l insulin, 10 μM dexamethasone, 10% FBS) for 2 h to allow attachment. After this period, media and unattached cells were removed and replaced with fresh William's E (control) or William's E supplemented with 6% intralipid (Baxter) for 24 h to establish the steatotic condition. Groups were termed WT control, WT intralipid, KO control, and KO intralipid.
Oil red O staining.
Isolated hepatocytes were assessed visually for fat accumulation by oil red O (ORO) staining as described (14) with slight modification for use in vitro. Plates were washed with PBS, permeabilized with 70% isopropyl alcohol for 3 min, and stained with ORO for 30 min at room temperature. Hepatocytes were washed with 70% isopropyl alcohol, stained with hematoxylin, and washed with lithium carbonate buffer. Cells were immediately photomicrographed at ×20.
Gas chromatography.
Hepatocytes were removed in 0.5 ml of ice-cold saline, and 200 μg of heptadecanoic acid was added as an internal standard. Samples were homogenized, and aliquots were taken for protein analysis. Lipids were extracted twice by the addition of 1 ml of chloroform:methanol (2:1 vol/vol) and centrifuged at 16,000 g for 2 min, and the chloroform layers were removed and combined. The chloroform samples were flushed with N2, and then 1 ml of boron trifluoride (12% wt/wt) in methanol was added. The samples were then heated to 85°C for 45 min to form FA methyl esters. Samples were centrifuged, and the chloroform samples were removed and brought up to 2 ml total volume with chloroform. FAs were quantified using a Hewlett-Packard 5890 gas chromatograph. The gas chromatograph was equipped with a 30-m DB-23 megabore column (J&W Scientific), and the oven temperature program for analysis was 35°C for 2 min followed by an increase to 150°C at a rate of 5°C/min with a 25-min hold at 150°C, followed by an increase to 200°C at a rate of 5°C/min with a 5-min hold. FA methyl esters as both a 37-component FA methyl ester mix and individual compounds (Supelco) were used for FA identification and quantification. FA concentrations were standardized to total protein content for each sample.
H/R.
After the initial 24-h culture period, cells were washed, and medium was replaced with Krebs-Ringer's HEPES buffer (KRH, pH 7.4); plates were placed in a hypoxia chamber (37°C, Po2 < 50 mmHg in medium, COY Laboratories) for 4 h. Samples were collected, and fresh medium was replaced (reoxygenation). Cells were placed back in the air-5% CO2 incubator for 1 or 24 h for reoxygenation (Po2 in media was >140 mmHg). For viability analysis, detached cells were collected, adhered cells were released using trypsin, and all cells were immediately observed for Trypan blue staining.
Mitochondrial labeling and imaging.
Hepatocytes were plated on collagen-coated 35-mm dishes with 1.5 thickness glass coverslip wells (MatTek) and cultured for the initial 24-h period. Hepatocytes were incubated with medium containing 200 nM tetramethylrhodamine methyl ester (TMRM) for 30 min at 37°C, and the media was replaced with KRH, 7.4 pH, containing 50 nM TMRM. Dishes were mounted on a Zeiss 510 laser scanning confocal microscope (Carl Zeiss), and 12-bit fluorescence intensity images of hepatocytes were acquired as described previously (23). After correcting for background intensity, electrical potential was estimated for every pixel of the acquired images using ImageJ 1.37v (NIH). Calculated potentials were used to map the distribution of electrical potential in pseudocolor for each image using Photoshop CS. After the background was subtracted, the fractions of pixels at categorical membrane potentials from these images were calculated. Three or four cells were analyzed from each dish and averaged. Three dishes, each seeded from separate hepatocyte isolations, were included per group.
UCP2 immunoblotting.
Liver mitochondria were isolated using an isolation kit from Sigma (MITO-ISO1), as described. Hepatocytes (50 mg) were homogenized using an ice-cold polytetrafluoroethylene pestle. For a purified “heavy” mitochondrial fraction, the low- and high-speed centrifugation steps were run at 1,000 g and 3,500 g, respectively. Samples were diluted in a 50 mM HEPES buffer, and protein concentrations were measured by BCA assay. Standardized samples were solubilized in lithium dodecyl sulfate, heat denatured, and run on an SDS-PAGE gel. Anti-mouse UCP2 primary antibody (Alpha Diagnostic International) and cyclooxygenase IV primary antibody (Cell Signaling) were incubated at concentrations of 1:1,000 and 1:10,000, respectively, followed by anti-rabbit horseradish peroxidase-linked secondary antibody (Cell Signaling).
Measurement of cellular ATP.
Hepatocytes were lysed with ice-cold buffer (150 mM NaCl, 50 mM Tris, 1% Triton X-100, 0.1% SDS, and 1% deoxycholate, pH 7.5) and then acidified with 1.5% trichloroacetic acid (TCA, for ATPase inhibition). Lysates were centrifuged at 20,000 g, and supernatants were diluted 1:100 in Tris acetate buffer (0.1 mM, containing sodium acetate). Each diluted sample was mixed with reconstituted luciferin-luciferase solution (Enliten, Promega), and the ATP concentration was measured luminometrically. For normalization, total cellular protein from each sample of lysate was determined by BCA assay.
Measurement of lipid peroxidation.
Lipid peroxidation in hepatocytes was monitored as the production of malondialdehyde with slight modifications (32). Cells were scraped in 500 μl of ice-cold PBS. Homogenates were treated with an equal volume of 10% TCA and then centrifuged at 14,000 g. The supernatant was removed, combined with 2-thiobarbituric acid (0.76%), boiled for 10 min, and cooled, and absorbance was measured at 532 nm with 1,1,3,3-tetraethoxypropane as a standard. Results were normalized to cell protein (BCA).
Statistical analysis.
All values are expressed as means + SD, or in box plot format. An α value of 0.05 was established before experimentation as the limit for statistical significance. Each n value represents a plating of cells from an individual animal. For a single pairwise comparison, a two-tailed t-test was used. For multiple independent groups, one-way ANOVA was used, and the posttest correction was done with the Tukey-Kramer multiple-comparison test using JMP 4 statistical software.
RESULTS
Establishment and characterization of hepatocyte steatosis in vitro.
For this study, we established a method for inducing and maintaining steatosis in vitro. In our experience, viability of ob/ob mouse hepatocytes is low compared with WT hepatocytes. Moreover, an in vitro system will eliminate the tedious process of breeding double KO mice that are obese, enabling the study of more proteins. It was reported that treatment of hepatocytes with medium supplemented with 6% intralipid induced UCP2 expression, and this response was not specific to any single FA component of the treatment (9). Intralipid is a pyrogen-free emulsification of FAs (soybean oil- palmitic, steric, oleic, and linoleic acids) designed for intravenous administration. We adopted this method and treated our primary hepatocytes with medium supplemented with 6% intralipid for 24 h. To confirm the phenotype, we measured the profile of fat visually by ORO staining and by gas chromatography mass spectrometry. The treated hepatocytes developed a steatotic phenotype consistent histologically with acute onset steatosis in vivo, including significant microvesicular fat accumulation. Microvesicular fat accumulation was substantial (Fig. 1A), and a broad-spectrum increase in the concentrations of both saturated and unsaturated FAs (i.e., arachidonic, linoleic, oleic palmitic, and steric) was observed (Table 1). A total of 36 FAs were measured, and all were greater (P < 0.05) in the intralipid-treated group compared with the control group with the exception of cis-8,11,14-eicosatrienoic acid, which was near zero in the control group and not detected after intralipid treatment. The steatotic phenotype was not affected by UCP2 deficiency either visually or by gas chromatography mass spectrometry. In WT hepatocytes, 24-h treatment with intralipid increased UCP2 protein 2.9-fold over hepatocytes in control medium (P < 0.05, Fig. 1B). Additionally, intralipid-treated cells maintained their lipid content for up to 24 h following the removal of intralipid, mimicking isolated hepatocytes from Ob/Ob mice (Fig. 2).
Fig. 1.
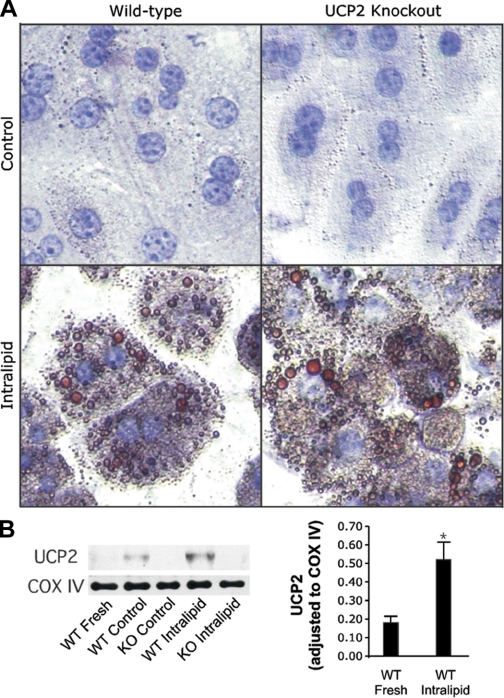
Intralipid treatment induces steatosis and uncoupling protein 2 (UCP2) upregulation in hepatocytes in vitro. Wild-type (WT) and UCP2 knockout (KO) mouse hepatocytes were cultured in the absence (control) or presence of 6% intralipid (emulsified fatty acids) for 24 h. A: cells were stained with oil red O (ORO) to visually assess the development of the steatotic phenotype and were counterstained with hematoxylin. Photomicrographs were captured at ×200. B: mitochondrial isolates were probed for UCP2, and cytochrome c oxidase subunit IV was detected to confirm equivalent loading. Mean values standardized to cyclooxygenase (COX) IV + SD are represented. n = 3/group, *P < 0.05.
Table 1.
Fatty acid concentrations in fresh and cultured (24 h control vs. intralipid) WT and UCP2 KO hepatocytes
| Fresh |
Control Media |
Intralipid Media |
||||
|---|---|---|---|---|---|---|
| Fatty Acids | WT | KO | WT | KO | WT | KO |
| Arachidonic | 5343 ± 1273 | 4507 ± 2015 | 5569 ± 2258 | 5230 ± 1759 | 11716 ± 1190 | 11025 ± 1848 |
| Caprylic | 196 ± 139 | 179 ± 91 | 327 ± 145 | 260 ± 84 | 840 ± 145 | 1010 ± 214 |
| Cis-Docosahexaenoic | 3153 ± 706 | 2765 ± 1607 | 3065 ± 1613 | 3097 ± 1638 | 7866 ± 1425 | 9811 ± 1633 |
| Cis-Eicosatrienoic | 208 ± 288 | 272 ± 241 | 739 ± 298 | 449 ± 257 | 0 ± 0 | 0 ± 0 |
| α Linolenic | 0 ± 0 | 0 ± 0 | 0 ± 0 | 80 ± 180 | 2584 ± 711 | 1982 ± 1094 |
| Linoleic | 8337 ± 1672 | 7743 ± 2684 | 9336 ± 2801 | 8988 ± 2108 | 39065 ± 7387 | 41572 ± 8086 |
| Oleic | 4383 ± 1347 | 4340 ± 1314 | 5036 ± 1882 | 5042 ± 1398 | 22681 ± 3667 | 27839 ± 2646 |
| Palmitic | 9485 ± 1955 | 8494 ± 3104 | 11303 ± 3271 | 9432 ± 2173 | 22918 ± 2793 | 25091 ± 3029 |
| Palmitoleic | 537 ± 292 | 364 ± 158 | 490 ± 293 | 529 ± 242 | 2629 ± 1279 | 2974 ± 1187 |
| Stearic | 5599 ± 821 | 4227 ± 1849 | 6286 ± 1882 | 5773 ± 1750 | 11162 ± 1685 | 13107 ± 1994 |
Data are means ± SD and represent ng/mg protein. For all fatty acids except cis-eicosatrienoic, concentrations are greater in the intralipid treated vs. freshly isolated and control cultured hepatocytes, P < 0.05. Wild-type (WT) vs. knockout (KO) intralipid for all groups, no difference. n = 5/group. UCP2, uncoupling protein-2.
Fig. 2.

Steatosis induced by intralipid is maintained in cultured hepatocytes. WT mouse hepatocytes are cultured in the presence or absence of 6% intralipid for 24 h along with Ob/Ob mouse hepatocytes. Cells are stained with ORO to show fat content.
UCP2 decreases MMP in steatotic hepatocytes.
In isolated mitochondria, UCP2-mediated proton leak is conditional. Little is known in hepatocytes concerning the role of physiological levels of UCP2 on mitochondrial function. To determine the effect of steatosis and UCP2, we labeled hepatocytes with TMRM, converted the fluorescence intensities to ΔΨ values for each cell, and plotted the distribution of ΔΨ (Fig. 3) (23). WT and KO control groups exhibited greater MMP compared with the WT intralipid (steatotic) hepatocytes. The steatotic UCP2 KO hepatocytes showed an intermediate density of highly polarized mitochondria compared with WT and UCP2-expressing steatotic hepatocytes. Estimation of the fraction of surface area with potential <−175 mV revealed that UCP2 KO steatotic hepatocytes had decreased MMP compared with WT controls but was greater than WT steatotic hepatocytes (Table 2). No differences were observed at polarization less than −175 mV. Steatosis decreased the number of highly polarized mitochondria in hepatocytes by 50%, and UCP2 expression accounted for 60% of the decrease.
Fig. 3.
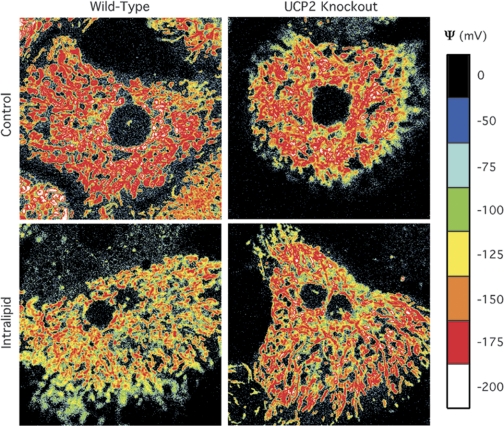
Assessment of UCP2 deficiency on mitochondrial polarization in steatotic hepatocytes. Following the initial 24-h culture in control or intralipid-supplemented medium, hepatocytes were stained with tetramethylrhodamine methyl ester, imaged, and pseudocolor mapped for mitochondrial membrane potential in mV.
Table 2.
Mitochondrial polarization profiles in WT and UCP2 KO hepatocytes
| Control |
Intralipid |
|||
|---|---|---|---|---|
| WT |
KO |
WT |
KO |
|
| ψ, mV | Fraction of Polarized Area, % | |||
| ≤−200 | 3.7 ± 0.7 | 3.6 ± 0.8 | 0.5 ± 0.4* | 1.8 ± 0.5† |
| ≤−175 to −200 | 20.8 ± 0.5 | 20.6 ± 1.7 | 11.2 ± 2.4* | 17.5 ± 0.5† |
| ≤−150 to −175 | 21.6 ± 0.9 | 20.6 ± 1.2 | 22.4 ± 2.1 | 20.2 ± 0.3 |
| ≤−125 to −150 | 18.3 ± 1.4 | 17.4 ± 0.8 | 17.3 ± 4.9 | 20.3 ± 2.9 |
| ≤−100 to −125 | 12.8 ± 0.1 | 9.9 ± 2.5 | 13.3 ± 2.1 | 15.3 ± 2.7 |
| ≤−75 to −100 | 9.9 ± 0.1 | 9.1 ± 0.5 | 10.9 ± 3.6 | 12.7 ± 3.3 |
| ≤−50 to −75 | 11.6 ± 1.5 | 10.4 ± 3.5 | 10.8 ± 3.9 | 9.9 ± 0.1 |
Data are means ± SD.
P < 0.05 vs. KO intralipid and media control,
P < 0.05 vs. media control, n = 3/group.
UCP2 deficiency protects against H/R-induced cell death in steatotic hepatocytes.
UCP2 possesses the ability to protect against acute oxidative stressors, including ischemia in some models (8, 27, 34). Our previous reports showed that steatotic livers are more susceptible to injury following I/R than lean livers, correlating directly to the expression of UCP2 (6, 17). To determine the effect of UCP2 on hepatocellular tolerance to H/R, we subjected our cultured (24-h with or without intralipid) hepatocytes to 4 h of hypoxia followed by 24 h of reoxygenation. After the initial culture, the development of steatosis with or without UCP2 had no effect on baseline viability (Fig. 4), nor did it decrease viability 1 h after reoxygenation (not shown). At 24 h following reoxygenation, cell death increased in all groups over baseline and 1 h. Percent death in the WT control hepatocytes was 20%, which was not different than the KO control group. In the WT steatotic hepatocytes, an increased percentage of cell death was observed (32%, P < 0.05), whereas cell death in steatotic UCP2 KO hepatocytes was not different than that of WT or UCP2 KO controls at 24 h.
Fig. 4.

The effect of UCP2 deficiency on steatotic hepatocyte viability following hypoxia/reoxygenation. WT and UCP2 KO hepatocytes were incubated in control or intralipid-supplemented medium for 24 h (baseline) and subjected to 4 h of hypoxia, and survival was measured by Trypan exclusion at baseline and at 24 h following reoxygenation, as represented by box plot. n = 5/group; a,b,cmeans with different superscripts are significantly different; P < 0.05.
Steatotic hepatocyte ATP concentrations are increased with UCP2 deficiency following H/R.
We have shown that ATP levels in steatotic livers are reduced compared with lean livers and that ATP levels do not recover following the ischemic event (6, 17). Other reports suggest that UCP2 may reduce ATP production in stress settings where UCP2 becomes fully activated (18, 25). At baseline, no differences in ATP levels were observed (Fig. 5A). At the end of hypoxia, ATP concentrations decreased equally in all groups. One hour following reoxygenation, ATP concentrations in the control groups and the steatotic UCP2 KO group recovered to baseline levels, which was maintained at 24 h (Fig. 5A). At 1 h, ATP concentrations in the steatotic WT group did not increase above those observed at the end of hypoxia and remained at this level at 24 h. ATP levels in control and UCP2 KO steatotic hepatocytes were ∼2.5-fold higher than in those observed in steatotic WT hepatocytes following 1 h of reoxygenation.
Fig. 5.

The effect of UCP2 deficiency on ATP concentrations and lipid peroxidation following hypoxia/reoxygenation in steatotic hepatocytes. At the end of the initial 24-h incubation (baseline), at the end of hypoxia (immediate), and 1 and 24 h after reoxygenation, cellular ATP concentrations were measured by chemiluminescent assay, and lipid peroxidation was monitored by thiobarbituric acid reactive substances (TBARS) measurement. A: levels of ATP are plotted over time on the line graph as means + SD, and the 1-h time point is expanded on the right in box plot format. n = 6/group. B: lipid peroxidation was analyzed by TBARS measurement, and illustrated as in A. n = 3/group (baseline) and 5/group (all other points); a,b,cmeans with different superscripts are significantly different; P < 0.05.
Lipid peroxidation is increased in steatotic hepatocytes following H/R but is not affected by UCP2.
To determine the effects of steatosis and UCP2 on lipid peroxidation, we measured thiobarbituric acid reactive substances before and throughout H/R. At baseline, lipid peroxidation was very low and not different between groups. Lipid peroxidation did not increase immediately following hypoxia; however, it increased following 1 h of reoxygenation (Fig. 5B). Lipid peroxidation increased 4.7-fold in the WT and KO control hepatocytes over baseline. Lipid peroxidation was 9.4-fold higher in WT steatotic hepatocytes compared with baseline levels and was not different from steatotic UCP2 KO hepatocytes. We suggest that, although steatosis itself increases lipid peroxidation after H/R and that lipid peroxidation plays a role in H/R-induced cell death, UCP2 does not affect H/R-induced lipid peroxidation. By 24 h, the levels of lipid peroxidation recovered in all groups.
DISCUSSION
We demonstrated the development of steatosis in primary cultured hepatocytes using intralipid. Interestingly, we noted an increase in the concentrations of FAs that are not significant components of the intralipid including palmitoleic, arachidonic, docosadienoic, and linolenic acid derivatives. This indicates that the phenotype is not a product of simple fat uptake and that de novo synthesis and/or FA metabolism occurred. Such profiles have not been described when comparing forms, extents, or models of liver steatosis, so we cannot comment on the significance of any single FA concentration or the profile as a whole. However, the visible fat accumulation, along with the FA profiles represent the definition of steatosis. In addition, FAs such as arachidonic acid have been shown to be bioactive. For example, arachidonic acid is associated with mitochondrial permeability transition (MPT) in succinate-fed liver mitochondria (13). The FA concentrations were not different between the WT and UCP2 KO steatotic hepatocytes, suggesting that outcomes attributable to UCP2 were not FA dependent.
Using this model, we show that hepatocyte steatosis and UCP2 expression do not affect viability, ATP concentrations, or lipid peroxidation in resting hepatocytes. Although UCP2 had no effect on the development of lipid peroxidation in hepatocytes, UCP2 expression may be a beneficial adaptation in the steatotic liver under basal conditions for the maintenance of low-level ROS. However, steatosis reduced survival and ATP levels following H/R, and UCP2 expression affected both. This is consistent with the idea that the upregulation of UCP2 associated with the steatosis leads to a decline in mitochondrial function in response to hypoxia. In the steatotic hepatocytes, ATP levels were unable to rebound after hypoxia, whereas ATP concentrations in the steatotic UCP2 KO and control hepatocytes recovered equally. Steatosis resulted in a greater development of lipid peroxidation after reoxygenation, whereas UCP2 expression was inconsequential. We previously established that UCP2 is upregulated in fatty livers and that these livers are more sensitive to I/R (6, 17). In livers of ob/ob mice, UCP2 deficiency resulted in increased levels of ATP, increased survival, and no differences in lipid peroxidation after I/R (17). Thus the results from this study suggest that the effects are hepatocyte related.
Because the number of mitochondria with very high MMP was higher in steatotic UCP2 KO hepatocytes compared with WT steatotic hepatocytes, it was possible that increased oxidative damage and decreased ATP levels could result. Neither was observed at baseline, indicating that UCP2 has little effect on ATP availability and that ROS production is not sufficient to cause damage. The baseline trends are likely not coincidental, as the activation of UCP2 is dependent on mitochondrial ROS. UCP2 deficiency did not affect the observed lipid peroxidation within hepatocytes after reoxygenation, and cell death was greater in the steatotic hepatocytes. Therefore, the lack of recovery of ATP in WT steatotic hepatocytes after H/R may be a response to full UCP2 activation attributable to increased ROS exposure during reoxygenation. Products of lipoperoxidation have been shown to activate UCP2 and support its uncoupling activity (20, 28). Alternatively, the lack of ATP in these cells may hinder the activity of cellular antioxidative defense although one would expect greater lipid peroxidation in the steatotic WT group compared with the UCP2 KO group after H/R, which was not observed.
In other tissues and models, studies conflict on the role of UCP2. For example, UCP2 overexpression was shown to be protective in models of oxidative stress in brain, cardiomyocytes, and HepG2 cells (8, 27, 34). Protection was primarily attributed to decreases in proapoptotic signaling secondary to UCP2 decreasing ROS production and indirectly reducing Ca2+ exposure. In contrast, UCP2 deficiency protected against brain ischemia in one model of cerebral infarction, and overexpression resulted in increased injury in a model of cardiomyocyte hypoxia (5, 12). In mesencephalic and cortical cells, upregulation of UCP2 was associated with necrosis as a result of cyanide toxicity, which depleted ATP and reduced MMP (24, 25). These examples illustrate that, under different conditions, UCP2 can have opposing effects in different tissues under different stressors. However, these examples are mainly based on induced UCP2 protein overexpression, whereas our model is one of deficiency and steatosis-induced upregulation.
Using whole cell systems and isolated mitochondria, including those from liver, Trenker et al. (35) have recently shown that UCP2 can act as a mitochondrial Ca2+ uniporter. UCP2 overexpression had no effect on mitochondrial Ca2+ uptake, MMP, or ATP production, but mitochondrial Ca2+ sequestration was increased following histamine stimulation, independent of ROS exposure. Histamine stimulation in UCP2 overexpressing cells caused an increase in ATP synthesis, which was thought to be Ca2+ induced. The consequence of Ca2+ transport by UCP2 during oxidant stress is unknown with respect to the development of Ca2+-dependent signaling in apoptosis. In other systems, UCP2 was shown to regulate apoptosis in stress settings. In brain, UCP2 overexpression protected against ischemic stress, and it was concluded that reductions in ATP led to decreases in cellular Ca2+ import, thus protecting against apoptosis (27). When cellular transport of Ca2+ is subordinate to endoplasmic reticulum Ca2+ release for the initiation of apoptosis, loss of UCP2 could be beneficial if it is responsible for mitochondrial Ca2+ uptake. In the case of necrosis, UCP2-mediated mitochondrial Ca2+ uptake could accelerate MPT activation, and the loss of UCP2 could be protective (19). However, the effects of UCP2 on MPT activation during stress are difficult to predict, as MPT is stimulated by changes in MMP as well as free radicals, which are both regulated by UCP2 through its better described function as a H+ channel (22). If UCP2 in hepatocytes does act as a Ca2+ transporter, it is possible that this function may be responsible for some of the effects we observed.
In the steatotic liver, it was also shown that UCP2 deficiency in ob/ob mice reduced apoptosis and alanine transferase release after challenge with Jo2 (Fas agonist) (18). Fas ligand has been identified as an apoptotic signal in livers after I/R (10). The reduced level of apoptosis in the Jo2 study was mirrored by increases in ATP in the UCP2-deficient livers after challenge, providing evidence for a link between UCP2 and apoptosis in the steatotic liver. The cell death induced by H/R can be both apoptotic and necrotic and is linked to the MPT (21). The conclusions from this study were much like those from our in vivo studies. The results of these in vitro studies show that the beneficial effects of UCP2 deficiency in the liver apply directly to the hepatocyte.
GRANTS
This work was funded by a grant from the National Institutes of Health, number DK069369.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
We thank Kathy Haines and Zainab Amani for laboratory support.
REFERENCES
- 1. Baffy G, Zhang CY, Glickman JN, Lowell BB. Obesity-related fatty liver is unchanged in mice deficient for mitochondrial uncoupling protein 2. Hepatology 35: 753–761, 2002 [DOI] [PubMed] [Google Scholar]
- 2. Behrns KE, Tsiotos GG, DeSouza NF, Krishna MK, Ludwig J, Nagorney DM. Hepatic steatosis as a potential risk factor for major hepatic resection. J Gastrointest Surg 2: 292–298, 1998 [DOI] [PubMed] [Google Scholar]
- 3. Berry MN, Barritt GJ, Edwards AM. Monolayer Culture of Hepatocytes. New York: Elsevier, 1991 [Google Scholar]
- 4. Birsner JH, Wan C, Cheng G, Evans ZP, Polito CC, Fiorini RN, Gilbert G, Haines JK, Schmidt MG, Chavin KD. Steatotic liver transplantation in the mouse: a model of primary nonfunction. J Surg Res 120: 97–101, 2004 [DOI] [PubMed] [Google Scholar]
- 5. Bodyak N, Rigor DL, Chen YS, Han Y, Bisping E, Pu WT, Kang PM. Uncoupling protein-2 modulates cell viability in adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol 293: H829–H835, 2007 [DOI] [PubMed] [Google Scholar]
- 6. Chavin KD, Yang S, Lin HZ, Chatham J, Chacko VP, Hoek JB, Walajtys-Rode E, Rashid A, Chen CH, Huang CC, Wu TC, Lane MD, Diehl AM. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J Biol Chem 274: 5692–5700, 1999 [DOI] [PubMed] [Google Scholar]
- 7. Cheng G, Polito CC, Haines JK, Shafizadeh SF, Fiorini RN, Zhou X, Schmidt MG, Chavin KD. Decrease of intracellular ATP content downregulated UCP2 expression in mouse hepatocytes. Biochem Biophys Res Commun 308: 573–580, 2003 [DOI] [PubMed] [Google Scholar]
- 8. Collins P, Jones C, Choudhury S, Damelin L, Hodgson H. Increased expression of uncoupling protein 2 in HepG2 cells attenuates oxidative damage and apoptosis. Liver Int 25: 880–887, 2005 [DOI] [PubMed] [Google Scholar]
- 9. Cortez-Pinto H, Zhi Lin H, Qi Yang S, Odwin Da Costa S, Diehl AM. Lipids upregulate uncoupling protein 2 expression in rat hepatocytes. Gastroenterology 116: 1184–1193, 1999 [DOI] [PubMed] [Google Scholar]
- 10. Cursio R, Filippa N, Miele C, Colosetti P, Auberger P, Van Obberghen E, Gugenheim J. Fas ligand expression following normothermic liver ischemia-reperfusion. J Surg Res 125: 30–36, 2005 [DOI] [PubMed] [Google Scholar]
- 11. D'Alessandro AM, Kalayoglu M, Sollinger HW, Hoffmann RM, Reed A, Knechtle SJ, Pirsch JD, Hafez GR, Lorentzen D, Belzer FO. The predictive value of donor liver biopsies for the development of primary nonfunction after orthotopic liver transplantation. Transplantation 51: 157–163, 1991 [DOI] [PubMed] [Google Scholar]
- 12. de Bilbao F, Arsenijevic D, Vallet P, Hjelle OP, Ottersen OP, Bouras C, Raffin Y, Abou K, Langhans W, Collins S, Plamondon J, Alves-Guerra MC, Haguenauer A, Garcia I, Richard D, Ricquier D, Giannakopoulos P. Resistance to cerebral ischemic injury in UCP2 knockout mice: evidence for a role of UCP2 as a regulator of mitochondrial glutathione levels. J Neurochem 89: 1283–1292, 2004 [DOI] [PubMed] [Google Scholar]
- 13. Di Paola M, Lorusso M. Interaction of free fatty acids with mitochondria: coupling, uncoupling and permeability transition. Biochim Biophys Acta 1757: 1330–1337, 2006 [DOI] [PubMed] [Google Scholar]
- 14. Drury R, Wallington E. Carleton's Histological Technique. New York: Oxford University, 1980 [Google Scholar]
- 15. Echtay KS, Murphy MP, Smith RA, Talbot DA, Brand MD. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem 277: 47129–47135, 2002 [DOI] [PubMed] [Google Scholar]
- 16. Echtay KS, Roussel D, -Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature 415: 96–99, 2002 [DOI] [PubMed] [Google Scholar]
- 17. Evans ZP, Ellett JD, Schmidt MG, Schnellmann RG, Chavin KD. Mitochondrial uncoupling protein-2 mediates steatotic liver injury following ischemia/reperfusion. J Biol Chem 283: 8573–8579, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fulop P, Derdak Z, Sheets A, Sabo E, Berthiaume EP, Resnick MB, Wands JR, Paragh G, Baffy G. Lack of UCP2 reduces Fas-mediated liver injury in ob/ob mice and reveals importance of cell-specific UCP2 expression. Hepatology 44: 592–601, 2006 [DOI] [PubMed] [Google Scholar]
- 19. Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 195: 460–467, 1979 [DOI] [PubMed] [Google Scholar]
- 20. Jaburek M, Miyamoto S, Di Mascio P, Garlid KD, Jezek P. Hydroperoxy fatty acid cycling mediated by mitochondrial uncoupling protein UCP2. J Biol Chem 279: 53097–53102, 2004 [DOI] [PubMed] [Google Scholar]
- 21. Kim JS, Qian T, Lemasters JJ. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology 124: 494–503, 2003 [DOI] [PubMed] [Google Scholar]
- 22. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 87: 99–163, 2007 [DOI] [PubMed] [Google Scholar]
- 23. Lemasters JJ, Ramshesh VK. Imaging of mitochondrial polarization and depolarization with cationic fluorophores. Methods Cell Biol 80: 283–295, 2007 [DOI] [PubMed] [Google Scholar]
- 24. Li L, Prabhakaran K, Mills EM, Borowitz JL, Isom GE. Enhancement of cyanide-induced mitochondrial dysfunction and cortical cell necrosis by uncoupling protein-2. Toxicol Sci 86: 116–124, 2005 [DOI] [PubMed] [Google Scholar]
- 25. Li L, Prabhakaran K, Zhang X, Borowitz JL, Isom GE. PPARalpha-mediated upregulation of uncoupling protein-2 switches cyanide-induced apoptosis to necrosis in primary cortical cells. Toxicol Sci 93: 136–145, 2006 [DOI] [PubMed] [Google Scholar]
- 26. Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology 43: S31–S44, 2006 [DOI] [PubMed] [Google Scholar]
- 27. Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med 9: 1062–1068, 2003 [DOI] [PubMed] [Google Scholar]
- 28. Murphy MP, Echtay KS, Blaikie FH, Asin-Cayuela J, Cocheme HM, Green K, Buckingham JA, Taylor ER, Hurrell F, Hughes G, Miwa S, Cooper CE, Svistunenko DA, Smith RA, Brand MD. Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: studies using a mitochondria-targeted spin trap derived from alpha-phenyl-N-tert-butylnitrone. J Biol Chem 278: 48534–48545, 2003 [DOI] [PubMed] [Google Scholar]
- 29. Negre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Penicaud L, Casteilla L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J 11: 809–815, 1997 [PubMed] [Google Scholar]
- 30. Pecqueur C, Alves-Guerra MC, Gelly C, Levi-Meyrueis C, Couplan E, Collins S, Ricquier D, Bouillaud F, Miroux B. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J Biol Chem 276: 8705–8712, 2001 [DOI] [PubMed] [Google Scholar]
- 31. Rashid A, Wu TC, Huang CC, Chen CH, Lin HZ, Yang SQ, Lee FYJ, Diehl AM. Mitochondrial proteins that regulate apoptosis and necrosis are induced in mouse fatty liver. Hepatology 29: 1131–1138, 1999 [DOI] [PubMed] [Google Scholar]
- 32. Schnellmann RG. Mechanisms of t-butyl hydroperoxide-induced toxicity to rabbit renal proximal tubules. Am J Physiol Cell Physiol 255: C28–C33, 1988 [DOI] [PubMed] [Google Scholar]
- 33. Selzner M, Rudiger HA, Sindram D, Madden J, Clavien PA. Mechanisms of ischemic injury are different in the steatotic and normal rat liver. Hepatology 32: 1280–1288, 2000 [DOI] [PubMed] [Google Scholar]
- 34. Teshima Y, Akao M, Jones SP, Marban E. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ Res 93: 192–200, 2003 [DOI] [PubMed] [Google Scholar]
- 35. Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol 9: 445–452, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, Zheng XX, Wheeler MB, Shulman GI, Chan CB, Lowell BB. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105: 745–755, 2001 [DOI] [PubMed] [Google Scholar]