

Abstract
Background
Malaria continues to be one of the most severe global infectious diseases, responsible for 1-2 million deaths yearly. The rapid evolution and spread of drug resistance in parasites has led to an urgent need for the development of novel antimalarial targets. Proteases are a group of enzymes that play essential roles in parasite growth and invasion. The possibility of designing specific inhibitors for proteases makes them promising drug targets. Previously, combining a comparative genomics approach and a machine learning approach, we identified the complement of proteases (degradome) in the malaria parasite Plasmodium falciparum and its sibling species [1-3], providing a catalog of targets for functional characterization and rational inhibitor design. Network analysis represents another route to revealing the role of proteins in the biology of parasites and we use this approach here to expand our understanding of the systems involving the proteases of P. falciparum.
Results
We investigated the roles of proteases in the parasite life cycle by constructing a network using protein-protein association data from the STRING database [4], and analyzing these data, in conjunction with the data from protein-protein interaction assays using the yeast 2-hybrid (Y2H) system [5], blood stage microarray experiments [6-8], proteomics [9-12], literature text mining, and sequence homology analysis. Seventy-seven (77) out of 124 predicted proteases were associated with at least one other protein, constituting 2,431 protein-protein interactions (PPIs). These proteases appear to play diverse roles in metabolism, cell cycle regulation, invasion and infection. Their degrees of connectivity (i.e., connections to other proteins), range from one to 143. The largest protease-associated sub-network is the ubiquitin-proteasome system which is crucial for protein recycling and stress response. Proteases are also implicated in heat shock response, signal peptide processing, cell cycle progression, transcriptional regulation, and signal transduction networks.
Conclusions
Our network analysis of proteases from P. falciparum uses a so-called guilt-by-association approach to extract sets of proteins from the proteome that are candidates for further study. Novel protease targets and previously unrecognized members of the protease-associated sub-systems provide new insights into the mechanisms underlying parasitism, pathogenesis and virulence.
Background
Malaria remains a major threat to health and economic development in endemic countries, infecting 300-500 million people yearly and claiming 1-2 million deaths, primarily of young children. Symptoms of malaria include high fever, shaking chills, headache, vomiting, and anemia. If left untreated, malaria can quickly become life threatening by disrupting the blood supply to vital organs. Malaria is caused by a group of parasites from the genus Plasmodium. Five species, P. falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi, are known to cause the disease in humans. P. falciparum is the most devastating and widespread species.
No effective anti-malaria vaccines are available for use in humans [13]. For decades, the management of malaria has relied heavily on chemotherapy, which uses a limited number of drugs. However, the rapid evolution and spread of drug resistance in parasites has led to an increase in morbidity and mortality rates in malaria endemic regions. The development of new drug/vaccine targets is urgently needed.
Thanks to the completion of the genome sequencing projects for P. falciprum and its sibling species [14-19], a novel array of proteins have been proposed as potential drug targets, including (1) proteins like 1-deoxy-D-xylulose 5-phosphate (DOXP) reductoisomerase [20,21], and apicoplast gyrase [22] that are located in the apicoplast, an organelle with its origin close to the chloroplast; (2) kinases such as cyclin-dependent protein kinases (Pfmrk) [23] and the plant-like calcium-dependent protein kinase (PfCDPK5) [24]; (3) transporters involved in drug resistance and nutrient acquisition from the host [25-30], and (4) proteases.
Proteases are a group of enzymes that degrade proteins by breaking peptide bonds. They are attractive antimalarial targets due to their indispensible roles in parasite development and invasion [31,32]. Previously we predicted the protease complement (degradome) in the malaria parasite P. falciparum and its four sibling species using a comparative genomics approach and a support vector machine (SVM)-based, supervised machine learning approach [1-3]. This catalog revealed a new line of novel proteases for functional characterization. Studies on malarial proteases have been focused on biochemical and molecular characterization [33-46], structural modeling and analysis [47,48], and inhibitor design and screening [49-59]. Although significant progress has been made, much remains to be learned about the roles played by these proteins, including how they interact with other proteins in space and time to coordinate important aspects of growth, transmission, invasion, response to drug treatment and pathogenesis of this devastating pathogen.
One approach to gaining wider views on the roles of proteins in biological systems relies on network biology. Known and inferred protein associations are used to build a network of proteins, thus establishing a map of all the associations in the organism and allowing deductions to be made as to the role of proteins that are poorly understood and poorly annotated. Clearly, both proposed and demonstrated protein-protein associations could aid us in understanding the role of a protease in the parasite. Therefore, we constructed a network of P. falciparum proteins using the protein-protein association data from STRING database [4], and analyzed these data, in conjunction with the data from protein-protein interaction assays using the yeast 2-hybrid (Y2H) system [5], blood stage microarray experiments [6-8], proteomics [9-12], literature text mining, and sequence homology analysis. The topology of the protein-protein association network was analyzed and the results examined for information as to how the proteases may function within the parasite. Sets of proteins associated with specific proteases or protease families were extracted from the whole-cell network to create protease-associated subnetworks and five of these subnetworks were examined in detail. Novel protease targets and previously unrecognized members of some sub-systems could be postulated; these insights help us to better understand the mechanisms underlying parasite metabolism, cell cycle regulation, invasion and infection.
Results and discussion
Proteases are involved in complex networks
We downloaded and mined the protein-protein association data from the STRING database [4] involving proteins from P. falciparum. Seventy-seven (77) out of 124 predicted proteases were found in this set and were associated with at least one other protein, constituting 2,431 associations (Additional Files 1 and 2). Each association between a pair of proteins has a confidence score (S) ranging from 0.15 to 0.999 that was inferred from the evidence used to establish the association: 221 associations (9.1%) have high confidence scores (S>0.7), 432 associations (17.8%) have medium confidence scores (0.4≤S≤0.7), and strikingly, 1,778 associations (73.1%) have relative low confidence scores (0.15≤S<0.4). The large proportion of low-scored associations arises from the paucity of annotation data. Before the genome of P. falciparum was sequenced, only about 20 proteins had been characterized; after genome sequencing this number increased by two orders of magnitude, but over 60% of the predicted gene products in the genome still had no functional assignment [18] and ten years of subsequent effort have reduced this number to roughly 45% [60]. Consequently, information such as KEGG pathway assignments, PDB protein structures and reactome data, which tend to improve association scores, is scarce for P. falciparum. Therefore, our subsequent analysis will not exclude the associations with low confidence scores as they may well represent associations that have not been previously recognized.
The degrees of connectivity vary among the 77 predicted proteases with protein-protein associations, ranging from one to 143 (Additional File 1). Twenty-four (24) putative proteases have less than five association partners, 13 have 5-10 partners, and 40 are highly connected with more than 11 partners, suggesting that proteases are involved in complex cellular networks. Functional enrichment analysis [61] revealed that 120 Gene Ontology (GO) terms were over-represented in these protease associations (p < 0.05) (Additional File 3). Figure 1 shows the distribution of functional categories in a hierarchical order: proteolysis (GO 6508) is, not surprisingly, enriched (p = 8.29*10-6), while the other most highly represented GO biological processes (p < 10-5) are related to cellular catabolic processes (GO 44248), protein metabolic processes (GO 19538), macromolecule metabolic processes (GO 43170), and cofactor and coenzyme metabolic processes (GO 51186 and 6732). This result reflects the involvement of proteases in fundamental biological processes, many of which have been established in the wet lab. Other processes that are moderately enriched (10-5<p < 0.05) included gene expression and response to endogenous and external stimuli such as heat, abiotic stimulus, organic substances, unfolded proteins, and protein stimuli. Five of the potentially most significant protease-associated sets of proteins are discussed in the following sections. They include the ubiquitin-proteasome system, the stress response system, the regulated intramembrane proteolysis system, the parasite egress network, and the signal peptidase network. These subnetworks were chosen because: (1) proteases are the central players in these networks; (2) These networks play crucial roles in parasite life cycle and are closely associated with adaptive phenotypes such as stress response, transcriptional regulation, pathogenesis, and virulence; (3) These networks are considered to be potential antimalarial targets as their disruption would cause deleterious effects on the growth or infectivity of the parasites.
Figure 1.

A graphical representation of the results of a Gene Ontology analysis done using BiNGO. The node size is proportional to the number of proteins represented by that GO term. The color represents the P-value for each enriched GO term as shown in the scale; white nodes are not enriched. The nodes are positioned to approximate their level in the Gene Ontology.
The ubiquitin-proteasome system (UPS)
The largest protease-associated network in P. falciparum is the ubiquitin-proteasome protein degradation system (UPS). The UPS is responsible for degrading unwanted or misfolded proteins and is believed to execute important roles in protein turnover and cell cycle regulation in a wide variety of organisms [62]. We previously identified a group of threonine proteases that form α- and β- subunits of the proteasome complex and two families of ubiquitin-specific hydrolases (C12 and C19) [1,63] (Additional File 1). The UPS pathway in P. falciparum has been deduced by Dr. Hagai Ginsburg (http://sites.huji.ac.il/malaria/maps/proteaUbiqpath.html), and involves two consecutive steps: (1) tagging the ubiquitin molecules to target proteins and (2) degradation of the tagged protein by the proteasome complex with release and recycling of ubiquitin. The major components of the UPS in P. falciparum are conserved with other eukaryotes. However, a growing body of evidence suggests that the UPS plays a critical role in the parasite-specific life style and it is therefore intriguing to unveil the proteins and pathways that are associated with or regulated by the UPS [64,65], as they may carry out functions specific to pathogenesis or virulence. We identified 1,148 associations in P. falciparum that involved 11 threonine proteases in the T1 family, two proteases in the C12 ubiquitin C-terminal hydrolase family, and six proteases in the C19 ubiquitin-specific protease family. One hundred and twenty-four (124) associations are protease-protease associations, and the remaining 1,024 associations involve non-protease partners. One hundred and sixty-four (164) of these associations have high confidence scores (S>0.7), the majority of which involve the association between catalytic components and regulatory components in the proteasome complex (Additional File 4).
The protease with the highest connectivity is PF10_0111, a putative 20S proteasome beta subunit, which has 143 association partners (Figure 2). In addition to the proteasome components and ubiquitin conjugation enzymes, the other interacting proteins appear to be involved in a variety of activities (Table 1): (1) a nucleotide binding activity involving a Tat binding protein homolog (PFL2345c) which has an AAA ATPase domain; (2) cell cycle regulation involving MAL13P1.337, which is a putative protein in the Skp1 (S-phase kinase-associated protein 1) family. The Skp1 protein is an important component of the cyclin A-CDK2 S phase kinase complex in baker's yeast (Saccharomyces cerevisiae) [66] and directs cell cycle-regulated proteins to the kinetochore; (3) translation involving a number of ribosomal proteins such as 60S ribosomal proteins L40/UBI (PF13_0346) and L10 (PF14_0141), a putative translation initiation factor eIF-1A (PF11_0447), and a putative elongation factor 1 (EF-1) (PFC0870w); (4) transcriptional regulation involving a putative multiprotein bridging factor type 1 (MBF1) (PF11_0293). MBF1 is a transcriptional cofactor that bridges the TATA box-binding protein (TBP) and its specific regulatory proteins for transcriptional activation [67]; (5) membrane traffic regulation involving a putative rab specific GDP dissociation inhibitor (PFL2060c) [68].
Figure 2.

A graph showing the proteins associated with PF10_0111. This protease is the most highly connected member of the set of proteases found in the P. falciparum ubiquitin-proteasome protein degradation system. Square nodes represent proteases. Node size is proportional to the degree of the node. Nodes are colored according to their functional classification in the eggNOG database [122]. The COG categories are [123] (J) Translation, ribosomal structure and biogenesis, (A) RNA processing and modification, (K) Transcription, (L) Replication, recombination and repair, (B) Chromatin structure and dynamics, (D) Cell cycle control, cell division, chromosome partitioning, (Y) Nuclear structure, (V) Defense mechanisms, (T) Signal transduction mechanisms, (M) Cell wall/membrane/envelope biogenesis, (N) Cell motility, (Z) Cytoskeleton, (W) Extracellular structures, (U) Intracellular trafficking, secretion, and vesicular transport, (O) Posttranslational modification, protein turnover, chaperones, (C) Energy production and conversion, (G) Carbohydrate transport and metabolism, (E) Amino acid transport and metabolism, (F) Nucleotide transport and metabolism, (H) Coenzyme transport and metabolism, (I) Lipid transport and metabolism, (P) Inorganic ion transport and metabolism, (Q) Secondary metabolites biosynthesis, transport and catabolism, (R) General function prediction only, and (S) Function unknown. Confidence scores for the interactions among the nodes (S values from STRING) were divided into three groups - low (0.150-0.399), medium (0.400-0.700) and high (0.701-0.999); the groups are represented by thin, medium and heavy lines, respectively.
Table 1.
Representative P. falciparum proteins that are associated with PF10_0111, a putative 20S proteasome beta subunit with the highest connectivity. Protein-protein interactions revealed by yeast 2-hybrid assays are italicized.
| Functional description | Protein accession number | Annotation |
|---|---|---|
| Nucleotide binding | PFL2345c | tat-binding protein homolog |
| Cell cycle regulation | MAL13P1.337 | putative Skp1 family protein |
| Transcriptional regulation | PF11_0293 | putative multiprotein bridging factor type 1 |
| PF11_0477 | CCAAT-box DNA binding protein subunit B | |
| Translation | PF13_0346 | putative 60S ribosomal protein L40/UBI |
| PF14_0141 | putative 60S ribosomal protein L10 | |
| PF11_0447 | putative translation initiation factor eIF-1A | |
| PFC0870w | putative elongation factor 1 (EF-1) | |
| PFE0350c | 60S ribosomal protein L4 | |
| PF14_0270 | putative apicoplast ribosomal protein L15 precursor | |
| PF11_0245 | putative translation elongation factor EF-1, subunit alpha | |
| Protein transport | PFL2060c | rab specific GDP dissociation inhibitor |
| Protein modification | MAL7P1.19 | putative ubiquitin transferase |
| Surface antigens | PF10_0345 | merozoite surface protein 3 |
| PF10_0345 | merozoite surface protein 3 | |
| PF10_0348 | duffy binding-like merozoite surface protein | |
| PFE0060w | parasite-infected erythrocyte surface protein | |
| Unknown | MAL7P1.170 | Plasmodium exported protein, unknown function |
Moreover, the yeast 2-hybrid assay using PF10_0111 as a bait revealed 15 PPI preys (Table 1), confirming that it is associated with (1) transcriptional regulation involving a CCAAT-box DNA binding protein subunit B (PF11_0477) containing a histone-like transcription factor domain, and (2) translation involving a putative translation elongation factor EF-1 subunit alpha (PF11_0245), a putative 60S ribosomal protein L4 (PFE0350c), and a putative ribosomal protein L15 precursor predicted to localize to the apicoplast (PF14_0270), a specific organelle of prokaryotic origin found in Apicomplexa parasites. PF10_0111 may also be associated with protein modifications involving a putative ubiquitin transferase (MAL7P1.19) [69] and chromatin fluidity involving a putative nucleosome assembly protein (PFI0930c).
Interestingly, PF10_0111 is shown to have PPI with three predicted surface antigens: (1) merozoite surface protein 3 (PF10_0345), which was shown by global RNA decay and nuclear run-on assays to serve a role in transcriptional regulation and RNA stabilization [70,71]; (2) a merozoite surface protein (PF10_0348). Domain analysis revealed a N-terminus Duffy binding domain that is present in the Duffy receptors expressing blood group surface determinants and a C-terminus SPAM (secreted polymorphic antigen associated with merozoites) domain, both of which have been implicated in parasite immune evasion, cytoadherence and pathogenesis [72,73]; (3) a parasite-infected erythrocyte surface protein (PFE0060w). The microarray and proteomics assays show that these three surface proteins are expressed at the invasive merozoite stage [6,8,10,11].
These results reflect much that is known about the UPS, but also suggest that it may also be associated with a variety of processes ranging from transcriptional regulation, translation, cell cycle progression, invasion, protein trafficking, and immune evasion. Not surprisingly, the UPS has become a promising antimalarial target. Various independent studies have shown that inhibition of proteasome activity can arrest parasite growth, and yet show limited toxicity to human cell lines [64,74,75].
Stress response network
The adaptation of the malaria parasite to the host environment requires a rapid and effective response to diverse physiological signals and stress conditions, such as changes in temperature within hosts, nutritional challenges, host immune responses, antimalarial administration, and so on. One such adaptive network in the malaria parasite is the robust heat shock response system. During its life cycle, the parasite is transmitted from the mosquito vector (~25°C) to the human host (37°C), resulting in heat shock. Periodic fever, the patient's response to infection, also presents recurrent heat shock to the parasite. A comprehensive chaperone system has been identified in P. falciparum genome, accounting for 2% of the open reading frames (ORFs) [76]. The system is comprised of various chaperone proteins [77] and includes proteases that degrade misfolded proteins. We identified 344 associations involving five putative proteases in the ClpP endopeptidase family (S14), a lon protease PF14_0147 (S16), and an hslV protease PFL1465c (T1B). As shown in Figure 3, these proteases are associated with a large number of heat shock proteins (HSPs) including Hsp90, Hsp70, Hsp40, and DnaJ proteins. The protease having the highest degree of connectivity (80) in the heat shock response network is PFL1465c, a threonine protease hslV. In addition to the classical HSPs, it is associated with a wide variety of enzymes such as ubiE/COQ5 methyltransferase, rRNA methyltransferase, multiple tRNA synthetases, various phosphate isomerases, amino transferase, aldolase, and a number of kinases, suggesting it may have an important role in parasite metabolism. It is also associated with three other heat-shock response proteases (PFC0310c and PF08_0063 in the S14 ClpP endopeptidase family, and PF14_0147 in the S16 ATP-dependent protease lon family), an organelle processing peptidase in the M16 pitrilysin family (PFI1625c), a leucyl aminopeptidase in the M17 family (PF14_0439), and aminopeptidase P in the M24 family (PF14_0517); together, they form a complex protease degradation network. It is also interesting to note that this putative hslV protease appears to be linked to a second very important stress system in the malaria parasite that acts against oxidative challenges: Protease hslV is associated with the thioredoxin 1 protein (PF14_0545), a member of the thioredoxin system which controls cell redox homeostasis, and a putative Fe-superoxide dismutase (Fe-SOD, PF08_0071) which is critical for antioxidant defense. Because the malaria parasite is sensitive to oxidative stress, both the thioredoxin system and SOD have been considered as potential antimalaria targets [78]. Finally, our phylogenetic analysis revealed that this hslV protease (PFL1465) is of prokaryotic origin and there is no homolog in the human host, a desirable feature for drug targets [79-81]. A second heat shock response protease PfClpP (PFC0310c) was recently characterized [82,83]; protease inhibition assays have shown that it, along with other ATP-dependent chaperones, plays a crucial role in parasite growth and development. Furthermore, PfClpP is localized to the apicoplast, which is of cyanobacterial origin, making this protein an apicoplast-targeting antimalarial candidate. This protein is indeed highly connected with 69 association partners, including Hsp70, Hsp60, Hsp40, co-chaperones, and proteins involved in proteasome acitivities, replication, translation, protein biosynthesis, metabolism, and heat shock response, implying that its inactivation would have devastating consequences for the parasite.
Figure 3.
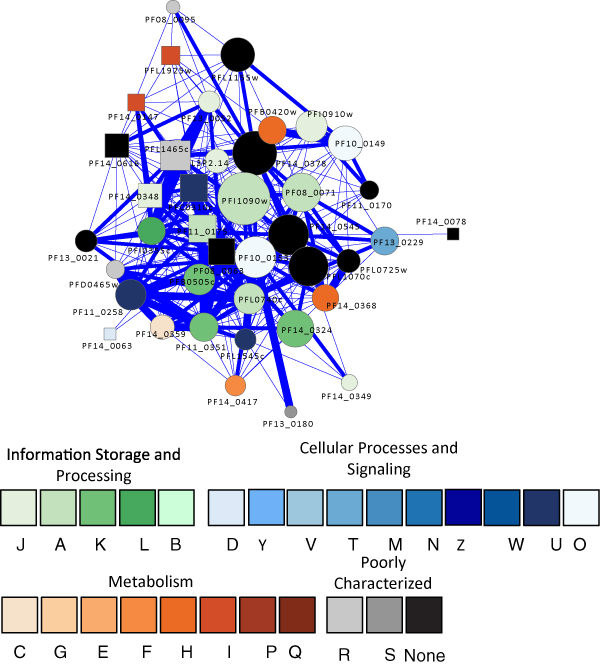
The graph shows a subnetwork of proteins linked to stress responses in P. falciparum. It was detected using the MINE plug in for Cytoscape, which uses an agglomerative approach to search the topology of large networks for significant clusters. The visualization is as for Figure 2.
Signal transduction via the regulated intramembrane proteolysis (RIP) network
The common belief that proteases cleave peptide bonds in a water environment was challenged by the discovery of a set of proteases that conduct hydrolysis in the hydrophobic environment of cellular membranes [84]. During RIP, intramembrane proteases cleave transmembrane-spanning helical (TMH) segments of the substrates and release soluble effectors, many of which are signaling molecules, thereby triggering cascades of signal transduction pathways [85,86]. RIP is now believed to be a ubiquitous signaling mechanism in a wide variety of organisms from bacteria to humans [87]. The roles of RIP in the parasite life cycle have begun to be unraveled. Three families of membrane-tethered proteases involved in RIP have been identified in P. falciparum, including an aspartic signal peptide peptidase (PfAPP, PF14_0543) in the A22 presenilin family, eight rhomboid serine proteases (PfROMs) in the S54 family, and two putative Site-2 metallo proteases (S2Ps, PF13_0028 and PF10_0317) in the M50 family[1,88-93].
The first family, PfAPP (PF14_0543), has 54 association partners (Figure 4 andTable 2). The association partner with the highest confidence score is a putative Rer1 (retrieval receptor for endoplasmic reticulum (ER) membrane proteins, PFI0150c) that is important for localizing proteins to the ER. Another related partner for PfAPP is a putative ER lumen protein retaining receptor (PF13_0280), which contains a signal sequence that facilitates the protein transport between the cis side of the Golgi apparatus and the ER [94]. It is believed that parasite invasion of erythrocytes requires the export of proteins to the ER and the cell surface. They then traverse the parasitophorous vacuole membrane (PVM) into the erythrocyte or parasite-derived membranous structures known as Maurer's clefts. PfAPP, along with the ER-localization proteins, may play a role in protein trafficking, cell-cell communication and remodeling of the host erythrocyte for parasite entry. Other proteins that are associated with PfAPP include secretory proteins, translation initiation and elongation factors, splicing factors and the spliceosome unit, peptide chain release factor, and various enzymes, suggesting it is involved in diverse networks related to transport, translation, posttranslational processing and metabolism. Recent gene disruption assays showed that PfAPP is essential for merozoite invasion and parasite growth [92,93]; the versatile associations of this protease underscore its potential as a drug target.
Figure 4.

The set of proteins associated with proteases that carry out regulated intramembrane proteolysis (RIP). This novel form of proteolysis is linked to signaling and the associated proteins may be targets for, or mediators of, this system. The red edges indicate experimentally validated interactions; other details of the visualization are as for Figure 2.
Table 2.
Representative P. falciparum proteins that are associated with the regulated intramembrane proteolysis (RIP) network.
| Protease family | Accession number of protease | Associated Protein accession number | Annotation |
|---|---|---|---|
| A22 (presenilin family) | PF14_0543 | PFI0150c | putative retrieval receptor for endoplasmic reticulum membrane proteins |
| PF13_0280 | ER lumen protein retaining receptor | ||
| MAL13P1.231 | Sec61 alpha subunit, PfSec61 | ||
| PFB0450w | secretory complex protein 61 gamma subunit | ||
| PF11_0447 | putative translation initiation factor eIF-1A | ||
| PF10_0077 | putative eukaryotic translation initiation factor 3 subunit 7 | ||
| PFL0310c | putative eukaryotic translation initiation factor 3 subunit 8 | ||
| PFL0335c | putative eukaryotic translation initiation factor 5 | ||
| PFE1405c | putative eukaryotic translation initiation factor 3, subunit 6 | ||
| PFC0870w | putative elongation factor 1 (EF-1) | ||
| MAL8P1.48 | putative splicing factor | ||
| PFB0550w | putative peptide chain release factor subunit 1 | ||
| PF07_0034 | cloroquine resistance associated protein Cg3 protein | ||
| PF13_0022 | cyclin | ||
| S54 (Rhomboid family) | PF11_0150 | PF11_0344 | apical membrane antigen 1 |
| PF11_0486 | merozoite adhesive erythrocytic binding protein | ||
| PFA0125c | erythrocyte binding antigen-181 | ||
| MAL13P1.60 | erythrocyte binding antigen-140 | ||
| MAL7P1.176 | erythrocyte binding antigen-175 | ||
| MAL13P1.176 | reticulocyte binding protein 2 homolog b | ||
| M50 (S2P protease family) | PF13_0028 | PF13_0287 | adenylosuccinate synthetase |
| PF10_0317 | PFB0260w | putative proteasome 26S regulatory subunit | |
| PFF0940c | putative cell division cycle protein 48 homolog | ||
The second family, PfROM, includes a group of serine proteins with demonstrated roles in parasite invasion [90,91,95,96]. Only one out of the ten rhomboid protease homologs in P. falciparum, PfRom1 (PF11_0150), was predicted to have protein-protein associations. Most interestingly, all the six proteins associated with it are antigens that have been considered as vaccine candidates; they belong to three families of adhesins that are essential for parasite invasion (Figure 4 andTable 2): (1) the apical membrane antigen 1 (AMA1, PF11_0344) is an adhesin required for merozoite invasion and it plays an indispensible role in the proliferation and survival of the malaria parasite [97]. PfRom1 was shown to be able to cleave AMA1 [88]; (2) the erythrocyte binding-like (EBL) family is involved in binding to a host chemokine receptor, the Duffy antigen [98]. Among the four EBAs with predicted association with PfRom1, EBA-175(MAL7P1.176) is proven a natural substrate for PfRom1 [88], but it remains unclear whether PfRom1 can cleave EBA-140 (MAL13P1.60), EBA-181 (MAL1P1.16), and a putative merozoite adhesive erythrocytic binding protein (PF11_0486); (3) a reticulocyte binding protein 2 homolog b protein (MAL13P1.176) in the reticulocyte binding-like (RBL) family. PfRom1 is able to cleave the RBL proteins [88]. Apparently, PfRom1 plays a central role in the RIP network that is tightly linked to the invasion process [86] and as such merits further investigation as a drug target.
S2Ps in the third family, PF10_0317 and PF13_0028, have two and one associations, respectively (Figure 4 andTable 2). PF10_0317 is associated with a proteasome 26S regulatory subunit and a cell division cycle (CDC) protein 48 homolog, which is implicated by GO analysis in ER localization and cell cycle regulation. Our previous domain analysis showed that PF10_0317 contains a Der-1 like domain, which was implicated in proteolysis associated with the ER [99-102]. PF13_0028 is associated with an adenylosuccinate synthetase AdsS (PF13_0287), which is important for the de novo biosynthesis of purine nucleotides. This association was predicted based on the genome synteny analysis, which revealed that the homologs of S2P and AdsS are located in the same chromosomal neighborhood in a variety of Actinobacteria. The functions of these S2Ps in malaria parasites are yet to be defined.
Parasite egress mediated by proteolysis
Egress, the parasite's emergence from host erythrocytes, is a well-coordinated process involving the rupture of the parasitophorous membrane (PVM) and the erythrocyte membrane (EM). Proteases that have been implicated in parasite egress [31,36] include (1) aspartic proteases (plasmepsins PMI, PMII, and PMIII, also known as histo-aspartic protease (HAP)) in the A1 family, (2) cystein proteases in the A1 papain family including falcipain 2a, 2b, and 3, dipeptidyl peptidase 3 (PfDPAP3), and a series of Serine Repeat Antigens (SERAs), and (3) a serine protease subtilase 1 (PfSUB1) in the subtilisin S8 family. We analyzed the protein-association network (Figure 5) involving proteases mediating egress and found that a central player in the network is SERA5 (PFB0340c), which has 28 associations. SERA5 is associated with PfSUB1 (PFE0370c) and PfDPAP3 (PFD0230c). Both these proteases can proteolytically activate SERA5, which triggers downstream processing of cellular substrates [103,104]. SERA5 is also associated with several erythrocyte membrane antigens such as PfEMP2 and EBA-175. It is abundantly expressed in the blood stage, especially in the schizont stage, as revealed by microarray and proteomic analysis. SERA5 has an in vitro catalytic activity and it is refractory to gene disruption [105], suggesting its vital role in the parasite life cycle.
Figure 5.

The protein associations of proteases involved in egress (exit from the erythrocyte). SERA5 (PFB0340c) is the most highly connected protease and appears to be a key player. It is linked with two proteases known to activate it, as well as potential substrates (see text). The red edges indicate experimentally validated interactions; other details of the visualization are as for Figure 2.
Signal peptidase network
As an adaptive survival strategy, the malaria parasite harbors a powerful secretion system that transports parasite-encoded virulence proteins to their subcellular locations. The central players in this secretion system are a group of signal peptidases that are capable of cleaving signal sequences from the target proteins that can then be routed to their destinations. Five signal peptidases have been predicted and characterized, constituting the signal peptidase complex (SPC) in P. falciparum [3,106,107]. Three of these peptidases have association partners: PfSPC21 (MAL13P1.167) has 120 associations; the putative microsomal signal peptidase (PF14_0317) has five associations; and the putative SPC22 (PFI0215c) has five associations (Figure 6). The associated proteins are part of the secretion pathway and include secretory complex protein 61 (Pf61) alpha and gamma subunits, a signal recognition particle (SRP) and an SRP receptor, an ER lumen protein retaining receptor, and a transport protein particle (TRAPP) component. These signal peptidases are also associated with members of the ubiquitin-proteasome system and the heat shock response system, with the translational machinery, and with metabolic networks.
Figure 6.

Proteins associated with three signal peptidases. As these proteases process signaling sequences on proteins, it is not surprising that they have a wide array of associations. PfSPC21 (MAL13P1.167) is associated with a large number of proteins from COG category O (Posttranslational modification, protein turnover, chaperones). The visualization is as for Figure 2.
Other potentially important protease-associated networks
Proteases in P. falciparum may play other roles important for parasite biology. We previously identified a single copy of calpain PfCalp (MAL13P1.310) in P. falciparum genome [3,106,107]. Calpain is crucial for signal transduction, cell cycle regulation, differentiation, development, and cell-cell communication from bacteria to humans. Very little is known about its role in P. falciparum. Only four proteins seemed to be associated with calpain: including a putative protein with a C3HC4 type zinc finger, the motif commonly present in transcriptional regulators, a ribosomal protein, and two proteins with unknown function. However, partial knockdown assays recently suggested that PfCalp is essential for the parasite's optimal growth and cell cycle progression [108]. Phylogenetic analysis revealed that PfCalp is a unique type of calpain confined to alveolates (a group of protists) with distant relatedness to human calpains [63,108], adding it to a new line of promising drug target. Another class of proteases that mediate cell cycle regulation and programmed cell death is comprised of the three metacaspases from the C14 protease family [63,109]. Only one association partner was identified for PF13_0289 and PF14_0363, (polyubiquitin and a hypothetical protein with unknown function respectively), and no associations were found for PF14_0160, reflecting our limited knowledge about their functions in malaria parasite.
Conclusions
Our network analysis of proteases from P. falciparum uses a so-called guilt-by-association approach to extract sets of proteins from the proteome that are candidates for further study. The network biology approach is readily adapted to any system for which a genome sequence exists and for which some type of protein-protein association is available, although there are limitations. Some of these stem from missing data, and/or noisy data, which lead to underestimation of the S value for a pair of associated proteins, but this problem becomes less significant with each release of data. A second problem is the lack of any dynamic element in evaluating the associations. A more formal integration of expression data could help to ameliorate this situation, especially expression data sets gathered under different conditions. Despite these limitations, our results produced known associations, which serve as positive controls such as the ubiquitin-proteasome system (UPS). It also indicated that proteases are playing previously unrecognized role in the biology of the parasite, such as the proteases that mediate the stress responses. Our results also imply that certain of these proteases, such as the proteases that mediate regulated intramembrane proteolysis, parasite egress, and signal peptide processing and protein secretion, may be good candidates for antimalarial targeting, as they are highly connected in the network. Furthermore, some of these candidates are known to have no or only distantly related homologs in humans, which reduces the probability of adverse effects resulting from their inactivation. Finally, our analysis has identified new components of previously recognized systems in the parasite, such as the protein(s) involved in transcriptional regulation, cell cycle progression, invasion, protein trafficking, and immune evasion in the UPS, or the antioxidant defense proteins associated with the heat shock response systems.
Methods
The protease data
The proteases in P. falciparum were predicted using a comparative genomics approach and a support vector machine (SVM)-based, supervised machine learning approach [1-3]. The classification and annotation were according to the MEROPS protease nomenclature, which is based on intrinsic evolutionary and structural relationships [110].
Network data and analysis
The complete set of protein-protein associations for P. falciparum was extracted from the downloaded STRING database [4]; each association between a pair of proteins has a confidence score (S) ranging from 0.15 to 0.999 that was inferred from the evidence used to establish the association, such as homology transfer, KEGG pathway assignments, conserved chromosome synteny, phylogenetic co-occurence, and literature co-occurence [111]. This set of associations was visualized in Cytoscape [112] and converted to an undirected weighted graph, where there is a single edge between any pair of proteins and the S value is used as the weight. The network was characterized using NetworkAnalyzer [113] and significant modules were detected using MINE [114] and MCODE [115]. The default values were used for all three plugins. The set of proteins directly associated with the 77 proteases in the association set were screened using BiNGO [116] to determine if any categories of proteins, as identified by their Gene Ontology terms, were over-represented. The hypergeometric test was used with the Benjamini and Hochberg false discovery date correction. A significance level of 0.05 was selected.
The omics data mining
We downloaded the P. falciparum genomic sequence and annotation data [18], transcriptomic microarray data [6-8], mass-spectrometry proteomic data [9-12], and protein-protein interactome [5] data for network associated proteins from PlasmoDB, the Plasmodium Genome resource center (http://www.plasmodb.org) [117]. Conserved domains/motifs in P. falciparum sequences were identified by searching InterPro [118]. Multiple alignments were obtained using the ClustalX program [119] and T-coffee [120], followed by manual inspection and editing. Phylogenetic trees were inferred by the neighbor-joining, maximum-parsimony and maximum-likelihood methods, using MEGA5 [121].
List of abbreviations used
AMA: apical membrane antigen; CDPK: calcium-dependent protein kinase; DOXP: 1-deoxy-D-xylulose 5-phosphate; EBL: erythrocyte binding-like; EF: elongation factor; EM: erythrocyte membrane; ER: endoplasmic reticulum; GO: Gene Ontology; HAP: histo-aspartic protease; HSP: heat shock protein; MBF: multiprotein bridging factor; ORF: open reading frame; PPI: protein-protein interaction; PVM: parasitophorous vacuole membrane; RBL: reticulocyte binding-like; RIP: regulated intramembrane proteolysis; SERA: Serine Repeat Antigen; SKP: S-phase kinase-associated protein; SOD: superoxide dismutase; SPAM: secreted polymorphic antigen associated with merozoites; SPC: signal peptidase complex; SVM: support vector machine; TBP: TATA box-binding protein; TMH: transmembrane-spanning helical; UPS: ubiquitin-proteasome system; Y2H: yeast 2-hybrid
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TGL and YW conceived and designed the study, performed bioinformatics data analysis, and drafted the manuscript. HC wrote scripts, and ZZ helped with data analysis. All authors read and approved the final manuscript.
Supplementary Material
P. falciparum proteases and their degrees of connectivity in protein association networks.
The protein-protein associations involving proteases in Plasmodium falciparum.
Functional categories involving protease associations identified by Gene Ontology enrichment analysis.
The graph shows the set of proteins associated with the proteases thought to be part of the P. falciparum ubiquitin-proteasome protein degradation system. Nodes are colored according to their functional classification in the eggNOG database [122] (key is shown). Node size is proportional to the degree of the node. Confidence scores for the interactions among the nodes (S values from STRING) were divided into three groups - low (0.150-0.399), medium (0.400-0.700) and high (0.701-0.999); the groups are represented by thin, medium and heavy lines, respectively.
Contributor Information
Timothy G Lilburn, Email: tlilburn@atcc.org.
Hong Cai, Email: hong.cai@utsa.edu.
Zhan Zhou, Email: zhan.zhou@utsa.edu.
Yufeng Wang, Email: yufeng.wang@utsa.edu.
Acknowledgements
We thank PlasmoDB for providing an all-in-one portal for malaria omic data. This work is supported by NIH grants GM081068 and AI080579 to Y. Wang. ZZ is supported by the government scholarship from China Scholarship Council. YW is also supported by NIH grant RR013646. We thank the Computational Biology Initiative at UTSA for providing computational support. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences, National Institute of Allergy and Infectious Diseases, National Center for Research Resources, or the National Institutes of Health.
References
- Kuang R, Gu J, Cai H, Wang Y. Improved prediction of malaria degradomes by supervised learning with SVM and profile kernel. Genetica. 2009;136(1):189–209. doi: 10.1007/s10709-008-9336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu Y. Computer assisted searches for drug targets with emphasis on malarial proteases and their inhibitors. Curr Drug Targets Infect Disord. 2004;4(1):25–40. doi: 10.2174/1568005043480952. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang X, Liu X, Wang Y. Data-mining approaches reveal hidden families of proteases in the genome of malaria parasite. Genome Res. 2003;13(4):601–616. doi: 10.1101/gr.913403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2010. pp. D561–568. [DOI] [PMC free article] [PubMed]
- LaCount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, Hesselberth JR, Schoenfeld LW, Ota I, Sahasrabudhe S, Kurschner C, et al. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature. 2005;438(7064):103–107. doi: 10.1038/nature04104. [DOI] [PubMed] [Google Scholar]
- Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, DeRisi JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003;1(1):E5. doi: 10.1371/journal.pbio.0000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdech Z, Zhu J, Joachimiak MP, Cohen FE, Pulliam B, DeRisi JL. Expression profiling of the schizont and trophozoite stages of Plasmodium falciparum with a long-oligonucleotide microarray. Genome Biol. 2003;4(2):R9. doi: 10.1186/gb-2003-4-2-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, De La Vega P, Holder AA, Batalov S, Carucci DJ, et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 2003;301(5639):1503–1508. doi: 10.1126/science.1087025. [DOI] [PubMed] [Google Scholar]
- Florens L, Liu X, Wang YF, Yang SG, Schwartz O, Peglar M, Carucci DJ, Yates JR, Wu YM. Proteomics approach reveals novel proteins on the surface of malaria-infected erythrocytes. Mol Biochem Parasit. 2004;135(1):1–11. doi: 10.1016/j.molbiopara.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, et al. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419(6906):520–526. doi: 10.1038/nature01107. [DOI] [PubMed] [Google Scholar]
- Lasonder E, Ishihama Y, Andersen JS, Vermunt AMW, Pain A, Sauerwein RW, Eling WMC, Hall N, Waters AP, Stunnenberg HG, et al. Analysis of the Plasmodium falciparum proteome by high-accuracy mass spectrometry. Nature. 2002;419(6906):537–542. doi: 10.1038/nature01111. [DOI] [PubMed] [Google Scholar]
- Lasonder E, Janse CJ, van Gemert GJ, Mair GR, Vermunt AM, Douradinha BG, van Noort V, Huynen MA, Luty AJ, Kroeze H, et al. Proteomic profiling of Plasmodium sporozoite maturation identifies new proteins essential for parasite development and infectivity. PLoS Pathog. 2008;4(10):e1000195. doi: 10.1371/journal.ppat.1000195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton PD, Pierce SK, Miller LH. Advances and challenges in malaria vaccine development. J Clin Invest. 2010;120(12):4168–4178. doi: 10.1172/JCI44423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton J. The Plasmodium vivax genome sequencing project. Trends Parasitol. 2003;19(5):227–231. doi: 10.1016/S1471-4922(03)00066-7. [DOI] [PubMed] [Google Scholar]
- Carlton J, Silva J, Hall N. The genome of model malaria parasites, and comparative genomics. Curr Issues Mol Biol. 2005;7(1):23–37. [PubMed] [Google Scholar]
- Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, Crabtree J, Angiuoli SV, Merino EF, Amedeo P, et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455(7214):757–763. doi: 10.1038/nature07327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, Silva JC, Ermolaeva MD, Allen JE, Selengut JD, Koo HL. et al. Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature. 2002;419(6906):512–519. doi: 10.1038/nature01099. [DOI] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S. et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419(6906):498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pain A, Bohme U, Berry AE, Mungall K, Finn RD, Jackson AP, Mourier T, Mistry J, Pasini EM, Aslett MA. et al. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature. 2008;455(7214):799–803. doi: 10.1038/nature07306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Turbachova I, Eberl M, Zeidler J, Lichtenthaler HK. et al. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999;285(5433):1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- Wiesner J, Jomaa H. Isoprenoid biosynthesis of the apicoplast as drug target. Curr Drug Targets. 2007;8(1):3–13. doi: 10.2174/138945007779315551. [DOI] [PubMed] [Google Scholar]
- Dahl EL, Rosenthal PJ. Apicoplast translation, transcription and genome replication: targets for antimalarial antibiotics. Trends Parasitol. 2008;24(6):279–284. doi: 10.1016/j.pt.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Jana S, Paliwal J. Novel molecular targets for antimalarial chemotherapy. Int J Antimicrob Agents. 2007;30(1):4–10. doi: 10.1016/j.ijantimicag.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Dvorin JD, Martyn DC, Patel SD, Grimley JS, Collins CR, Hopp CS, Bright AT, Westenberger S, Winzeler E, Blackman MJ. et al. A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science. 2010;328(5980):910–912. doi: 10.1126/science.1188191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker A, Lakshmanan V, Sinnis P, Coppens I, Fidock DA. Evidence that mutant PfCRT facilitates the transmission to mosquitoes of chloroquine-treated Plasmodium gametocytes. J Infect Dis. 2011;203(2):228–236. doi: 10.1093/infdis/jiq036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joet T, Eckstein-Ludwig U, Morin C, Krishna S. Validation of the hexose transporter of Plasmodium falciparum as a novel drug target. Proc Natl Acad Sci USA. 2003;100(13):7476–7479. doi: 10.1073/pnas.1330865100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DJ, Fidock DA, Mungthin M, Lakshmanan V, Sidhu AB, Bray PG, Ward SA. Evidence for a central role for PfCRT in conferring Plasmodium falciparum resistance to diverse antimalarial agents. Mol Cell. 2004;15(6):867–877. doi: 10.1016/j.molcel.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk K, Howitt SM, Broer S, Saliba KJ, Downie MJ. Purine uptake in Plasmodium: transport versus metabolism. Trends Parasitol. 2009;25(6):246–249. doi: 10.1016/j.pt.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Kirk K, Saliba KJ. Targeting nutrient uptake mechanisms in Plasmodium. Curr Drug Targets. 2007;8(1):75–88. doi: 10.2174/138945007779315560. [DOI] [PubMed] [Google Scholar]
- Patel AP, Staines HM, Krishna S. New antimalarial targets: the example of glucose transport. Travel Med Infect Dis. 2008;6(1-2):58–66. doi: 10.1016/j.tmaid.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Blackman MJ. Malarial proteases and host cell egress: an 'emerging' cascade. Cell Microbiol. 2008;10(10):1925–1934. doi: 10.1111/j.1462-5822.2008.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PJ. Cysteine proteases of malaria parasites. Int J Parasitol. 2004;34(13-14):1489–1499. doi: 10.1016/j.ijpara.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Wang F, Krai P, Deu E, Bibb B, Lauritzen C, Pedersen J, Bogyo M, Klemba M. Biochemical characterization of Plasmodium falciparum dipeptidyl aminopeptidase 1. Mol Biochem Parasitol. 2011;175(1):10–20. doi: 10.1016/j.molbiopara.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silmon de Monerri NC, Flynn HR, Campos MG, Hackett F, Koussis K, Withers-Martinez C, Skehel JM, Blackman MJ. Global identification of multiple substrates for Plasmodium falciparum SUB1, an essential malarial processing protease. Infect Immun. 2011;79(3):1086–1097. doi: 10.1128/IAI.00902-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SU, Kang JM, Kim TS, Kong Y, Sohn WM, Na BK. Plasmodium vivax: collaborative roles for plasmepsin 4 and vivapains in hemoglobin hydrolysis. Exp Parasitol. 2011;128(2):127–132. doi: 10.1016/j.exppara.2011.02.015. [DOI] [PubMed] [Google Scholar]
- Sologub L, Kuehn A, Kern S, Przyborski J, Schillig R, Pradel G. Malaria proteases mediate inside-out egress of gametocytes from red blood cells following parasite transmission to the mosquito. Cell Microbiol. 2011;13(6):897–912. doi: 10.1111/j.1462-5822.2011.01588.x. [DOI] [PubMed] [Google Scholar]
- Boddey JA, Hodder AN, Gunther S, Gilson PR, Patsiouras H, Kapp EA, Pearce JA, de Koning-Ward TF, Simpson RJ, Crabb BS. et al. An aspartyl protease directs malaria effector proteins to the host cell. Nature. 2010;463(7281):627–631. doi: 10.1038/nature08728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer PW, Simon GM, Cravatt BF, Bogyo M. Global profiling of proteolysis during rupture of Plasmodium falciparum from the host erythrocyte. Mol Cell Proteomics. 2010;10(5) doi: 10.1074/mcp.M110.001636. M110 001636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey KC, Barkan DT, Sali A, Rosenthal PJ. Regulatory elements within the prodomain of Falcipain-2, a cysteine protease of the malaria parasite Plasmodium falciparum. PLoS One. 2009;4(5) doi: 10.1371/journal.pone.0005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Hardt M, Choe Y, Niles RK, Johansen EB, Legac J, Gut J, Kerr ID, Craik CS, Rosenthal PJ. Hemoglobin cleavage site-specificity of the Plasmodium falciparum cysteine proteases Falcipain-2 and Falcipain-3. PLoS One. 2009;4(4) doi: 10.1371/journal.pone.0005156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaccapelo R, Janse CJ, Caterbi S, Franke-Fayard B, Bonilla JA, Syphard LM, Di Cristina M, Dottorini T, Savarino A, Cassone A. et al. Plasmepsin 4-deficient Plasmodium berghei are virulence attenuated and induce protective immunity against experimental malaria. Am J Pathol. 2010;176(1):205–217. doi: 10.2353/ajpath.2010.090504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner-Adams TS, Stack CM, Trenholme KR, Brown CL, Grembecka J, Lowther J, Mucha A, Drag M, Kafarski P, McGowan S. et al. Plasmodium falciparum neutral aminopeptidases: new targets for anti-malarials. Trends Biochem Sci. 2010;35(1):53–61. doi: 10.1016/j.tibs.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Russo I, Babbitt S, Muralidharan V, Butler T, Oksman A, Goldberg DE. Plasmepsin V licenses Plasmodium proteins for export into the host erythrocyte. Nature. 2010;463(7281):632–636. doi: 10.1038/nature08726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan S, Oellig CA, Birru WA, Caradoc-Davies TT, Stack CM, Lowther J, Skinner-Adams T, Mucha A, Kafarski P, Grembecka J. et al. Structure of the Plasmodium falciparum M17 aminopeptidase and significance for the design of drugs targeting the neutral exopeptidases. Proc Natl Acad Sci USA. 2010;107(6):2449–2454. doi: 10.1073/pnas.0911813107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na BK, Bae YA, Zo YG, Choe Y, Kim SH, Desai PV, Avery MA, Craik CS, Kim TS, Rosenthal PJ. et al. Biochemical properties of a novel cysteine protease of Plasmodium vivax, vivapain-4. PLoS Negl Trop Dis. 2010;4(10) doi: 10.1371/journal.pntd.0000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FW, Patra KP, Yowell CA, Dame JB, Chin K, Vinetz JM. Apical surface expression of aspartic protease Plasmepsin 4, a potential transmission-blocking target of the plasmodium ookinete. J Biol Chem. 2010;285(11):8076–8083. doi: 10.1074/jbc.M109.063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruprasad L, Tanneeru K, Guruprasad K. Structural rationale for the recognition of arginine at P3 in PEXEL motif containing proteins of Plasmodium falciparum by plasmepsin V. Protein Pept Lett. 2011;18(6):634–641. doi: 10.2174/092986611795222786. [DOI] [PubMed] [Google Scholar]
- Rawat M, Vijay S, Gupta Y, Dixit R, Tiwari PK, Sharma A. Sequence homology and structural analysis of plasmepsin 4 isolated from Indian Plasmodium vivax isolates. Infect Genet Evol. 2011;11(5):924–933. doi: 10.1016/j.meegid.2011.02.024. [DOI] [PubMed] [Google Scholar]
- de Farias Silva N, Lameira J, Alves CN. Computational analysis of aspartic protease plasmepsin II complexed with EH58 inhibitor: a QM/MM MD study. J Mol Model. 2011;17:2631–2638. doi: 10.1007/s00894-011-0963-1. [DOI] [PubMed] [Google Scholar]
- Tastan Bishop O, Kroon M. Study of protein complexes via homology modeling, applied to cysteine proteases and their protein inhibitors. J Mol Model. 2011. [DOI] [PubMed]
- Shah F, Mukherjee P, Gut J, Legac J, Rosenthal PJ, Tekwani BL, Avery MA. Identification of novel malarial cysteine protease inhibitors using structure-based virtual screening of a focused cysteine protease inhibitor library. J Chem Inf Model. 2011;51(4):852–864. doi: 10.1021/ci200029y. [DOI] [PubMed] [Google Scholar]
- Ettari R, Bova F, Zappala M, Grasso S, Micale N. Falcipain-2 inhibitors. Med Res Rev. 2010;30(1):136–167. doi: 10.1002/med.20163. [DOI] [PubMed] [Google Scholar]
- Bova F, Ettari R, Micale N, Carnovale C, Schirmeister T, Gelhaus C, Leippe M, Grasso S, Zappala M. Constrained peptidomimetics as antiplasmodial falcipain-2 inhibitors. Bioorg Med Chem. 2010;18(14):4928–4938. doi: 10.1016/j.bmc.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Coteron JM, Catterick D, Castro J, Chaparro MJ, Diaz B, Fernandez E, Ferrer S, Gamo FJ, Gordo M, Gut J. et al. Falcipain inhibitors: optimization studies of the 2-pyrimidinecarbonitrile lead series. J Med Chem. 2010;53(16):6129–6152. doi: 10.1021/jm100556b. [DOI] [PubMed] [Google Scholar]
- Shah F, Mukherjee P, Desai P, Avery M. Computational approaches for the discovery of cysteine protease inhibitors against malaria and SARS. Curr Comput Aided Drug Des. 2010;6(1):1–23. doi: 10.2174/157340910790980142. [DOI] [PubMed] [Google Scholar]
- Gibbons P, Verissimo E, Araujo NC, Barton V, Nixon GL, Amewu RK, Chadwick J, Stocks PA, Biagini GA, Srivastava A. et al. Endoperoxide carbonyl falcipain 2/3 inhibitor hybrids: toward combination chemotherapy of malaria through a single chemical entity. J Med Chem. 2010;53(22):8202–8206. doi: 10.1021/jm1009567. [DOI] [PubMed] [Google Scholar]
- Ahmed W, Rani M, Khan IA, Iqbal A, Khan KM, Haleem MA, Azim MK. Characterisation of hydrazides and hydrazine derivatives as novel aspartic protease inhibitors. J Enzyme Inhib Med Chem. 2010;25(5):673–678. doi: 10.3109/14756360903508430. [DOI] [PubMed] [Google Scholar]
- Miura T, Hidaka K, Uemura T, Kashimoto K, Hori Y, Kawasaki Y, Ruben AJ, Freire E, Kimura T, Kiso Y. Improvement of both plasmepsin inhibitory activity and antimalarial activity by 2-aminoethylamino substitution. Bioorg Med Chem Lett. 2010;20(16):4836–4839. doi: 10.1016/j.bmcl.2010.06.099. [DOI] [PubMed] [Google Scholar]
- Gupta D, Yedidi RS, Varghese S, Kovari LC, Woster PM. Mechanism-based inhibitors of the aspartyl protease plasmepsin II as potential antimalarial agents. J Med Chem. 2010;53(10):4234–4247. doi: 10.1021/jm100233b. [DOI] [PubMed] [Google Scholar]
- Florent I, Marechal E, Gascuel O, Brehelin L. Bioinformatic strategies to provide functional clues to the unknown genes in Plasmodium falciparum genome. Parasite. 2010;17(4):273–283. doi: 10.1051/parasite/2010174273. [DOI] [PubMed] [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Review Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Wu YM, Wang XY, Liu X, Wang YF. Data-mining approaches reveal hidden families of proteases in the genome of malaria parasite. Genome Res. 2003;13(4):601–616. doi: 10.1101/gr.913403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreidenweiss A, Kremsner PG, Mordmuller B. Comprehensive study of proteasome inhibitors against Plasmodium falciparum laboratory strains and field isolates from Gabon. Malar J. 2008;7:-. doi: 10.1186/1475-2875-7-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponts N, Yang JF, Chung DWD, Prudhomme J, Girke T, Horrocks P, Le Roch KG. Deciphering the ubiquitin-mediated pathway in apicomplexan parasites: a potential strategy to interfere with parasite virulence. PLoS One. 2008;3:e2386. doi: 10.1371/journal.pone.0002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmann O, Lechner J. The Saccharomyces cerevisiae kinetochore contains a cyclin-CDK complexing homologue, as identified by in vitro reconstitution. EMBO J. 1996;15(14):3611–3620. [PMC free article] [PubMed] [Google Scholar]
- Takemaru K, Li FQ, Ueda H, Hirose S. Multiprotein bridging factor 1 (MBF1) is an evolutionarily conserved transcriptional coactivator that connects a regulatory factor and TATA element-binding protein. Proc Natl Acad Sci USA. 1997;94(14):7251–7256. doi: 10.1073/pnas.94.14.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O, Stenmark H, Alexandrov K, Huber LA, Kaibuchi K, Sasaki T, Takai Y, Zerial M. Rab GDP dissociation inhibitor as a general regulator for the membrane association of rab proteins. J Biol Chem. 1993;268(24):18143–18150. [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Beaudenon S, Howley PM. A family of proteins structurally and functionally related to the E6-AP ubiquitin protein ligase. Proc Natl Acad Sci USA. 1995;92(7):2563–2567. doi: 10.1073/pnas.92.7.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shock JL, Fischer KF, DeRisi JL. Whole-genome analysis of mRNA decay in Plasmodium falciparum reveals a global lengthening of mRNA half-life during the intra-erythrocytic development cycle. Genome Biol. 2007;8(7) doi: 10.1186/gb-2007-8-7-r134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JS, Militello KT, Sims PA, Patel VP, Kasper JM, Wirth DF. Patterns of gene-specific and total transcriptional activity during the Plasmodium falciparum intraerythrocytic developmental cycle. Eukaryot Cell. 2009;8(3):327–338. doi: 10.1128/EC.00340-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JH, Hudson DE, Torii M, Ward GE, Wellems TE, Aikawa M, Miller LH. The Duffy receptor family of Plasmodium knowlesi is mocated within the micronemes of invasive malaria merozoites. Cell. 1990;63(1):141–153. doi: 10.1016/0092-8674(90)90295-P. [DOI] [PubMed] [Google Scholar]
- McColl DJ, Silva A, Foley M, Kun JFJ, Favaloro JM, Thompson JK, Marshall VM, Coppel RL, Kemp DJ, Anders RF. Molecular variation in a novel polymorphic antigen associated with Plasmodium falciparum merozoites. Mol Biochem Parasit. 1994;68(1):53–67. doi: 10.1016/0166-6851(94)00149-9. [DOI] [PubMed] [Google Scholar]
- Chung DW, Le Roch KG. Targeting the Plasmodium ubiquitin/proteasome system with anti-malarial compounds: promises for the future. Infect Disord Drug Targets. 2010;10(3):158–164. doi: 10.2174/187152610791163345. [DOI] [PubMed] [Google Scholar]
- Gantt SM, Myung JM, Briones MRS, Li WD, Corey EJ, Omura S, Nussenzweig V, Sinnis P. Proteasome inhibitors block development of Plasmodium spp. Antimicrob Agents Ch. 1998;42(10):2731–2738. doi: 10.1128/aac.42.10.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya P, Kumar R, Tatu U. Chaperoning a cellular upheaval in malaria: heat shock proteins in Plasmodium falciparum. Mol Biochem Parasit. 2007;153(2):85–94. doi: 10.1016/j.molbiopara.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Botha M, Pesce ER, Blatch GL. The Hsp40 proteins of Plasmodium falciparum and other apicomplexa: regulating chaperone power in the parasite and the host. Int J Biochem Cell Biol. 2007;39(10):1781–1803. doi: 10.1016/j.biocel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Kanzok SM, Schirmer RH, Turbachova I, Iozef R, Becker K. The thioredoxin system of the malaria parasite Plasmodium falciparum. Glutathione reduction revisited. J Biol Chem. 2000;275(51):40180–40186. doi: 10.1074/jbc.M007633200. [DOI] [PubMed] [Google Scholar]
- Tschan S, Kreidenweiss A, Stierhof YD, Sessler N, Fendel R, Mordmuller B. Mitochondrial localization of the threonine peptidase PfHslV, a ClpQ ortholog in Plasmodium falciparum. Int J Parasitol. 2010;40(13):1517–1523. doi: 10.1016/j.ijpara.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Powers JC, Asgian JL, Ekici OD, James KE. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev. 2002;102(12):4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- Ramasamy G, Gupta D, Mohmmed A, Chauhan VS. Characterization and localization of Plasmodium falciparum homolog of prokaryotic ClpQ/HslV protease. Mol Biochem Parasitol. 2007;152(2):139–148. doi: 10.1016/j.molbiopara.2007.01.002. [DOI] [PubMed] [Google Scholar]
- El Bakkouri M, Pow A, Mulichak A, Cheung KL, Artz JD, Amani M, Fell S, de Koning-Ward TF, Goodman CD, McFadden GI, The Clp chaperones and proteases of the human malaria parasite Plasmodium falciparum. J Mol Biol. pp. 456–477. [DOI] [PubMed]
- Rathore S, Sinha D, Asad M, Bottcher T, Afrin F, Chauhan VS, Gupta D, Sieber SA, Mohmmed A. A cyanobacterial serine protease of Plasmodium falciparum is targeted to the apicoplast and plays an important role in its growth and development. Mol Microbiol. 2010;77(4):873–890. doi: 10.1111/j.1365-2958.2010.07251.x. [DOI] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100(4):391–398. doi: 10.1016/S0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler SF, Haass C, Steiner H. Regulated intramembrane proteolysis--lessons from amyloid precursor protein processing. J Neurochem. 2011;117(5):779–796. doi: 10.1111/j.1471-4159.2011.07248.x. [DOI] [PubMed] [Google Scholar]
- Urban S. Making the cut: central roles of intramembrane proteolysis in pathogenic microorganisms. Nat Rev Microbiol. 2009;7(6):411–423. doi: 10.1038/nrmicro2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinch LN, Ginalski K, Grishin NV. Site-2 protease regulated intramembrane proteolysis: sequence homologs suggest an ancient signaling cascade. Protein Sci. 2006;15(1):84–93. doi: 10.1110/ps.051766506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RP, Wijetilaka R, Urban S. Two Plasmodium rhomboid proteases preferentially cleave different adhesins implicated in all invasive stages of malaria. PLoS Pathog. 2006;2(10):e113. doi: 10.1371/journal.ppat.0020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brossier F, Jewett TJ, Sibley LD, Urban S. A spatially localized rhomboid protease cleaves cell surface adhesins essential for invasion by Toxoplasma. Proc Natl Acad Sci USA. 2005;102(11):4146–4151. doi: 10.1073/pnas.0407918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowse TJ, Pascall JC, Brown KD, Soldati D. Apicomplexan rhomboids have a potential role in microneme protein cleavage during host cell invasion. Int J Parasitol. 2005;35(7):747–756. doi: 10.1016/j.ijpara.2005.04.001. [DOI] [PubMed] [Google Scholar]
- O'Donnell RA, Hackett F, Howell SA, Treeck M, Struck N, Krnajski Z, Withers-Martinez C, Gilberger TW, Blackman MJ. Intramembrane proteolysis mediates shedding of a key adhesin during erythrocyte invasion by the malaria parasite. J Cell Biol. 2006;174(7):1023–1033. doi: 10.1083/jcb.200604136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chen H, Bahamontes-Rosa N, Kun JF, Traore B, Crompton PD, Chishti AH. Plasmodium falciparum signal peptide peptidase is a promising drug target against blood stage malaria. Biochem Biophys Res Commun. 2009;380(3):454–459. doi: 10.1016/j.bbrc.2009.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chen H, Oh SS, Chishti AH. A Presenilin-like protease associated with Plasmodium falciparum micronemes is involved in erythrocyte invasion. Mol Biochem Parasitol. 2008;158(1):22–31. doi: 10.1016/j.molbiopara.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley FM, Wilson DW, Pelham HR. Mutational analysis of the human KDEL receptor: distinct structural requirements for Golgi retention, ligand binding and retrograde transport. EMBO J. 1993;12(7):2821–2829. doi: 10.1002/j.1460-2075.1993.tb05943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowse TJ, Soldati D. Rhomboid-like proteins in Apicomplexa: phylogeny and nomenclature. Trends Parasitol. 2005;21(6):254–258. doi: 10.1016/j.pt.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Singh S, Plassmeyer M, Gaur D, Miller LH. Mononeme: a new secretory organelle in Plasmodium falciparum merozoites identified by localization of rhomboid-1 protease. Proc Natl Acad Sci USA. 2007;104(50):20043–20048. doi: 10.1073/pnas.0709999104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triglia T, Healer J, Caruana SR, Hodder AN, Anders RF, Crabb BS, Cowman AF. Apical membrane antigen 1 plays a central role in erythrocyte invasion by Plasmodium species. Mol Microbiol. 2000;38(4):706–718. doi: 10.1046/j.1365-2958.2000.02175.x. [DOI] [PubMed] [Google Scholar]
- Miller LH, Mason SJ, Dvorak JA, McGinniss MH, Rothman IK. Erythrocyte receptors for (Plasmodium knowlesi) malaria: Duffy blood group determinants. Science. 1975;189(4202):561–563. doi: 10.1126/science.1145213. [DOI] [PubMed] [Google Scholar]
- Kalanon M, Tonkin CJ, McFadden GI. Characterization of two putative protein translocation components in the apicoplast of Plasmodium falciparum. Eukaryot Cell. 2009;8(8):1146–1154. doi: 10.1128/EC.00061-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Finger A, Braun T, Hellmuth K, Wolf DH. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 1996;15(4):753–763. [PMC free article] [PubMed] [Google Scholar]
- Spork S, Hiss JA, Mandel K, Sommer M, Kooij TW, Chu T, Schneider G, Maier UG, Przyborski JM. An unusual ERAD-like complex is targeted to the apicoplast of Plasmodium falciparum. Eukaryot Cell. 2009;8(8):1134–1145. doi: 10.1128/EC.00083-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dooren GG, Tomova C, Agrawal S, Humbel BM, Striepen B. Toxoplasma gondii Tic20 is essential for apicoplast protein import. Proc Natl Acad Sci USA. 2008;105(36):13574–13579. doi: 10.1073/pnas.0803862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arastu-Kapur S, Ponder EL, Fonovic UP, Yeoh S, Yuan F, Fonovic M, Grainger M, Phillips CI, Powers JC, Bogyo M. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat Chem Biol. 2008;4(3):203–213. doi: 10.1038/nchembio.70. [DOI] [PubMed] [Google Scholar]
- Yeoh S, O'Donnell RA, Koussis K, Dluzewski AR, Ansell KH, Osborne SA, Hackett F, Withers-Martinez C, Mitchell GH, Bannister LH. et al. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell. 2007;131(6):1072–1083. doi: 10.1016/j.cell.2007.10.049. [DOI] [PubMed] [Google Scholar]
- McCoubrie JE, Miller SK, Sargeant T, Good RT, Hodder AN, Speed TP, de Koning-Ward TF, Crabb BS. Evidence for a common role for the serine-type Plasmodium falciparum serine repeat antigen proteases: implications for vaccine and drug design. Infect Immun. 2007;75(12):5565–5574. doi: 10.1128/IAI.00405-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Pradhan A, Chauhan VS, Tuteja R. Isolation and characterization of type I signal peptidase of different malaria parasites. J Biomed Biotechnol. 2005;2005(4):301–309. doi: 10.1155/JBB.2005.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuteja R, Pradhan A, Sharma S. Plasmodium falciparum signal peptidase is regulated by phosphorylation and required for intra-erythrocytic growth. Mol Biochem Parasitol. 2008;157(2):137–147. doi: 10.1016/j.molbiopara.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Russo I, Oksman A, Vaupel B, Goldberg DE. A calpain unique to alveolates is essential in Plasmodium falciparum and its knockdown reveals an involvement in pre-S-phase development. Proc Natl Acad Sci USA. 2009;106(5):1554–1559. doi: 10.1073/pnas.0806926106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Chat L, Sinden RE, Dessens JT. The role of metacaspase 1 in Plasmodium berghei development and apoptosis. Mol Biochem Parasitol. 2007;153(1):41–47. doi: 10.1016/j.molbiopara.2007.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND. A large and accurate collection of peptidase cleavages in the MEROPS database. Database (Oxford) 2009;2009:bap015. doi: 10.1093/database/bap015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, Jouffre N, Huynen MA, Bork P. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005. pp. D433–437. [DOI] [PMC free article] [PubMed]
- Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2010;27(3):431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assenov Y, Ramirez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24(2):282–284. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- Rhrissorrakrai K, Gunsalus KC. MINE: Module Identification in Networks. BMC Bioinformatics. 2011;12:192. doi: 10.1186/1471-2105-12-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009. pp. D539–543. [DOI] [PMC free article] [PubMed]
- Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Multiple sequence alignment with Clustal x. Trends Biochem Sci. 1998;23(10):403–405. doi: 10.1016/S0968-0004(98)01285-7. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302(1):205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011. [DOI] [PMC free article] [PubMed]
- Muller J, Szklarczyk D, Julien P, Letunic I, Roth A, Kuhn M, Powell S, von Mering C, Doerks T, Jensen LJ, eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 2010. pp. D190–195. [DOI] [PMC free article] [PubMed]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278(5338):631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
P. falciparum proteases and their degrees of connectivity in protein association networks.
The protein-protein associations involving proteases in Plasmodium falciparum.
Functional categories involving protease associations identified by Gene Ontology enrichment analysis.
The graph shows the set of proteins associated with the proteases thought to be part of the P. falciparum ubiquitin-proteasome protein degradation system. Nodes are colored according to their functional classification in the eggNOG database [122] (key is shown). Node size is proportional to the degree of the node. Confidence scores for the interactions among the nodes (S values from STRING) were divided into three groups - low (0.150-0.399), medium (0.400-0.700) and high (0.701-0.999); the groups are represented by thin, medium and heavy lines, respectively.