

Abstract
We use least absolute shrinkage and selection operator (LASSO) regression to select genetic markers and phenotypic features that are most informative with respect to a trait of interest. We compare several strategies for applying LASSO methods in risk prediction models, using the Genetic Analysis Workshop 17 exome simulation data consisting of 697 individuals with information on genotypic and phenotypic features (smoking, age, sex) in 5-fold cross-validated fashion. The cross-validated averages of the area under the receiver operating curve range from 0.45 to 0.63 for different strategies using only genotypic markers. The same values are improved to 0.69–0.87 when both genotypic and phenotypic information are used. The ability of the LASSO method to find true causal markers is limited, but the method was able to discover several common variants (e.g., FLT1) under certain conditions.
Background
Recent advances have enabled researchers to study genetic associations with familial diseases in remarkable detail. Genome-wide association studies (GWAS) of common variants have revealed numerous genetic loci that significantly modulate phenotypes for a wide assortment of important clinical phenotypes, ranging from the expected risk of certain malignancies [1,2] to commonly measured clinical traits, such as lipid levels [3]. Nevertheless, it is increasingly evident that the common variants found in GWAS provide an incomplete picture of the underlying genetic risk for many of the familial diseases that have been studied [4-6]. Thanks to the increased availability of sequencing technologies and to large-scale efforts such as the 1000 Genomes Project, exome scans are becoming increasingly popular in complex disease genetics. These studies represent several new challenges in genetic analysis.
Although a variety of machine learning methods have been used in GWAS [7], penalized regression methods are among the most flexible and are thus well suited for analysis of data sets such as exome scans, which may contain both common and rare effects. Numerous penalized regression methods have been shown to be effective for both common and rare variants [4,8-10]. Zhou et al. [4] proposed a combination of group and least absolute shrinkage and selection operator (LASSO) penalties to find both rare and common variants using sets of markers grouped by pathway and gene. However, their method was evaluated using family breast cancer registry data, and its performance is unclear for larger scale data from GWAS.
To improve accuracy, some studies have imposed an arbitrary p-value cutoff to limit the number of genetic variants in the LASSO model [9], whereas others have applied the model across all variants using the LASSO penalty and a group penalty for the gene or pathway [4]. In this study, we propose an approach using a LASSO model that first selects sets of genetic variants for each pathway and gene and then generates an optimized LASSO model based on the selected marker sets. Taking advantage of information provided in the Genetic Analysis Workshop 17 (GAW17) exome data set, we can build two LASSO models for each pathway or gene based on regression on either disease status or a quantitative trait. This approach is more time-consuming than optimization of a LASSO model for the full set of variants. However, our strategy permits us to build individual optimal models on each variant set related to the pathway and gene, allowing a more flexible and accurate model determination. In the remainder of this paper, we examine the performance of this new approach using the GAW17 exome data set.
Methods
LASSO regression
We compare several LASSO models that incorporate gene, pathway, and phenotypic information in this study. For a response vector Y = (y1,…,yn) containing case-control labels coded as 0 or 1 for a set of n subjects, a genotype matrix G = (X1,…,Xn), with each vector Xi consisting of m single-nucleotide polymorphisms (SNPs) coded as 0, 1, or 2, and a coefficient vector β, the standard logistic regression model:
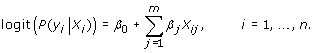 |
(1) |
can be fitted using Y and G. However, this model is not well suited for large genetic studies with far more variables than samples, and it often results in inaccuracies as a result of model instabilities, colinearities, and overfitting. Several penalized regression methods have become popular in the analysis of large-scale genetic data sets [7,9] for their improved variable selection. In this study, we use the L1 LASSO penalty method, which selects β based on the maximization of:
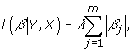 |
(2) |
where l(β | Y, X) is the logistic log-likelihood and λ is the shrinkage parameter. The LASSO-penalized regression model can also be defined for a linear regression for a continuous response vector [11]. In this study, we evaluate several different strategies for applying a LASSO regression that incorporates gene, pathway, and phenotypic information into the model.
Data description
The GAW17 data set contains 697 unrelated individuals from the 1000 Genomes Project genotyped at 24,487 autosomal SNPs from 3,205 genes [12]. Two hundred six pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG) [13] are represented, spanning 7,929 different SNPs and 1,100 different genes. We restrict our analysis to the 13,572 nonsynonymous variants in the study. Each of the 200 simulated data sets includes the following information for each individual: case-control status, three continuous quantitative traits (Q1, Q2, Q4), and three phenotypic features (Age, Smoking status, and Sex). We use a multidimensional scaling analysis based on genome-wide pairwise identity-by-state distances computed in PLINK [14] to determine three main continental population strata: African (Luhya, Luhya-additional, Yoruba-1, Yoruba-2, Yoruba-additional), Asian (Denver Chinese, Denver Chinese-additional, Han Chinese-1, Han Chinese-2, Han Chinese-additional, Japanese-1, Japanese-2, Japanese-additional), and European (CEPH-1, CEPH-2, Tuscan, and Tuscan-additional) [15,16]. We then generate three binary features to include in our model, assigning patients to their corresponding Asian, European, and African populations. Two main population outliers were removed from our analysis.
Analysis
We use the R software package glmnet in our analysis for LASSO regression [11] and evaluate our models using a 5-fold cross-validation procedure for each simulation data set. More specifically, we split the data sets into five independent folds of approximately equal size such that the case-control ratios in each population are maintained in each fold. Models are trained using four folds of the data and then tested using the remaining fold. This procedure is repeated for each of the five training and testing fold combinations. To determine an optimal value λ* for each training set, we apply an inner loop of 10-fold cross-validation. Then λ* is used on the entire training set to build the final model for the evaluation of the testing fold. Finally, the averaged evaluation measures over the five testing folds are reported as the testing accuracy. In our analysis the evaluation measures are the area under the receiver operating curve (AROC) for logistic models and the mean-square error for continuous linear regression models.
We consider three basic models: (1) LASSO logistic regression with all genetic variants included; (2) LASSO logistic regression for each of the (a) 3,205 genes or (b) 206 pathways, followed by a LASSO regression using the combined set of selected variants from all genes or pathways; and (3) three separate LASSO linear regression models for each of the continuous quantitative traits Q1, Q2, and Q4 for each pathway, followed by a LASSO logistic regression over the entire set of selected variants across all pathways.
For each of these strategies, we consider a genotypes-only model, a combined model that includes phenotype information (Age, Smoking, and Sex), and a restricted model that is limited to a fixed number of variables. In this study, the restricted models are limited to have a maximum of 50 variables.
Model 1 is similar to most other applications of the LASSO regression model, in which a single regularization parameter is used. This model is convenient and computationally efficient, but its ability to detect local effects within biologically meaningful subsets of genes that are of interest in an exome study may be limited. Models 2 and 3 first determine optimized models for each gene or pathway and then run a LASSO regression over the combined set of variants selected for each gene or pathway.
Results
Performance of the models
Results for all the models are shown in Table 1. Each of the 200 simulated data sets was analyzed separately. Because model 2 had a substantially longer running time, it was evaluated for only 50 (model 2a) and 150 (model 2b) randomly selected data sets. To determine the baseline performance for our models, we sampled several simulation data sets using 180 random variants (corresponding to the average size of the basic genotypes-only model 1 result). The expected average AROC for a randomly selected set of variants was 0.49. Similarly, we used glmnet to compute optimal models from the set of 160 causal simulation markers and determined that the average AROC of this optimal set of genotypes was 0.59. This value represents the average predictive accuracy of an optimized subset of the genetic variants responsible for assigning disease status in the simulation and is considered the target value of our models that use only genotype data. As observed in Table 1, the purely genetic models have AROC values closer to 0.55 for all models considered. The combined models with phenotypic features had an AROC of 0.82, a universally higher average testing AROC value independent of any genotypic combination. Because of the high marginal effect sizes of the phenotypic variables (Age, Sex, and Smoking status), these effects frequently overpowered the effect sizes of genetic markers included in the LASSO models. The unrestricted LASSO models often resulted in solutions with a large number of variables, limiting the practical utility of these models. The testing AROC values of the restricted models were often the same as or better than those of the unrestricted models, indicating better generalization ability for the restricted models. However, the predictive performance of the genetic component did not reach the best possible level, and the models included larger numbers of noncausal variants. The use of gene and pathway information did not result in meaningful improvements in the regression models with respect to predictive capability.
Table 1.
Prediction results for various model types
| Model | Model type | Training AROC | Testing AROC | Number of truea | Sizeb | N |
|---|---|---|---|---|---|---|
| 1 | Genotypes only | 0.57 | 0.55 | 3.57 | 179.43 | 200 |
| Genotypes restricted | 0.56 | 0.55 | 0.84 | 22.07 | 200 | |
| Combined model | 0.82 | 0.82 | 1.27 | 28.38 | 200 | |
| Combined model restricted | 0.82 | 0.82 | 1.06 | 18.70 | 200 | |
| 2a | Genotypes only | 0.61 | 0.54 | 9.98 | 545.33 | 50 |
| Genotypes restricted | 0.56 | 0.55 | 0.86 | 21.66 | 50 | |
| Combined model | 0.83 | 0.81 | 2.78 | 94.32 | 50 | |
| Combined model restricted | 0.83 | 0.82 | 1.14 | 20.57 | 50 | |
| 2b | Genotypes only | 0.73 | 0.54 | 11.65 | 348.86 | 150 |
| Genotypes restricted | 0.58 | 0.56 | 2.01 | 29.57 | 150 | |
| Combined model | 0.85 | 0.78 | 9.35 | 228.43 | 150 | |
| Combined model restricted | 0.83 | 0.82 | 2.48 | 29.26 | 150 | |
| 3 | Genotypes only | 0.62 | 0.54 | 11.32 | 294.68 | 200 |
| Genotypes restricted | 0.58 | 0.56 | 1.75 | 22.84 | 200 | |
| Combined model | 0.83 | 0.82 | 3.94 | 64.17 | 200 | |
| Combined model restricted | 0.83 | 0.82 | 2.04 | 20.40 | 200 |
a Average number of causal simulation markers included.
b Average number of variables in each model.
Averaged results from a 5-fold evaluation procedure on N simulation data sets. Training AROC values were obtained from the internal 10-fold cross-validation on the training sets, as implemented in the R package glmnet. Testing AROC values were determined by applying each of the trained models to the five independent testing sets.
Variables selected by the models
Table 2 shows results from each experiment for the most frequent variables that were selected in at least four out of five trained models within a simulation data set for models 1 and 3. These results reveal that the true variants detected were predominantly common variants, but our model may also have some capacity to identify true rare variants. The gene- and pathway-based regression approaches did not seem to produce substantially different AROC values or find different casual variants than those found using the simpler LASSO approach. However, as shown in Table 2, the proportion of those casual variant occurring was higher in model 3, indicating a more robust model.
Table 2.
Feature selection
| Model type | Model 1 | Model 3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | SNP | Counta | MAFb | Causalc | Gene | SNP | Counta | MAFb | Causalc | ||
| Gene only | FLT1 | C13S523 | 35 | 0.0667 | Y | FLT1 | C13S523 | 71 | 0.0667 | Y | |
| ADAMTS7 | C15S3360 | 22 | 0.0029 | N | SRPR | C11S6885 | 63 | 0.0014 | N | ||
| TG | C8S4379 | 17 | 0.0050 | N | TG | C8S4379 | 61 | 0.0050 | N | ||
| MDN1 | C6S4146 | 15 | 0.0050 | N | RPA3 | C7S297 | 58 | 0.0007 | N | ||
| GOLGA1 | C9S4013 | 13 | 0.0308 | N | LAMB3 | C1S10178 | 54 | 0.0007 | N | ||
| FLT1 | C13S522 | 12 | 0.0280 | Y | RPL27 | C17S2981 | 52 | 0.0007 | N | ||
| Gene restricted | FLT1 | C13S523 | 19 | 0.0667 | Y | FLT1 | C13S523 | 44 | 0.0667 | Y | |
| TEX14 | C17S3819 | 9 | 0.0043 | N | FLT1 | C13S522 | 24 | 0.0280 | Y | ||
| FLT1 | C13S522 | 8 | 0.0280 | Y | CYP3A43 | C7S2324 | 21 | 0.0976 | N | ||
| UBA3 | C3S2197 | 7 | 0.0108 | N | TG | C8S4379 | 18 | 0.0050 | N | ||
| GOLGA1 | C9S4013 | 7 | 0.0308 | N | PRKCA | C17S4578 | 16 | 0.1664 | Y | ||
| CYP3A43 | C7S2324 | 7 | 0.0976 | N | PIK3C2B | C1S9189 | 15 | 0.0065 | Y | ||
| Combined | Age | Age | 200 | NA | Y | Age | Age | 200 | NA | Y | |
| Smoke | Smoke | 163 | NA | Y | Smoke | Smoke | 185 | NA | Y | ||
| FLT1 | C13S523 | 49 | 0.0667 | Y | FLT1 | C13S523 | 81 | 0.0667 | Y | ||
| FLT1 | C13S522 | 16 | 0.0280 | Y | FLT1 | C13S522 | 34 | 0.0280 | Y | ||
| PIK3C3 | C18S2492 | 7 | 0.0172 | Y | PIK3C3 | C18S2492 | 18 | 0.0172 | Y | ||
| HFE | C6S853 | 3 | 0.0036 | N | PRKCA | C17S4578 | 8 | 0.1664 | Y | ||
| ARNT | C1S6533 | 3 | 0.0115 | Y | ARNT | C1S6533 | 8 | 0.0115 | Y | ||
| ACP1 | C2S1 | 2 | 0.0093 | N | UBA3 | C3S2197 | 7 | 0.0108 | N | ||
| Combined restricted | Age | Age | 200 | NA | Y | Age | Age | 200 | NA | Y | |
| Smoke | Smoke | 163 | NA | Y | Smoke | Smoke | 180 | NA | Y | ||
| FLT1 | C13S523 | 49 | 0.0667 | Y | FLT1 | C13S523 | 75 | 0.0667 | Y | ||
| FLT1 | C13S522 | 17 | 0.0280 | Y | FLT1 | C13S522 | 32 | 0.0280 | Y | ||
| PIK3C3 | C18S2492 | 7 | 0.0172 | Y | PIK3C3 | C18S2492 | 17 | 0.0172 | Y | ||
| ARNT | C1S6533 | 3 | 0.0115 | Y | UBA3 | C3S2197 | 6 | 0.0108 | N | ||
| LARGE | C22S1540 | 3 | 0.0201 | N | ARNT | C1S6533 | 6 | 0.0115 | Y | ||
| MMS19 | C10S4869 | 3 | 0.0050 | N | KDR | C4S1861 | 5 | 0.0022 | Y | ||
a Number of times a given variable was observed in four out of five trained models.
b Minor allele frequency.
c Variables used to determine disease risk by the GAW17 simulators.
The top most frequent variables occurred in at least four out of five trained models for models 1 and 3. All models were run for the 200 simulation data sets.
Discussion
In this paper, we assessed the utility of several different strategies for analyzing exome simulation data with a range of causal allele frequencies in the presence of quantitative and phenotypic information. A comparison of the three proposed approaches indicates that the simple LASSO regression model may be an efficient means to determine truly associated variants, but it must be modified to reduce the number of variables to avoid unreasonably large models and overfitting. As discussed in other studies of these data at GAW17, the primary genetic effects that were expected to be observed in this study were those from common variants, such as C13S523 and C13S522 in FLT1. As shown in Table 2, individual genetic variants were identified consistently in four out of five training models in only a minority of simulation analyses. For example, FLT1 C13S523 occurred in at most 81 out of 200 simulations in the combined analysis for model 3. Some loss of power was expected in our analysis, because we developed our models using 80% of a simulation data set to obtain an independent evaluation of our methods’ predictive ability. However, if we consider the same model calculated on all 200 replicates using the entire set of patients (no training set), then FLT1 C13S523 is included in 132 of 200 data sets. In larger studies or in studies that have a preexisting independent sample to validate the predictive model, this diminished power will not affect our method as strongly and our model may be better able to discern genetic predictors.
Some variants, for example, PIK3C3, appeared much more frequently in the models that combined genotypic and phenotypic effects than in models that considered only genotypes. To further investigate this finding, we built logistic regression models for Y and PIK3C3, adjusting for either only population variables or both population and phenotypic variables. PIK3C3 was significant (α = 0.01) in 22 out of 200 data sets for the model adjusted for population only and in 105 out of 200 data sets for the model adjusted for both population and phenotypic variables, providing an explanation for this observation. Our analysis also indicates a significant relationship in the linear regression model for Q1 and PIK3C3 adjusted for population only (184 out of 200 data sets) and adjusted for both population and phenotypic variables (197 out of 200 data sets) at α = 0.01. This may also explain the more frequent occurrence of PIK3C3 in model 3 than in model 1 for the combined models.
Our method was able to reliably ascertain some true variants using subsets of the data for training. In addition, the signs of the regression coefficients for the frequently selected variants were highly consistent (about 99%) over different simulation data sets. However, the ability of our model to find true variants was also accompanied by a large number of noncausal variants. Because several long-range correlations exist within the GAW17 data set, a portion of the variants classified as noncausal in our study may actually be truly associated with the disease state or phenotypic traits. The predictive ability of the LASSO model using only genetic information is limited because none of the examined genomic subsets have a predictive ability that is comparable to that of the phenotypic variables. Nevertheless, incorporating these phenotypic variables into our model increases the proportion of causal genetic variants found using our method.
Conclusion
Although our method is able to detect some causal rare variants, the results do not indicate that this is a promising approach for the general analysis of exome sequencing data that include causal rare variants. Identifying optimal sets of genetic variants for every gene and pathway in a data set may take considerably higher computation time than the standard LASSO model and is expected to generate robust predictive models only when there are several adequately powered common causal variants to distinguish case subjects from control subjects.
Competing interests
The authors declare that there are no competing interests.
Authors’ contributions
JF and YD collaborated on the design of the study. JF carried out the data analysis and drafted the manuscript. YD oversaw the project, participated in the data analysis, and helped to draft the manuscript. Both authors read and approved the final manuscript.
Contributor Information
Joel B Fontanarosa, Email: jfonta3@uic.edu.
Yang Dai, Email: yangdai@uic.edu.
Acknowledgments
The Genetic Analysis Workshop is supported by National Institutes of Health grant R01 GM031575.
This article has been published as part of BMC Proceedings Volume 5 Supplement 9, 2011: Genetic Analysis Workshop 17. The full contents of the supplement are available online at http://www.biomedcentral.com/1753-6561/5?issue=S9.
References
- Meyer KB, Maia AT, O’Reilly M, Teschendorff AE, Chin SF, Caldas C, Ponder BA. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008;6:e108. doi: 10.1371/journal.pbio.0060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BL, Cramer SD, Wiklund F, Isaacs SD, Stevens VL, Sun J, Smith S, Pruett K, Romero LM, Wiley KE. et al. Fine mapping association study and functional analysis implicate a SNP in MSMB at 10q11 as a causal variant for prostate cancer risk. Hum Mol Genet. 2009;18:1368–1375. doi: 10.1093/hmg/ddp035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ. et al. Biological, clinical, and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Sehl ME, Sinsheimer JS, Lange K. Association screening of common and rare genetic variants by penalized regression. Bioinformatics. 2010;26:2375–2382. doi: 10.1093/bioinformatics/btq448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- Dasgupta A, Sun YV, König IR, Bailey-Wilson JE, Malley JD. Brief review of machine learning methods in genetic epidemiology: the GAW17 experience. Genet Epidemiol. 2011;X(suppl X):X–X. doi: 10.1002/gepi.20642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Lin S. Generalized linear modeling with regularization for detecting common disease rare haplotype association. Genet Epidemiol. 2009;33:308–316. doi: 10.1002/gepi.20382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooperberg C, LeBlanc M, Obenchain V. Risk prediction using genome-wide association studies. Genet Epidemiol. 2010;34:643–652. doi: 10.1002/gepi.20509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymczak S, Biernacka JM, Cordell HJ, Gonzalez-Recio O, Konig IR, Zhang H, Sun YV. Machine learning in genome-wide association studies. Genet Epidemiol. 2009;33(suppl 1):S51–S57. doi: 10.1002/gepi.20473. [DOI] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- Almasy LA, Dyer TD, Peralta JM, Kent JW Jr, Charlesworth JC, Curran JE, Blangero J. Genetic Analysis Workshop 17 mini-exome simulation. BMC Proc. 2011;5(suppl 8):S2. doi: 10.1186/1753-6561-5-S9-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan JS, He Y, Calixte R, Nyirabahizi E, Jiang Y, Zhang H. A LASSO-based approach to analyzing rare variants in genetic association studies. BMC Proc. 2011;5(suppl 9):S100. doi: 10.1186/1753-6561-5-S9-S100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Dantzer J, Liu Y. Identification of multiple rare variants associated with a disease. BMC Proc. 2011;5(suppl 9):S103. doi: 10.1186/1753-6561-5-S9-S103. [DOI] [PMC free article] [PubMed] [Google Scholar]