

Abstract
Many bacterial oxidoreductases depend on the Tat translocase for correct cell localization. Substrates for the Tat translocase possess twin-arginine leaders. System specific chaperones or redox enzyme maturation proteins (REMPs) are a group of proteins implicated in oxidoreductase maturation. DmsD is a REMP discovered in Escherichia coli, which interacts with the twin-arginine leader sequence of DmsA, the catalytic subunit of DMSO reductase. In this study, we identified several potential interacting partners of DmsD by using several in vitro protein–protein interaction screening approaches, including affinity chromatography, co-precipitation, and cross-linking. Candidate hits from these in vitro findings were analyzed by in vivo methods of bacterial two-hybrid (BACTH) and bimolecular fluorescence complementation (BiFC). From these data, DmsD was confirmed to interact with the general molecular chaperones DnaK, DnaJ, GrpE, GroEL, Tig and Ef-Tu. In addition, DmsD was also found to interact with proteins involved in the molybdenum cofactor biosynthesis pathway. Our data suggests that DmsD may play a role as a “node” in escorting its substrate through a cascade of chaperone assisted protein-folding maturation events.
Keywords: Chaperones, Molybdenum cofactor biosynthesis pathway, DmsD, Protein–protein interactions, Tat
1. Introduction
A number of bacterial oxidoreductases depend on the Tat (Twin-arginine translocase) system for correct subcellular localization [1–4]. In bacteria, this system represents a method of protein translocation, which differs from the general Sec system in that folded, cofactor-containing proteins are exported into the periplasm. The Tat translocase is thought to form a pore in the inner membrane, comprised of Tat A, B and C subunits [2,4,5]. The TatBC subunits are believed to be the “receptor complex”, and have been shown to be required for Tat substrate interaction with the membrane [6–9], whereas TatA is thought to form a very large oligomeric ring structure, presumed to be the protein-conducting channel itself [6,10].
A twin-arginine motif (S/TRRXFLK motif) in the leader peptides of Tat substrates was identified by Berks in 1996 [1]. This leader motif has been shown to be important in the transport and targeting of these proteins to the translocase [11; for a review see 6]. DmsD, from Escherichia coli, was shown to interact in vitro with the twin-arginine leader of the catalytic subunit from dimethyl sulfoxide (DMSO) reductase, DmsA [12]. DMSO reductase maturation requires the Tat translocase, but also DmsD, which plays a chaperone role in cytoplasmic processes. In support of this, DmsD was found to interact with the membrane in a TatBC dependent manner [9]. Furthermore, dmsD knockout mutants demonstrate an almost entire loss of DMSO reductase activity, suggesting that DmsD may not only be required for correct DMSO reductase localization, but also for proper enzyme maturation [6,13]. DmsD was designated a REMP (Redox Enzyme Maturation Protein) along with other proteins of similar proposed function [14]. In addition to DmsD, REMPs for other oxidoreductases have been implicated in oxidoreductase enzyme maturation. These include NarJ for nitrate reductase, TorD for TMAO reductase, and HyaE/HybE for hydrogenase-1 and 2, respectively [14]. In a review from Palmer et al. [15], a suggestion was put forward that all Tat substrates might have some form of a leader-binding specific chaperone.
REMPs may play a number of different roles in oxidoreductase maturation, including: interaction with the twin-arginine leader peptides, maintaining the oxidoreductases in a cofactor-competent state, Sec translocase avoidance, proofreading, protease protection, interaction with the ribosome, interaction with the membrane, and interaction with components of the Tat translocase [14,16].
Recent work is beginning to deconvolute the functions of REMPs. TorD has been suggested to be involved in the protection of TorA from protease degradation [17]. It was also demonstrated to play specific roles in both cofactor insertion as well as regulating Tat export [18]. Moreover, it has been suggested that GTP binding by REMP proteins themselves may play a role in the REMP/substrate maturation process [19]. This GTP binding to TorD has been suggested to in fact be binding to the mature molybdopterin-guanine dinucleotide form of the molybdenum cofactor (Moco) as part of the cofactor insertion event [20]. NarJ has been shown to facilitate NarGH interaction with Moco biosynthetic machinery and facilitate NarGHI assembly [21–23].
In addition to these system specific REMP chaperones, it is recognized that general molecular chaperones may also play a role in oxidoreductase maturation pathways. A study from DeLisa’s group and coworkers has shown that DnaK has stabilizing effects on Tat-dependent substrates [24] and that work from Brüser’s group has demonstrated roles of the chaperones SlyD and DnaK [25]. It is not known if general chaperones work together in parallel or in series with the system specific chaperones. Although evidence for REMP-RR-leader and DmsD-TatBC interactions exists, little else is known if other proteins are involved with DmsD, and how these may contribute to the redox enzyme maturation pathway. Many specific and low specificity transient protein interactions can occur from the time of the nascent polypeptide exiting from the ribosome to a targeted, assembled and functional mature protein. From this line of thought we asked the question: do the REMP proteins, in this case DmsD, interact with other proteins in the cell? Such an idea would see the REMP escorting its substrate protein through a cascade of each protein-folding/stabilizing machine.
Here, in order to answer this question, we screened using a variety of in vitro protein–protein interaction methods, including: affinity chromatography, co-precipitation, and cross-linking. Protein chaperones and related proteins identified from these experiments were subsequently tested by such in vivo techniques as bacterial two-hybrid (BACTH) and bimolecular fluorescence complementation (BiFC). BACTH is an in vivo approach for protein–protein interaction studies based on functional reconstitution of the two fragments of Bordetella pertussis adenylate cyclase (CyaA) [26,27]. BiFC is a biophysical technique based on the reassembly of the two nonfluorescent moieties of the yellow fluorescent protein (YFP) and the subsequent refolding of YFP when the two halves are brought together by the interacting partners fused to them [28,29]. The results presented here demonstrate that DmsD partners with other general chaperone systems and my also play a similar role to TorD in targeting to the Moco biosynthesis proteins and may lead its substrate through a protein chaperone cascade.
2. Materials and methods
The bacterial strains and plasmids used in this work are described in Table 1.
Table 1.
Bacterial strains and plasmids used in this study.
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| E. coli strain | ||
| DH5α | F′/endA1 hsdR17 ( Equation) glnV44 thi-1 recA1 gyrA (NalR) relA1 Δ(lacIZYA-argF)U160 deoR (Φ80dlacΔ(lacZ)M15) |
Laboratory collection |
| HB101 | F−, Δ(gpt-proA)62, leuB6, glnV44, supE44, ara14, galK2, lacY1, rpsL-20(StrR), xyl-5, Δ(mcrC-mrr), hsdS20, mtl-1, recA13 | Laboratory collection |
| C41(DE3) | F−, ompT gal hsdSB ( Equation) dcm lon λDE3 and an uncharacterized mutation described in the reference |
[30] |
| MC4100 | F− araD139 Δ(argF-lac)U169 rpsL150 (StrR) relA1 flbB5301 deoC1 ptsF25 rbsR | Laboratory collection |
| DADE | MC4100; ΔtatABCD/E | [31] |
| BTH101 | F−, cya-99, araD139, galE15, galK16, rpsL1(StrR), hsdR2, mcrA1, mcrB1 | D. Ladant |
| Plasmid | ||
| pTDMS28 | His6-T7-DmsD cloned into pRSETC | [12] |
| pTDMS124 | His6-T7-TehB cloned into pRSETC | [32] |
| pKNT25 | T25 fragment of the catalytic domain of Bordetella pertussis adenylate cyclase CyaA; KanR. | [26,27] |
| pUT18 | T18 fragment of the catalytic domain of Bordetella pertussis adenylate cyclase CyaA; AmpR. | [26,27] |
| pDmsDT25 | pKNT25 derivative, DmsD fused to T25 | [33] |
| pDmsALT18 | pUT18 derivative, DmsA leader fused to T18 | [33] |
| pTehBT18 | pKNT25 derivative, TehB fused to T25 | This work |
| pMogAT18 | pUT18 derivative, MogA–T18 fusion | This work |
| pMoeAT18 | pUT18 derivative, MoeA–T18 fusion | This work |
| pMoeBT18 | pUT18 derivative, MoeB–T18 fusion | This work |
| pMobAT18 | pUT18 derivative, MobA–T18 fusion | This work |
| pMobBT18 | pUT18 derivative, MobB–T18 fusion | This work |
| pDnaKT18 | pUT18 derivative, DnaK–T18 fusion | This work |
| pDnaJT18 | pUT18 derivative, DnaJ–T18 fusion | This work |
| pGrpET18 | pUT18 derivative, GrpE–T18 fusion | This work |
| pGroELT18 | pUT18 derivative, GroEL–T18 fusion | This work |
| pEf-TuT18 | pUT18 derivative, Ef-Tu–T18 fusion | This work |
| pTigT18 | pUT18 derivative, Tig–T18 fusion | This work |
| pDmsDYcK | pDmsDT25 derivative, DmsD fused to C-terminus of YFP (Yc) | This work |
| pDmsALYnK | pDmsALT18 derivative, DmsA leader fused to N-terminus of YFP (Yn) | This work |
| pYnK | pUT18 derivative, T18 replaced by Yn | This work |
| pMogAYnK | pDmsALYnK derivative, MogA–Yn fusion | This work |
| pMoeAYnK | pDmsALYnK derivative, MoeA–Yn fusion | This work |
| pMoeBYnK | pDmsALYnK derivative, MoeB–Yn fusion | This work |
| pMobAYnK | pDmsALYnK derivative, MobA–Yn fusion | This work |
| pMobBYnK | pDmsALYnK derivative, MobB–Yn fusion | This work |
| pDnaKYnK | pDmsALYnK derivative, DnaK–Yn fusion | This work |
| pDnaJYnK | pDmsALYnK derivative, DnaJ–Yn fusion | This work |
| pGrpEYnK | pDmsALYnK derivative, GrpE–Yn fusion | This work |
| pGroELYnK | pDmsALYnK derivative, GroEL–Yn fusion | This work |
| pEf-TuYnK | pDmsALYnK derivative, Ef-Tu–Yn fusion | This work |
| pTigYnK | pDmsALYnK derivative, Tig–Yn fusion | This work |
2.1. Preparation of bait and prey protein samples for in vitro assays
As the bait protein, the tagged His6-T7-DmsD was isolated from E. coli C41(DE3) cells containing pTDMS28 [12]. An unrelated protein, His6-T7-TehB, of a comparable molecular mass and pI, was generated from E. coli C41(DE3) containing pTWT124, and used as the negative control [32]. Prey proteins are from wild-type (WT) E. coli strain MC4100 and/or its ΔtatABCD/E mutant DADE [31]. The cell growth conditions and the preparation of bait and prey protein samples were addressed in detail in [34].
2.2. In vitro protein–protein interaction techniques
The in vitro protein–protein interaction methods used in this work include Ni2+-affinity chromatography, co-purification, co-precipitation, and cross-linking. These methods were in general performed based on stringency approaches optimized for transient protein–protein interactions. The Ni2+-affinity chromatography and co-precipitation methods were described previously [34]. Co-purification was carried out similarly as shown in Ni2+-affinity chromatography, however, both soluble cytosolic fraction and 2% (v/v) CHAPS-solubilized membrane fraction was obtained from anaerobically grown, uninduced MC4100 cells containing pTDMS28 or pTWT124 were incubated with Ni2+-NTA resin for 2.5–3 h at 4 °C.
Cross-linking experiments were performed according to the kit provider (Pierce Biotechnology Inc.). In all experiments, the Sulfo-SBED biotin UV-reactive label transfer reagent was used. Briefly, purified protein (DmsD or control) was reacted with Sulfo-SBED reagent (5 μg/μL of protein along with 1.12 mg of reagent per 25 μL in 125 mM DMSO) at room temperature for 30 min in the dark to allow labeling. Samples were applied to a 5-mL Hi-Trap desalting column to remove unreacted reagent. Samples (500 μL) were incubated with soluble or solubilized membrane fractions from anaerobically grown WT E. coli MC4100 suspended in Buffer B (8 mM NaH2PO4, 140 mM NaCl, 2 mM K2PO4, 10 mM KCl, pH7.4), and incubated at room temperature for 15 min in the dark. Samples were then exposed to 365 nm UV-irradiation for 20 min. Label transfer was accomplished with the addition of 50 mM DTT, followed by incubation of the samples on ice for 2–3 h with gentle, intermittent mixing. Samples were then applied to monomeric avidin columns (Pierce), and the columns were washed to reduced A280. Eluted protein fractions were then collected in 4–6 column volumes, following application of 2 mM biotin in PBS, and then 100 mM glycine (pH 2.8) to the columns. Eluted fractions were pooled, concentrated with Millipore Biomax filters (0.5 mL volume, 10 kDa cut-off), and assessed through standard SDS-PAGE procedures. Cross-linked, label transferred biotinylated proteins were detected through Western blotting using Streptavidin–HRP (Pierce).
2.3. Protein identification methods
Proteins isolated from various protein–protein interaction experiments were excised from stained SDS-PAGE gels and then subjected to a 16-h, 37 °C in-gel trypsin digestion. Peptide mass fingerprinting was performed using the SELDI-TOF mass spectrometry (MS) in our department’s biophysics suit or by MALDI-TOF MS at the Southern Alberta Mass Spectroscopy Centre (Faculty of Medicine, University of Calgary). Alternatively, LC–MS/MS analysis was carried out at the Genome BC Proteomics Centre, University of Victoria, BC, Canada. Masses were imported into Mascot, PeptIDent or ProFound databases for analysis.
2.4. BACTH assay
E. coli DH5α was used as the cloning host and a cya deficient E. coli strain BTH101 (from D. Ladant) was used as the recipient in all BACTH assays. Bacterial cultures were aerobically grown in LB-broth medium supplemented with 100 μg/mL ampicillin and 50 μg/mL kanamycin, where applicable. Genes of the various interacting proteins were individually amplified by PCR using E. coli HB101 genomic DNA as the template. Then, the PCR products were subcloned into the corresponding sites of pUT18 [26,27], providing a fusion to the T18 fragment of CyaA. pDmsDT25, constructed previously [33], was used to generate another fusion protein, DmsD-T25, in the BACTH assays. A control protein for this study, TehB, was also evaluated. This protein is involved in metal oxyanion resistance from E. coli, and is a protein of similar size and pI. We have found that it expresses and accumulates at ca. the same levels as DmsD upon expression from the same promoter and vector. We used this protein in the initial studies exploring different interaction methods for transient interactions and has been our control for the in vitro studies [34]. Table 1 shows the plasmids constructed for the BACTH assays. Efficiencies of protein–protein interactions were quantified by measuring β-galactosidase activity in Miller units [27,35,36].
2.5. BiFC assay
BiFC assays by fluorescence spectrometry were performed with a Fluorolog-3 spectrofluorometer (Jobin Yvon Inc.) as described previously [37] with minor modifications. After overnight incubation of E. coli transformants on the agar plates at 37 °C, the further incubation time at room temperature was shortened to 3 days. In addition, rather than excitation at the λmax for YFP at 513 nm, the excitation wavelength was set at 488 nm. Although this is far from the λmax for YFP, the interference of the emission spectrum by the excitation spectrum, which could be observed when excitation was at 513 nm, was significantly reduced. Thus the emission peak at 526 nm has a better resolution.
Plasmids used for the BiFC assays were generated based on the constructs for the BACTH assays. pDmsDYcK and pDmsALYnK were constructed by replacing the T25 and T18 fragments in pDmsDT25 and pDmsALT18 with PCR fragments encoding the C- (residues 155–238, Yc) and N-(residues 1–154, Yn) terminal moieties of YFP, respectively. The linker sequences in the fusion proteins were also changed to those used by Kerppola et al. [28]. These two plasmids were designated differently elsewhere [38]. The plasmids harbouring genes of the various interacting proteins were generated correspondingly by replacing the T18 fragment in those plasmids constructed for the BACTH assays with the Yn fragment, thus generating a fusion of the interacting proteins to Yn. For the negative control, a plasmid pYn was constructed by replacing T18 with Yn in pUT18 [26,27]. Plasmids constructed for this approach are listed in Table 1.
3. Results
The first question that was asked in this study is if DmsD interacts with other proteins other than the substrate twin-arginine leader peptide from DmsA. In order to evaluate this question, we used epitope tagged DmsD to probe proteins in the cell extracts separated by SDS-PAGE and transferred onto a nitrocellulose membrane for a far-Western analysis. Additionally, we performed protein chip experiments analyzed by SELDI-TOF MS. These preliminary experiments provided evidence that interacting proteins of DmsD exist beyond the twin-arginine leader substrate (data not shown; [34]); however, these approaches do not provide identification of the interacting proteins.
Given these positive preliminary results, we employed a set of in vitro protein–protein interaction techniques to isolate and identify proteins that interact with DmsD. In consideration of the expected transient nature of the protein–protein interactions being sought, we recognized that we would need multiple techniques to identify interacting proteins to overcome method specific limitations. The figures presented here are representative data from various multiple replicates, minimally 3 at each experimental condition explored.
In some experiments, a Δtat strain was used. Our reasoning was that substrates would not be terminally processed/translocated and thus may become enriched in the cell and potentially remain in a complex with other accessory proteins. Similarly, as DmsABC and many other Tat substrates are anaerobically expressed proteins, we performed a number of the experiments using cell extracts prepared from anaerobically grown cultures. We employed a His6-T7-tagged version of DmsD for our in vitro experiments for immobilization and identification. We have found this tagged version of DmsD to be functional in binding DmsA leader peptide [39,40] and interacting with TatBC [9].
For transient interaction studies, we found that varying stringency conditions and key controls are important. We developed a comparative approach where the experiments were performed under the same conditions as DmsD but with a system ‘silent’ protein (TehB) and the proteins from cell-free extracts of the two were compared and different proteins were then extracted and identified. For a more detailed description and background on the methods used here and the choice of controls, the reader is directed elsewhere [34]. Although a number of proteins were identified with our approaches, we chose to concentrate only on proteins identified by multiple methods (Table 2) for further validation of the interactions by two in vivo protein–protein interaction approaches.
Table 2.
Chaperones and molybdopterin cofactor biosynthesis proteins identified to interact with DmsD by in vitro methods.
| Interactor | Description | Technique |
|---|---|---|
| Ef-Tu | Translation elongation factor (TufA) | 1, 3, and 4 |
| DnaK | Hsp70 Homologue; Chaperone | 1, 2, 3, and 4 |
| GroEL | 60 kDa Chaperonin | 1, 2, 3, 4, and 5 |
| GrpE | GrpE protein; part of the DnaK/DnaJ/GrpE system | 1, 3, and 4 |
| Tig | Trigger factor; interplay with DnaK/GroEL as chaperones | 1, 3, and 4 |
| MoeB | Molybdopterin biosynthesis | 1, 3, and 4 |
Techniques:
- Cross-linking followed by direct LC–MS/MS for detection.
- Ni2+-affinity chromatography followed by SDS-PAGE and MALDI-TOF MS.
- Co-precipitation followed by SDS-PAGE and MALDI-TOF MS.
- Cross-linking followed by SDS-PAGE and MALDI-TOF MS
- Co-purification followed by SDS-PAGE and MALDI-TOF MS.
3.1. Identification of potential DmsD interactors by chromatography approaches
Affinity chromatographic methods provide a tool for examining low-abundance proteins interacting with a protein of interest. Large volumes of cell-free extracts containing prey protein can be applied onto a small column containing immobilized bait protein. In this fashion, the column can effectively immobilize and concentrate lower-abundance prey proteins in a small volume of affinity resin. Selection of experimental parameters that will permit specific protein–protein interactions to occur while minimizing background is of great importance (addressed in [34]). Previous studies using pre-immobilized His6-T7-DmsD to the resin resulted in the successful identification of pre-DmsA [12]. Here, purified His6-T7-DmsD was pre-equilibrated with the Ni-NTA agarose to provide a high concentration of bait, generating an effective affinity column for interacting proteins. Using this approach under a variety of experimental conditions, it was possible to identify several interactors using standard peptide mapping and subsequent MS analysis. Under almost all conditions, different chaperones were found to interact with the immobilized DmsD (Fig. 1, Table 2).
Fig. 1.

Representative findings from affinity chromatography approaches. (A) Co-purifying proteins with DmsD preparation (lane 1; lane 2. control protein preparation). Proteins with asterisk confirmed to interact with DmsD by far-western (not shown). (B) Potential His6-T7-DmsD-interacting species from anaerobic solubilized membrane prey fractions. Eluted fractions from Ni2+-NTA resin exposed to CHAPS-solubilized E. coli membrane prey fractions. Lane 1, His6-T7-DmsD-immobilized resin; lane 2, His6-T7-TehB control protein immobilized resin. (C) Co-purification experiments demonstrating potential His6-T7-DmsD-interacting species from anaerobic expressed DmsD soluble fractions. Lane 1, MC4100; lane 2, MC4100/pTDMS28. (D) Co-purification experiments demonstrating potential His6-T7-DmsD-interacting species from anaerobic solubilized membranes. Putative interacting proteins are indicated with asterisks. Arrows denote the His6-T7-DmsD or control protein. Molecular mass markers (in kDa) are indicated to the left.
In the second chromatographic approach, less aggressive wash conditions were performed in order to allow for co-purification of DmsD interactors. Under our media conditions for DmsD purification, we observed low levels of DmsD accumulation without induction, presumably due to promotor leakage. Therefore, we purified DmsD from these expression conditions on a Ni-NTA matrix under low stringency conditions. A key chaperone found to co-purify along with His6-T7-DmsD was GroEL, a bacterial chaperone involved in protein folding in the cytosol (Fig. 1A). GroEL antibody (mouse monoclonal anti-GroEL; StressGen BioReagents) was used to further confirm the identity of the co-purified GroEL in a far-western and in co-immunoprecipitation (not shown).
3.2. Identification of potential DmsD interactors through co-precipitation experiments
Purified His6-T7-DmsD was covalently immobilized to AminoLink resin, and was then exposed to anaerobically grown E. coli extract. Following SDS-PAGE separation, the lanes were cut out and subjected to LC–MS/MS analysis. By performing LC–MS/MS on a lane using the co-precipitation control resin and the TehB control protein, it was possible to subtract out proteins that were a system artifact. This method was successful in identifying MoeB, GroEL and other general chaperone proteins (Table 2).
3.3. Identification of potential DmsD interactors through cross-linking and label transfer experiments
Chemical cross-linkers have greatly enhanced the detection of protein interactions, especially in cases of protein complexes, as well as more transient or weak interactions that may not withstand standard protein interaction detection strategies (for a review, see [41]). Label transfer methods, as a subset of protein cross-linking strategies, are excellent tools used in the detection of protein–protein interactions [42]. We chose to use the newly developed Sulfo-SBED biotin UV-reactive label transfer reagent. Here, purified DmsD was reacted with the Sulfo-SBED reagent through amine groups. The labeled, purified bait protein was then incubated with prey protein fractions. Following UV-irradiation, the now-cross-linked bait and prey proteins were cleaved through a reducing agent, resulting in biotin label transfer to the cross-linked prey proteins. The now-biotinylated prey proteins were purified through the use of a monomeric avidin column, resulting in significantly reduced matrix-dependent background levels. The fact that there is a single purified source of labeled bait protein means that, following label transfer, biotinylated prey proteins must have been the result of close proximity to the bait. Fig. 2 depicts results from His6-T7-DmsD cross-linking studies. Protein bands of potential DmsD interactors were excised from SDS-PAGE gels, subjected to tryptic digestion and subsequent LC–MS/MS analysis (Table 2).
Fig. 2.
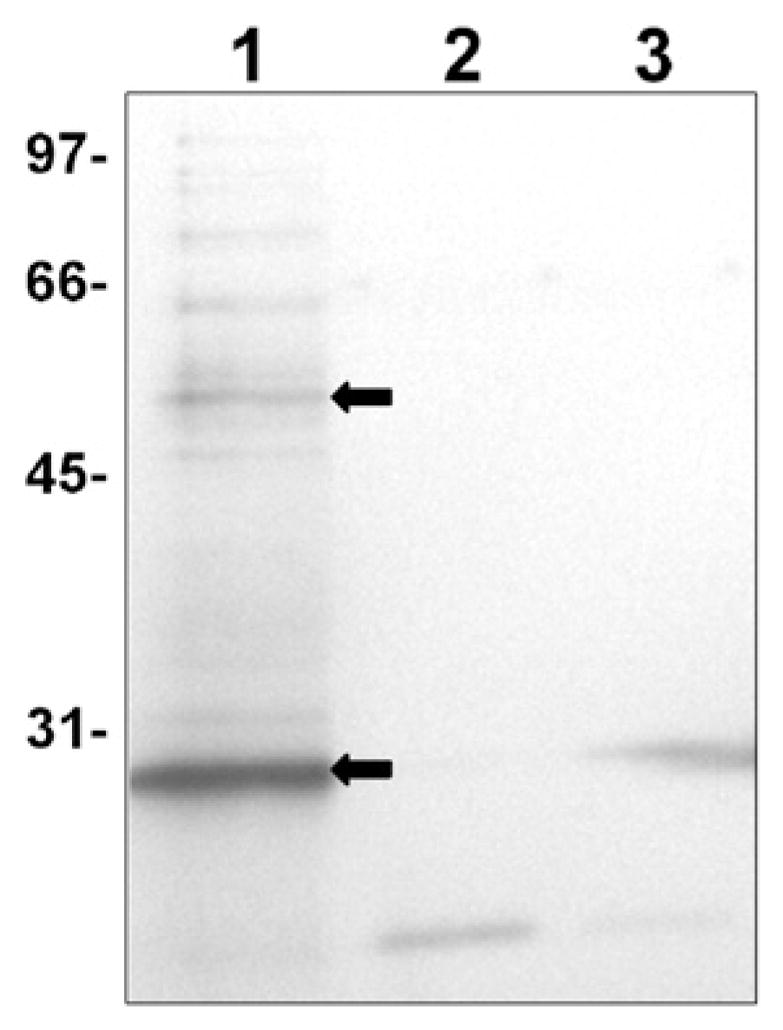
Representative interaction experiments using Sulfo-SBED cross-linker. Sulfo-SBED reacted purified His6-T7-DmsD was incubated with wild-type soluble prey protein fraction (~1.6 μg bait to prey extract protein ratio) and exposed to UV to permit cross-linking. Parallel experiments performed with non-reacted control. Cross-linked samples were cleaved with DTT to transfer the biotin label to the interacting partner and purified on an avidin column. After incubation the various soluble cell extract samples were run on a 12% SDS-PAGE gel and electroblotted. Following blocking in 10% milk/TBS/0.2% azide, the blot was initially probed with 100 ng/mL Streptavidin–HRP (Novagen) prior to development with Opti-4CN HRP developer reagent (Bio-Rad). Therefore bands in the figure contain biotin. Lane 1, Sulfo-SBED cross-linker reacted His6-T7-DmsD with soluble cell extract; lane 2, His6-T7-DmsD with no cross-linker exposed to cell extract; lane 3, no His6-T7-DmsD added to the cell extract. Molecular masses (in kDa) are indicated to the left. Non cross-linked biotin labeled DmsD bands are highlighted with an arrow indicating both monomer and dimer forms (confirmed by anti-DmsD antibody). Additional bands found in control lanes 2 and 3 are naturally occurring biotinylated or biotin-binding proteins.
To further verify that DmsD was interacting with GroEL, we used pure GroEL (StressGen Bioreagents) and purified DmsD were found to cross-linking leading to label transfer, as GroEL was found to immobilize to the avidin column via the biotin label transferred from the DmsD. Although this cannot be taken alone as evidence of DmsD works with GroEL for DMSO reductase maturation, it does provide some support for the DmsD interaction with GroEL and other chaperones not being the result of unfolded DmsD acting as a chaperone substrate.
3.4. In vivo BACTH investigation of DmsD-interacting proteins
We employed the BACTH method based on the use of two fragments of CyaA [26,27]. Fig. 3 demonstrates that the proteins identified as interactors by the above in vitro methods, also demonstrate interactions in vivo. The magnitude of the observed β-galactosidase activity resulted from the level of cAMP produced from the reconstitution of the T25 and T18 domains of CyaA. For a strong, permanent interaction between two proteins, there would be a large production of cAMP and thus a high β-galactosidase activity. We saw this in the interaction between DmsD and the DmsA-RR-leader peptide showing a very strong signal (Fig. 3A). Other interactions were found to have much lower signals.
Fig. 3.

DmsD interactions revealed by bacterial two-hybrid approach. Functional complementation of the two CyaA domains brought together by the interaction of DmsD and a given interacting partner result in cAMP synthesis and the subsequent induction of the Lac operon that is evaluated by the β-galactosidase activity. Miller unit activities represent the relative levels of hybrid protein–protein interaction. The background was established using the control system of pDmsDT25 interacting with pUT18-Zip, which is not expected to have any interaction. A: Interactions with general chaperones. For comparison DmsD interaction with its DmsA leader substrate is also shown. To show relative amounts the Tig and DmsA interactions are shown at 1/3 (×3) and 1/5 (×5) their values obtained. B: Interactions with proteins involved in the molybdopterin cofactor biosynthesis. Data was subjected to the T-Test and were found to be significantly different than background control for all samples except for MobA.
For more transient interactions, the amount of cAMP produced is an indication of the residence time of the DmsD interaction with a given partner. So for short interaction times a low Miller activity would be found. Additionally, it was important not to over express the genes involved, which could lead to concentration artifacts, which could lead to increased signals. The Miller assay is widely used in laboratories worldwide, however, the exact details of the assay are not generally stated in publications. Due to the many steps and reagents involved in the assay, variations in a single element can affect the results drastically. It has been recognized in an article reviewing the uses of lacZ for yeast two-hybrid studies that the assays are more useful when comparing results from internal controls that were treated identically during individual experiments [43]. Many of the results observed in Fig. 3 are lower than what is normally expected for such a reporter system. To investigate the validity of these weaker signals, we utilized a control protein, TehB, which was used as the background control in the in vitro experiments. Miller unit activities from TehBT25 with T18 constructs of the proteins investigated in Fig. 3 showed values ranging from 45 to 60 (Supplementary Fig. 1), which is within the same range as the Zip peptide control with DmsD. Therefore, although ‘weak’ signals are in some cases observed and likely a result of very weak or transient interaction, the values are still significantly different from the control based on a student T-Test and compared to the TehB control protein.
Fig. 3A also displays data investigating interactions with other general chaperones. As DnaK, GrpE and GroEL were identified by in vitro methods, in this study we also investigated DnaJ as there is a chaperone cascade of DnaK – DnaJ – GrpE – GroEL [44]. Our data suggests a weak, transient interaction with this chaperone cascade. It is intriguing that the trigger factor (Tig) showed strong interaction evidence with DmsD in this assay although no fluorescence signal could be detected in another in vivo assay (see below). Similar method-related discrepancies were also observed when two fluorescence-based in vivo techniques were employed in the studies on the associations of E. coli NarJ with its substrate NarG [37].
Like the thoughts for the chaperone cascade, since MoeB of the molybdopterin biosynthesis pathway was found to interact with DmsD by the in vitro methods, other members of this pathway were chosen as it has been shown that the final stages of molybdenum cofactor biosynthesis occur on a complex consisted of MogA, MoeA, MobA and MobB [21]. Our BACTH data shows reasonably strong interactions of DmsD with MoeA and MoeB, weaker interactions with MogA and MobB, and very weak or no interaction with MobA (Fig. 3B).
3.5. BiFC confirmation of DmsD interacting proteins
The in vivo interaction between DmsD and the DmsA-RR leader peptide was confirmed by using the BiFC method [38]. As the YFP reassembly leading to fluorophore generation is an irreversible process, any weakly or transiently interacting protein partners are trapped and the interaction becomes permanent. Therefore, this technique is specifically powerful in investigating weak or transient protein–protein interactions.
Fig. 4 shows the specific intensity values determined for E. coli MC4100 and DADE transformants expressing both DmsD-Yc and DmsAL-Yn as well as various other Yn-tagged proteins after a 3-day incubation at room temperature on nutrient agar. No data is available for Tig, since the expression of the Yn-tagged trigger factor (Tig–Yn construct) seemed to be lethal to the cells. Cell growth was inhibited but not ceased for DnaJ and Ef-Tu transformants, therefore data was still able to be collected but the lower signals may be due to this stress. Regardless this approach supports further that DmsD interacts with the chaperone cascade (Fig. 4A). This approach also further confirms that DmsD interacts with all the five proteins involved in the molybdenum cofactor biosynthesis pathway (Fig. 4B), perhaps in a way similar to the way NarJ interacts with NarG for molybdenum cofactor insertion [22] and TorD that facilitates an interaction of TorA with the cofactor biosynthetic system [20].
Fig. 4.

DmsD interactions revealed by the biomolecular fluorescence complementation assay. The specific intensity is the relative fluorescence from the reconstituted YFP protein ratio to the cell density. The negative control for these experiments is the data obtained from transformants containing both pDmsDYcK and pYnK which is based on the same parent plasmid for the other constructs used in the BiFC assay but only harbours the gene encoding the N-terminal moiety of YFP. A: Interactions with general chaperones. B: Interactions with proteins involved in the molybdopterin cofactor biosynthesis. For comparison DmsD interaction with its DmsA leader substrate is also shown in panel B. Shaded bars represent data obtained using a Δtat strain, whereas open bars are data for WT E. coli. Data was subjected to the T-Test and were found to be significantly different than background control for all samples except interaction with DnaJ in WT host.
Performing these experiments with a ΔtatABCD/E strain led to stronger signals, which is in agreement with our hypothesis that the maturation pathway cannot continue and thus ‘hangs up on hold’ allowing for more complexes to get trapped by the YFP chromophore cross-link. The difference in signals may be telling us about the ‘lifetime’ or kinetics of their interactions.
4. Discussion
The maturation of a multi-subunit enzyme with complex cofactors would involve a number of steps from translation at the ribosome, folding and cofactor insertion, targeting, translocation across the membrane, and finally assembly into an active functional complex. Here we asked the question what is involved in this process for DMSO reductase by examining if there is an interactome for this system specific chaperone, DmsD.
The existence of proteins that transiently interact with DmsD was explored using a variety of in vitro protein interaction approaches. Each technique reveals different bait–prey interaction information: protein chip and far-western approaches can be used to define that specific prey proteins do exist but only provide a list of molecular masses of targets. On the other hand, affinity chromatography, co-precipitation and cross-linking studies, in conjunction with protein identification methods such as peptide mapping/mass spectrometry revealed identities of ‘potential’ interactors. Because of the variations in physiochemical conditions, each technique in its isolation was not considered to provide conclusive evidence for a component of the DmsD interactors, nor a complete list of proteins in the interactome. The recurrence of a given prey protein in several protein–protein interaction approaches generates increased confidence in the potential bait–prey interaction, as well as ruling out the possibility that the interaction is simply a technique-specific artifact. The relative success of these experiments could be measured by the ability of these techniques to pull out expected DmsD interactors as well as a negative control protein not displaying a given interaction [34]. Affinity chromatography experiments with immobilized, purified His6-T7-DmsD exposed to crude subcellular prey protein extracts led to the co-purification of not only the DmsD substrate DmsA, but also the closely related homologues. DmsA was also identified in co-precipitation and cross-linking experiments. Thus, several of the in vitro studies undertaken here were able to identify known as well as expected interactors. To protect against false positives, we utilized several different methods to ‘sample’ as many conditions as possible and we have chosen to concentrate on only those proteins found with multiple methods. However, it would be naive to assume Table 2 provides a complete list of the DmsD interactome as with such an approach we are sure to have missed some interactors via false negatives.
Examination of DmsD interactors reveals several features in the context of redox enzyme maturation in vivo. GroEL, a general chaperone protein involved in protein folding, may represent a key protein contributing either directly or indirectly to the DmsD interactome. Within the context of redox enzyme maturation, evidence in the literature has implicated GroEL with the insertion of the molybdenum–iron cofactor into the nitrogenase enzyme of Azobacter vinelandii [45]. The authors proposed that GroEL could be more generally involved in metalloenzyme maturation. Thus, the presence of GroEL could be significant in the context of the maturation pathway involving DmsD, given that DMSO reductase is itself a metalloenzyme containing both molybdenum cofactors as well as iron–sulfur clusters. Moreover, GroEL, in cooperation with DnaK (a known in vitro RR-leader interactor; [12,24,25]), has been implicated in the folding of plant ferredoxin-NADP+ reductase [46]. Recent findings have also implicated GroEL in an interaction with NapD, the REMP protein for periplasmic nitrate reductase in E. coli [47].
Studies by Houry et al. [48] and Kerner et al. [49] generated a list of ~300 E. coli GroEL substrates and interacting proteins. These general high-throughput studies did not find any of the REMPs as GroEL substrates, supporting that the interaction observed here is not because DmsD is a substrate of GroEL. Several of the potential DmsD-interacting proteins identified through co-precipitation and cross-linking studies, such as Tig, GrpE, and Ef-Tu, have been identified as potential GroEL substrates/interactors in the cell [48,49]. As we also identified these proteins, it is possible that the above GroEL substrates may represent indirect DmsD interactors (As in protein X interacts with Y which has Z interacting with it) and/or that DmsD is ‘holding’ the RR-leader containing substrate for the chaperonin/foldase.
In prokaryotes, the ribosome-associated trigger factor (Tig) is the first chaperone nascent polypeptides encounter when they emerge the ribosomal exit tunnel and are released into the cytosol. Evidence showed that Tig forms a protective shield for nascent polypeptides on the ribosome [50]. Although no data is available from our BiFC assay because of an unclear lethal effect of the expression of the Tig–Yn fusion, the in vitro data from the co-precipitation and cross-linking experiments as well as the in vivo BACTH data showed a clear interaction between DmsD and Tig. Moreover, DmsD interaction with Tig has an interesting support as Tig has been shown to interact in vivo with translating RR-leader containing peptides, which may suggest direct participation in REMP substrate binding [51]. Together these data suggest that DmsD may interact with DmsA by attaching to its RR-leader immediately after the DmsA polypeptide leaves the ribosome and acts in concert with Tig to protect it from untimely degradation or aggregation processes. However, the weak and/or transient interaction between DmsD and the translation elongation factor Ef-Tu shown in the BiFC assay suggests that DmsD might approach to the freshly synthesized DmsA leader even prior to the termination of synthesis of the DmsA polypeptide chain. The above implies that DmsD may be part of a chaperone cascade complex facilitating a folding-maturation pathway for the substrate protein.
The proteins discussed above suggest a chaperone cascade in which DmsD participates or escorts its substrates along. In addition to these chaperones, links to cofactor biosynthesis are also suggested through the interactor hit of MoeB. The system specific chaperone NarJ has been shown to be required for the interaction of NarGH (nitrate reductase A) with a complex of proteins involved in the last stages of the molybdenum cofactor biosynthetic pathway including MobA, MobB, MogA and MoeA [21]. However, MoeB is considered to be involved earlier in the pathway [52], and is a sulfurase in a complex with MoaD [53]. Given the present understanding of molybdenum cofactor biosynthesis, our observation of DmsD interaction with MoeB was unanticipated. Whether NarG also interacts with MoeB or not was not reported [21,22]. However, a mutant in MoeB was found to affect DmsA maturation but not NarG or TorA [54], thus there may be a unique path related to DMSO reductase maturation requiring chaperone participation in a different fashion. Regardless we used the same two-hybrid method (BACTH) as the NarG study [21] and showed an interaction of DmsD with MoeA and MobB similar to that found with NarG, suggesting that the molybdenum cofactor biosynthesis may occur in a metabolon complex that the REMP chaperones target their substrates to. The in vivo BiFC data not only confirms our in vitro observation but also shows the interactions of DmsD with all the other four molybdopterin cofactor biosynthetic enzymes involved in the maturation of the nitrate reductase A. Furthermore, it was proposed that the maturation of the E. coli trimethylamine oxide reductase (TorA) requires TorD to make the enzyme competent to receive the molybdenum cofactor [55]. It was proposed that TorD acts as a platform connecting the last step of the synthesis of the molybdenum cofactor just before its insertion into the catalytic site of TorA [20]. Taken together, all these data suggest a general chaperone role of REMPs in the molybdenum cofactor insertion, although different systems with different REMPs may be interacting with the metabolon complex differently.
To seek transient and weak interactors of DmsD, we employed various in vitro protein–protein interaction techniques. These were then verified by two in vivo two-hybrid approaches. On this note, reliance on protein interaction prediction programs such as STRING is problematic, in that often not all interactors are presented, and much of the listed interactors are the result of automated literature text mining or inferred by co-expression. On the other hand, sites such as Bacteriome.org collect E. coli specific experimental data and rank based on confidence [56,57]. However, only a few of our observed interactions have been reported at this site, indicating many interactions still to be found, particularly those of a more transient nature. Compilation of evidence from the literature, further supported by this present study, points to not only the broad substrate specificity of certain general cytosolic chaperones, but also as interaction partners, thus representing ‘nodes’ at which several interactions occur [for example, see 51].
Given the above evidence, and based on the in vitro results summarized in Table 2 and in vivo data of Figs. 4 and 5, it is tempting to speculate that the process of redox enzyme maturation from the perspective of metalloprotein-containing Tat substrates involves a host of protein factors from the moment of translation to export through the Tat translocase. Proteins identified here would act in concert not only to stabilize the substrate protein, thereby preventing degradation and/or premature folding, but also to facilitate interaction with molybdopterin biosynthetic proteins and allow cofactor insertion. Some of the interaction results obtained here correlate with findings from the high-throughput in vitro evaluation of E. coli interaction networks using the random approach of Tandem-Affinity-Purification method, which identified protein–protein interactions of tighter interaction affinities [51,57]. Targeting to the Tat translocase would then be accomplished probably through transient and weak but specific interactions of the TatBC complex with the REMP proteins and the RR-leader itself.
Fig. 5.
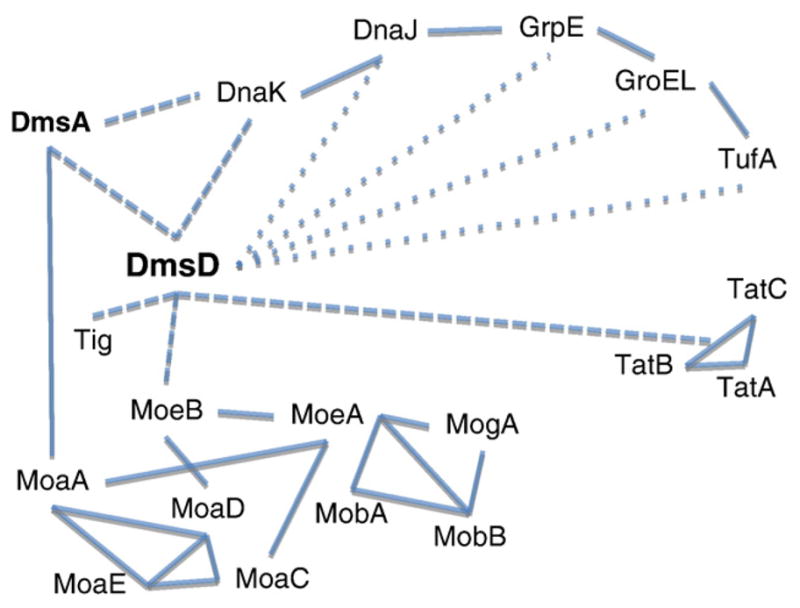
Interaction web for DmsD. Interactions are cartooned showing in solid line interactions described by the Bacterial Protein interaction database (Bacteriome.org). Dashed lines indicate strong interactions demonstrated by this study and previous work from our group. Dotted lines are weaker or more transient interactions suggested by this study. The interaction network of the molybdopterin cofactor biosynthesis is shown, however, for clearer viewing interactomes of the chaperones are not included.
Overall our findings suggest that DmsD acts as a central hub or linker for a network or pathway to facilitate maturation events of the substrate folding and cofactor incorporation as well as subsequent targeting.
Supplementary Material
Acknowledgments
We thank B.D. Haslam and A. Binding for protein purification conducted as summer undergraduate students. C.S. Chan and T. L. Winstone are gratefully acknowledged for useful discussions. J.M.H. was supported by a NSERC graduate scholarship. This work was supported by a Canadian Institutes of Health Research grant to RJT.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bbapap.2010.01.022.
References
- 1.Berks BC. A common export pathway for proteins binding complex redox cofactors. Mol Microbiol. 1996;22:393–404. doi: 10.1046/j.1365-2958.1996.00114.x. [DOI] [PubMed] [Google Scholar]
- 2.Sargent F, Bogsch EG, Stanley NR, Wexler M, Robinson C, Berks BC, Palmer T. Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J. 1998;17:3640–3650. doi: 10.1093/emboj/17.13.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santini CL, Ize B, Chanal A, Müller M, Giordano G, Wu LF. A novel sec-independent protein translocation pathway in Escherichia coli. EMBO J. 1998;17:101–112. doi: 10.1093/emboj/17.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiner JH, Bilous PT, Shaw GM, Lubitz SP, Frost L, Thomas GH, Cole JA, Turner RJ. A novel and ubiquitous system for membrane targeting and secretion of proteins in the folded state. Cell. 1998;93:93–101. doi: 10.1016/s0092-8674(00)81149-6. [DOI] [PubMed] [Google Scholar]
- 5.Berks BC, Sargent F, Palmer T. The Tat protein export pathway. Mol Microbiol. 2000;35:260–274. doi: 10.1046/j.1365-2958.2000.01719.x. [DOI] [PubMed] [Google Scholar]
- 6.Berks BC, Palmer T, Sargent F. The Tat protein translocation pathway and its role in microbial physiology. Adv Microb Physiol. 2003;47:187–254. doi: 10.1016/s0065-2911(03)47004-5. [DOI] [PubMed] [Google Scholar]
- 7.Cline K, Mori H. Thylakoid ΔpH-dependent precursor proteins bind to a cp TatC-Hcf106 complex before Tha4-dependent transport. J Cell Biol. 2001;154:719–729. doi: 10.1083/jcb.200105149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Leeuw E, Granjon T, Porcelli I, Alami M, Carr SB, Müller M, Sargent F, Palmer T, Berks BC. Oligomeric properties and signal peptide binding by Escherichia coli Tat protein transport complexes. J Mol Biol. 2002;322:1135–1146. doi: 10.1016/s0022-2836(02)00820-3. [DOI] [PubMed] [Google Scholar]
- 9.Papish A, Ladner C, Turner RJ. The twin-arginine leader binding protein, DmsD, is associated with the Escherichia coli membrane and the Tat translocase. J Biol Chem. 2003;278:32501–32506. doi: 10.1074/jbc.M301076200. [DOI] [PubMed] [Google Scholar]
- 10.Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, Palmer T, Saibil HR, Berks BC. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Natl Acad Sci U S A. 2005;102:10482–10486. doi: 10.1073/pnas.0503558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sambasivarao D, Scraba DG, Trieber C, Weiner JH. Organization of dimethyl sulfoxide reductase in the plasma membrane of Escherichia coli. J Bacteriol. 1990;172:5938–5948. doi: 10.1128/jb.172.10.5938-5948.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oresnik I, Ladner CL, Turner RJ. Identification of a twin-arginine leader binding protein. Mol Microbiol. 2001;40:323–331. doi: 10.1046/j.1365-2958.2001.02391.x. [DOI] [PubMed] [Google Scholar]
- 13.Ray N, Oates J, Turner RJ, Robinson C. DmsD is required for the biogenesis of DMSO reductase in Escherichia coli but not for the interaction of the DmsA signal peptide with the Tat apparatus. FEBS Lett. 2003;534:156–160. doi: 10.1016/s0014-5793(02)03839-5. [DOI] [PubMed] [Google Scholar]
- 14.Turner RJ, Papish AL, Sargent F. Sequence analysis of bacterial oxido-reductase enzyme maturation proteins (REMPs) Can J Microbiol. 2004;50:225–238. doi: 10.1139/w03-117. [DOI] [PubMed] [Google Scholar]
- 15.Palmer T, Sargent F, Berks BC. Export of complex cofactor-containing proteins by the bacterial Tat pathway. Trends Microbiol. 2005;13:175–180. doi: 10.1016/j.tim.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Sargent F. Constructing the wonders of the bacterial world: biosynthesis of complex enzymes. Microbiology. 2007;153:633–651. doi: 10.1099/mic.0.2006/004762-0. [DOI] [PubMed] [Google Scholar]
- 17.Genest O, Seduk F, Theraulaz L, Méjean V, Iobbi-Nivol C. Chaperone protection of immature molybdoenzyme during molybdenum cofactor limitation. FEMS Microbiol Lett. 2006;265:51–55. doi: 10.1111/j.1574-6968.2006.00468.x. [DOI] [PubMed] [Google Scholar]
- 18.Jack RL, Buchanan G, Dubini A, Hatzixanthis K, Palmer T, Sargent F. Coordinating assembly and export of complex bacterial proteins. EMBO J. 2004;23:3962–3972. doi: 10.1038/sj.emboj.7600409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatzixanthis K, Clarke TA, Oubrie A, Richardson DJ, Turner RJ, Sargent F. Signal peptide–chaperone interactions on the twin-arginine protein transport pathway. Proc Natl Acad Sci U S A. 2005;102:8460–8465. doi: 10.1073/pnas.0500737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genest O, Neumann M, Seduk F, Stöcklein W, Méjean V, Leimkühler S, Iobbi-Nivol C. Dedicated, metallochaperone connects apoenzyme and molybdenum cofactor biosynthesis components. J Biol Chem. 2008;283:21433–21440. doi: 10.1074/jbc.M802954200. [DOI] [PubMed] [Google Scholar]
- 21.Vergnes A, Gouffi-Belhabich K, Blasco F, Giordano G, Magalon A. Involvement of the molybdenum cofactor biosynthetic machinery in the maturation of the Escherichia coli nitrate reductase A. J Biol Chem. 2004;279:41398–41403. doi: 10.1074/jbc.M407087200. [DOI] [PubMed] [Google Scholar]
- 22.Vergnes A, Pommier J, Toci R, Blasco F, Giordano G, Magalon A. NarJ chaperone binds on two distinct sites of the aponitrate reductase of Escherichia coli to coordinate molybdenum cofactor insertion and assembly. J Biol Chem. 2006;281:2170–2176. doi: 10.1074/jbc.M505902200. [DOI] [PubMed] [Google Scholar]
- 23.Lanciano P, Vergnes A, Grimalki S, Guigliarelli B, Magalon A. Biogenesis of a respiratory complex is orchestrated by a single accessory protein. J Biol Chem. 2007;282:17468–17474. doi: 10.1074/jbc.M700994200. [DOI] [PubMed] [Google Scholar]
- 24.Pérez-Rodríguez R, Fisher AC, Perlmutter JD, Hicks MG, Chanal A, Santini CL, Wu LF, Palmer T, DeLisa MP. An essential role for the DnaK molecular chaperone in stabilizing over-expressed substrate proteins of the bacterial twin-arginine translocation pathway. J Mol Biol. 2007;367:715–730. doi: 10.1016/j.jmb.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 25.Graubner W, Schierhorn A, Bruser T. DnaK plays a pivotal role in Tat targeting of CueO and functions beside SlyD as a general Tat signal binding chaperone. J Biol Chem. 2007;282:7116–7124. doi: 10.1074/jbc.M608235200. [DOI] [PubMed] [Google Scholar]
- 26.Karimova G, Pidoux J, Ullmann A, Ladant D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A. 1998;95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karimova G, Dautin N, Ladant D. Interaction, network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J Bacteriol. 2005;187:2233–2243. doi: 10.1128/JB.187.7.2233-2243.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 29.Kerppola TK. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc. 2006;1:1278–1286. doi: 10.1038/nprot.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 31.Wexler M, Sargent F, Jack RL, Stanley NR, Bogsch EG, Robinson C, Berks BC, Palmer T. TatD is a cytoplasmic protein with DNase activity. J Biol Chem. 2000;275:16717–16722. doi: 10.1074/jbc.M000800200. [DOI] [PubMed] [Google Scholar]
- 32.Liu M, Turner RJ, Winstone TL, Saetre A, Dyllick-Brenzinger M, Jickling G, Tari LW, Weiner JH, Taylor DE. Escherichia coli TehB requires S-adenosylmethionine as a cofactor to mediate tellurite resistance. J Bacteriol. 2000;182:6509–6513. doi: 10.1128/jb.182.22.6509-6513.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan CS, Winstone TL, Workentine M, Chang L, Stevens C, Li H, Wei Y, Ondrechen MJ, Paetzel M, Turner RJ. Mutagenesis to DmsD, identifying regions required for twin-arginine leader binding. Biochemistry. 2008;47:2749–2759. doi: 10.1021/bi702138a. [DOI] [PubMed] [Google Scholar]
- 34.Howell JM, Winstone TL, Coorssen J, Turner RJ. An evaluation of in vitro protein–protein interaction techniques: assessing contaminating background proteins. Proteomics. 2006;6:2059–2069. doi: 10.1002/pmic.200500517. [DOI] [PubMed] [Google Scholar]
- 35.Chan CS, Chang L, Rommens K, Turner RJ. Differential interactions between Tat-specific redox enzyme peptides and their chaperones. J Bacteriol. 2009;191:2091–2101. doi: 10.1128/JB.00949-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller JH. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor; New York, NY: 1972. [Google Scholar]
- 37.Li H, Turner RJ. In vivo associations of Escherichia coli NarJ with a peptide of the first 50 residues of Nitrate reductase catalytic subunit NarG. Can J Microbiol. 2009;55:179–188. doi: 10.1139/w08-111. [DOI] [PubMed] [Google Scholar]
- 38.Kostecki JS, Li H, Turner RJ, DeLisa MP. Visualizing interactions along the Escherichia coli twin-arginine translocation pathway using protein fragment complementation. PLOS One. 2010;5:e9225. doi: 10.1371/journal.pone.0009225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarfo KJ, Winstone TL, Papish A, Howell JM, Kadir H, Vogel HJ, Turner RJ. Folding forms of Escherichia coli DmsD, a twin-arginine leader binding protein. Biochem Biophys Res Comm. 2004;315:397–403. doi: 10.1016/j.bbrc.2004.01.070. [DOI] [PubMed] [Google Scholar]
- 40.Winstone TL, Workentine ML, Sarfo K, Binding AJ, Haslam BD, Turner RJ. Characterization of Escherichia coli DmsD signal peptide binding. Arch Biochem Biophys. 2006;455:89–97. doi: 10.1016/j.abb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 41.Fancy DA. Elucidation of protein–protein interactions using chemical cross-linking or label transfer techniques. Curr Opin Chem Biol. 2000;4:28–33. doi: 10.1016/s1367-5931(99)00047-2. [DOI] [PubMed] [Google Scholar]
- 42.Phizicky E, Fields S. Protein–protein interactions: methods for detection and analysis. Microbiol Rev. 1995;59:94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serebriiskii IG, Golemis EA. Uses of lacZ to study gene function: evaluation of beta-galactosidase assays employed in the yeast two hybrid system. Anal Biochem. 2000;284:1–5. doi: 10.1006/abio.2000.4672. [DOI] [PubMed] [Google Scholar]
- 44.Mayhew M, Hartl F-U. Cellular and Molecular Biology. 2. ASM Press; Washington DC: 1996. Molecular Chaperone Proteins in Escherichia coli and Salmonella. [Google Scholar]
- 45.Ribbe MW, Burgess BK. The chaperone GroEL is required for the final assembly of the molybdenum–iron protein of nitrogenase. Proc Natl Acad Sci U S A. 2001;98:5521–5525. doi: 10.1073/pnas.101119498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dionisi HM, Checa SK, Krapp AR, Arakaki AK, Ceccarelli EA, Carrillo N, Viale AM. Cooperation, of the DnaK and GroE chaperone systems in the folding pathway of plant ferredoxin-NADP+ reductase expressed in Escherichia coli. Eur J Biochem. 1998;251:724–728. doi: 10.1046/j.1432-1327.1998.2510724.x. [DOI] [PubMed] [Google Scholar]
- 47.Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, Starostine A, Richards D, Beattie B, Krogan N, Davey M, Parkinson J, Greenblatt J, Emili A. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 48.Houry WA, Frishman D, Eckerskorn C, Lottspeich F, Hartl FU. Identification of in vivo substrates of the chaperonin GroEL. Nature. 1999;402:147–154. doi: 10.1038/45977. [DOI] [PubMed] [Google Scholar]
- 49.Kerner MJ, Naylor DJ, Ishihama Y, Maier T, Chang HC, Stines AP, Gerogopoulos C, Frishman D, Hayer-Hartl M, Mann M, Hartl FU. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell. 2005;122:209–220. doi: 10.1016/j.cell.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 50.Hoffmann A, Merz F, Rutkowska A, Zachmann-Brand B, Deuerling E, Bukau B. Trigger factor forms a protective shield for nascent polypeptides at the ribosome. J Biol Chem. 2006;281:6539–6545. doi: 10.1074/jbc.M512345200. [DOI] [PubMed] [Google Scholar]
- 51.Jong WS, ten Hagen-Jongman CM, Genevaus P, Brunner J, Oudega B, Luirink J. Trigger factor interacts with the signal peptide of nascent Tat substrates but does not play a critical role in Tat-mediated export. Eur J Biochem. 2004;271:4779–4787. doi: 10.1111/j.1432-1033.2004.04442.x. [DOI] [PubMed] [Google Scholar]
- 52.Schwarz G. Molybdenum cofactor biosynthesis and deficiency. Cell Mol Life Sci. 2005;62:2792–2810. doi: 10.1007/s00018-005-5269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leimkühler S, Wuebbens MM, Rajagopalan KV. Characterization of Escherichia coli MoeB and its involvement in the activation of molybdopterin synthase for the biosynthesis of the molybdenum cofactor. J Biol Chem. 2001;276:34695–34701. doi: 10.1074/jbc.M102787200. [DOI] [PubMed] [Google Scholar]
- 54.Sambasivarao D, Turner RJ, Bilous PT, Rothery RA, Shaw G, Weiner JH. Differential effects of an aryl sulfurylase (moeB) mutation on Escherichia coli molybdoenzyme maturation. Biochem Cell Biol. 2002;80:435–443. doi: 10.1139/o02-131. [DOI] [PubMed] [Google Scholar]
- 55.Ilbert M, Méjean V, Giudici-Orticoni MT, Samama JP, Iobbi-Nivol C. Involvement, of a mate chaperone (TorD) in the maturation pathway of molybdoenzyme TorA. J Biol Chem. 2003;278:28787–28792. doi: 10.1074/jbc.M302730200. [DOI] [PubMed] [Google Scholar]
- 56.Xenarios I, Rice DW, Salwinski L, Baron MK, Marcotte EM, Eisenberg D. DIP: the database of interacting proteins. Nucleic Acids Res. 2000;28:289–291. doi: 10.1093/nar/28.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arifuzzaman M, Maeda M, Itoh A, Nishikata K, Takita C, Saito R, Ara T, Nakahigashi K, Huang HC, Hirai A, Tsuzuki K, Nakamura S, Altaf-Ul-Amin M, Oshima T, Baba T, Yamamoto N, Kawamura T, Ioka-Nakamichi T, Kitagawa M, Tomita M, Kanaya S, Wada C, Mori H. Large-scale identification of protein–protein interaction of Escherichia coli K-12. Genome Res. 2006;16:686–691. doi: 10.1101/gr.4527806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.