

Abstract
Hsp90 is an ATP dependent molecular chaperone protein which integrates multiple oncogenic pathways. As such, Hsp90 inhibition is a promising anti-cancer strategy. Several inhibitors that act on Hsp90 by binding to its N-terminal ATP pocket have entered clinical evaluation. Robust pre-clinical data suggested anti-tumor activity in multiple cancer types. Clinically, encouraging results have been demonstrated in melanoma, acute myeloid leukemia, castrate refractory prostate cancer, non-small cell lung carcinoma and multiple myeloma. In breast cancer, proof-of-concept was demonstrated by first generation Hsp90 inhibitors in combination with trastuzumab mainly in human epidermal growth factor receptor 2 (HER2) + metastatic breast cancer. There are a multitude of second generation Hsp90 inhibitors currently under investigation. To date, however, there is no FDA approved Hsp90 inhibitor nor standardized assay to ascertain Hsp90 inhibition. This review summarizes the current status of both first and second generation Hsp90 inhibitors based on their chemical classification and stage of clinical development. It also discusses the pharmacodynamic assays currently implemented in clinic as well as other novel strategies aimed at enhancing the effectiveness of Hsp90 inhibitors. Ultimately, these efforts will aid in maximizing the full potential of this class of agents.
Keywords: heat shock protein 90, chaperone, cancer, targeted therapy
1. Introduction
Hsp90 is a chaperone that functions in the correct folding of its client proteins to their active conformation. Hsp90 is an ATPase and accomplishes its role through a complex cycle regulated by the binding and hydrolysis of ATP as well as by numerous co-chaperones (Hsp70, HOP, Cdc37, p23, Aha1). Inhibition of the Hsp90 chaperone cycle causes client proteins to undergo ubiquitination and subsequent degradation by the proteasome [1, 2]. Because many of its clients include oncoproteins with important functions in the development and promotion of cancer, Hsp90 is an important target in cancer therapy [3].
The discovery of Hsp90 as the target of anticancer activity of geldanamycin (GM) (Fig. 1) sparked much interest in the inhibition of Hsp90 as a strategy for the treatment of cancer. This interest has resulted in intense efforts from both industry and academic centers to develop clinically viable small molecule Hsp90 inhibitors [4]. As a result of these efforts, there are numerous small-molecule Hsp90 inhibitors that are currently undergoing clinical investigation in a wide range of malignancies. Of the numerous ways of potentially inhibiting the chaperone cycle, it should be noted that each of the molecules currently in clinical trials inhibit the ATPase activity of Hsp90 by binding to the N-terminal domain nucleotide binding pocket [5, 6].
Figure 1.

Structures of representative Hsp90 inhibitor chemical classes
The clinical development of Hsp90 inhibitors, from GM to the newest agents, has been and will continue to be driven by the optimization of pharmaceutical properties including pharmacokinetic, pharmacodynamic and toxicological. Even though there are currently no approved Hsp90 targeted agents, research during this past decade has been very promising as there has been considerable progress on many fronts including improved formulations and the introduction of chemically distinct Hsp90 inhibitors with improved properties. These advances together with the increased biological knowledge of Hsp90 can help to fulfill the promise of this target and potentially result in the approval of the first-in-class agent.
2. Clinical studies with Hsp90 inhibitors in cancers
While the molecules currently advanced to clinical trials constitute a diverse array of structures, a close inspection of them shows that they can in general be classified according to their similarity to GM, radicicol (RD) or to the purine-scaffold. Only SNX-5422 falls outside any of these designations (Fig. 1). As is often the case in drug discovery, natural products play a prominent role in the discovery of lead agents. In the case of Hsp90, GM, RD and ATP have each had a profound effect on the development of small-molecule Hsp90 inhibitors. While none of these are suitable as a clinical agent, they have each served as good lead molecules or starting points for most of the agents in clinical trials.
2.1. Geldanamycin derivatives
GM is a benzoquinone ansamycin first isolated from a fermentation broth of Streptomyces hygroscopicus in 1970 (Fig. 1) [7]. Originally pursued as an antibiotic, its anticancer properties were discovered following a phenotypic screening of compounds capable of reversing v-src oncogene transformed cells [8]. Initially, it was believed to be a direct inhibitor of src kinase, however, it was later shown to directly bind to Hsp90 and interfere with Hsp90-v-src heterocomplex formation [9]. Further work showed that GM inhibits the ATPase activity of Hsp90 by competing with ATP for binding to the N-terminal domain nucleotide binding pocket [10, 11], resulting in ubiquitin mediated proteasomal degradation of its client proteins [1, 2, 12].
Despite its potent anti-tumor effects, GM was never evaluated in clinical trials because of its poor “drug-like” properties including poor solubility, limited in vivo stability and significant hepatotoxicity in animals [13, 14]. Structural features of GM include a quinone ring (depicted in blue, Fig. 1), moiety contributing to the observed hepatotoxicity, along with a pendant macrocycle containing a carbamate group essential for binding (Fig. 1). Additionally, GM contains a non-essential methoxy group on C-17 of the quinone ring (depicted in red, Fig. 1) that can readily be substituted with amines. This approach was used in an attempt to overcome the liabilities associated with GM and resulted in the preparation of many analogs including those that have entered clinical trials (Table 1, entry 1–4).
Table 1.
Hsp90 inhibitors in clinical evaluation
| Inhibitor | Company | Structure | Class | Route | Phase | |
|---|---|---|---|---|---|---|
| 1. | Tanespimycin (17-AAG, KOS-953) | Kosan Biosciences/Bristol-Myers-Squibb |  |
GM | IV | III |
| 2. | Alvespimycin (17-DMAG) | Kosan Biosciences/Bristol-Myers-Squibb | 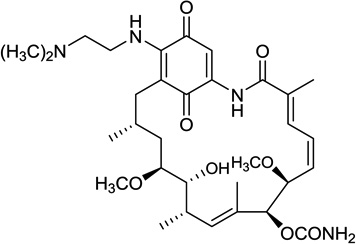 |
GM | IV Oral |
I |
| 3. | Retaspimycin (IPI-504) | Infinity Pharmaceuticals |  |
GM | IV | III |
| 4. | IPI-493 | Infinity Pharmaceuticals |  |
GM | Oral | I |
| 5. | CNF2024/BIIB 021 | Biogen Idec |  |
Purine | Oral | II |
| 6. | MPC-3100 | Myriad Pharmaceuticals/Myrexis |  |
Purine | Oral | I |
| 7. | Debio 0932 (CUDC-305) | DebioPharm |  |
Purine-like | Oral | I |
| 8. | PU-H71 | Samus Therapeutics |  |
Purine | IV | I |
| 9. | Ganetespib (STA-9090) | Synta Pharmaceuticals | Not reported | Resorcinol - Triazole | IV | II |
| 10. | NVP-AUY922 (VER-52269) | Novartis | 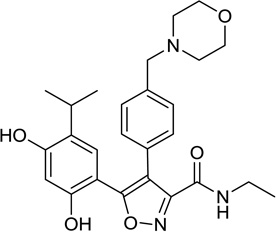 |
Resorcinol - Isoxazole | IV | II |
| 11. | HSP990 | Novartis | Not reported but claimed as a follow up compound to NVP-AUY922 | not reported | Oral | I |
| 12. | KW-2478 | Kyowa Hakko Kirin Pharma | 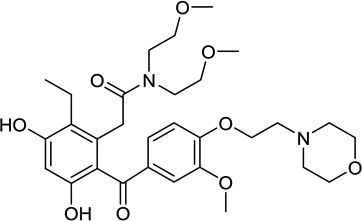 |
Resorcinol | IV | I |
| 13. | AT13387 | Astex | 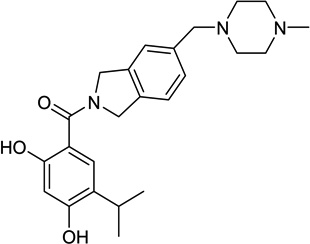 |
Resorcinol | IV Oral |
I |
| 14. | SNX-5422 | Serenex/Pfizer | 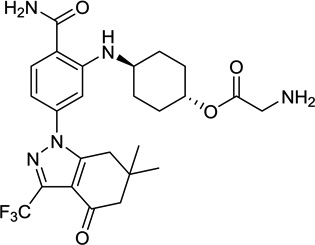 |
Indazol-4-one | Oral | I |
| 15. | DS-2248 | Daiichi Sankyo Inc | Not reported | not reported | Oral | I |
| 16. | XL888 | Exelixis | Not reported | not reported | Oral | I |
2.1.1. 17-AAG (17-allyl-17-desmethoxygeldanamycin)
Substitution in GM of the methoxy group with an allylamino group resulted in 17-AAG (Table 1, entry 1), a compound with biological activity similar to GM but with an improved toxicity profile [15]. In fact, 17-AAG was the first Hsp90 inhibitor selected by the Cancer Therapy Evaluation Program (CTEP) of the National Cancer Institute (NCI) for clinical studies in cancer and provided proof-of-concept for Hsp90 inhibition in humans. In these studies, 17-AAG has been evaluated using weekly, weekly for three weeks (28 day cycle), daily × 5 (21 day cycle), and daily × 3 (14 day cycle), twice weekly (day 1, 4), and on days 1, 4, 8, 11 (21 day cycle) schedules [16–23]. Toxicity of 17-AAG in these trials was dose and schedule dependent, with hepatotoxicity being more prominent with daily administration. Other toxicities were primarily diarrhea and fatigue. Because of its poor solubility and in order to administer 17-AAG intravenously, the NCI had developed a dimethyl sulfoxide and egg phospholipid vehicle, however, at doses greater than 100 mg/m2, the toxicities due to this vehicle (bad odor, nausea, anorexia) were as significant as the toxicities from the drug itself. Pharmacokinetic (PK) studies that were incorporated in these phase I trials suggested that serum concentrations greater than those required for depletion of Hsp90 client proteins in vitro and in xenograft models could be achieved at well-tolerated doses and schedules.
Pharmacodynamic (PD) assays in these trials included peripheral blood mononuclear cell (PBMC) studies and limited tumor biopsies, as discussed further in Section 3 of this review [16, 20]. Despite the evidence of at least partial target modulation in these phase I trials, there were no complete or partial responses (CR or PR respectively) by RECIST (Response Evaluation Criteria in Solid Tumors). Several patients with melanoma, breast cancer, prostate cancer and renal cell carcinoma did have stable disease (SD). Disappointingly, single agent phase II studies using 17-AAG once weekly (300–450 mg/m2) in prostate cancer or melanoma or twice weekly (220 mg/m2) in renal cell carcinoma patients also showed no objective responses with this dose and schedule of 17-AAG [24–26].
In patients heavily pretreated for relapsed/refractory multiple myeloma, single agent 17-AAG administered on days 1, 4, 8, and 11 every 3 weeks was well tolerated with one minimal response (MR), with a progression-free survival (PFS) of 3 months, a 41% decline in urine M protein, and a 33% decrease from baseline in serum M protein. Fifteen other patients had SD with a median PFS of 2.1 months [27].
Limited success of 17-AAG in these studies was attributed to poor solubility, poor patient enrichment for those most likely to benefit based on preclinical experience (for e.g. patients with HER2 amplified breast cancer), suboptimal target inhibition and off-target toxicities from 17-AAG and its DMSO formulation.
Improving the formulation and the delivery of 17-AAG in disease specific population have led to encouraging results. Kosan Biosciences developed a Cremophor containing formulation of 17-AAG (tanespimycin, KOS-953) as an injectable suspension. The first solid tumor trial with this formulation was a phase I/II study of 17-AAG and trastuzumab. The phase I portion was open to all tumor histologies. Of the 25 patients enrolled, 15 had HER2+ metastatic breast cancer. Patients were enrolled onto 4 dose cohorts (225 – 450 mg/m2) and treated with both agents on a weekly schedule. Dose limiting toxicity (DLT) included fatigue, nausea, anorexia and thrombocytopenia. With the weekly schedule, hepatotoxicity was minimal. Antitumor activity was seen only in patients with HER2+ metastatic breast cancer with 1 confirmed PR, 4 MR and 4 patients with SD for more than 4 months [28]. In order to obtain a more precise estimate of activity, the phase II trial enrolled 31 patients with HER2+ metastatic breast cancer who were previously refractory to trastuzumab therapy [29]. Patients were treated with 450 mg/m2 of tanespimycin with trastuzumab weekly. The primary endpoint was response rate (RR) by RECIST criteria. Most common toxicities were diarrhea, fatigue, nausea and headache. The overall RR was 22% and the clinical benefit rate (CR+PR+SD) was 59% with a median PFS of 6 months (95% CI: 4–9) and median overall survival (OS) of 17 months (95% CI: 16–28). The effectiveness noted in this phase II trial could be due to potent target degradation (i.e. HER2+) due to the combination of these drugs and perhaps because Hsp90 inhibitors can overcome or delay the initial resistance to trastuzumab [30].
Similar to these responses for tanespimycin in solid tumors, strong synergistic effect of anti-tumor activity were also noted with combination of tanespimycin and bortezomib in multiple myeloma. In a phase I/II study, 72 patients with relapsed and refractory multiple myeloma were treated with this combination (tanespimycin 100–340 mg/m2 and bortezomib 0.7 –1.3 mg/m2) [31]. The highest dose for tanespimycin tested in this study was 340 mg/m2 and no DLT was identified. Importantly, there were no cases of grade 3 peripheral neuropathy. Anti-tumor activity was observed with overall RR ≥ MR in 48% of bortezomib-naive, 22% of bortezomib-pretreated and 13% of bortezomib refractory patients; median duration of response for all responders was 12 months. The phase II study of this combination, although an extension of the phase I/II study, deployed a broader tanespimycin dosage range (50–340 mg/m2). Unlike the previously described phase I/II study, this phase II trial involved a slightly different population, in that they excluded bortezomib—naïve patients and patients with no measurable multiple myeloma [32]. Twenty-two patients were administered this combination regimen on days 1, 4 , 8 , 11 in each 3-week cycle. Most common adverse events were fatigue, diarrhea, nausea, constipation and vomiting. Liver toxicity related adverse events were noted in 4 patients, which was manageable and reversible, and thrombocytopenia was the most common hematological adverse event. Peripheral neuropathy is a significant toxicity of single agent bortezomib therapy. However, there were low rates of peripheral neuropathy, in particular grade 3 peripheral neuropathy, seen with combination of tanespimycin and bortezomib. This favorable effect of both decreased rate and severity of peripheral neuropathy correlated well with the pre-clinical observation that tanespimycin has a neuroprotective effect against bortezomib-induced peripheral neuropathy [33]. Overall RR was 14% (2 PR and 1 MR) and an additional 10 patients had SD. This led to further development of tanespimycin in phase III trials for potential treatment of multiple myeloma. However, Kosan, acquired by Bristol-Myers-Squibb in 2008, suspended this phase III trial for non-clinical reasons [34].
Because Hsp90 can protect cells under stress conditions, Hsp90 inhibitors have the ability to sensitize cells to the toxic effects of chemotherapy (including taxanes, anthracyclines, nucleoside analogs and topoisomerase I inhibitors and lead to synergistic activity) and radiation therapy. One possible mechanism by which Hsp90 inhibitors are thought to enhance the cytotoxicity of chemotherapy is by depleting the checkpoint kinase1 (chk1). Chk1 is a Hsp90 client protein that regulates progression through the cell cycle and is particularly critical for the S and G2-M phases [35]. Hsp90 inhibitors degrade chk1 which in turn abrogates G1/S arrest induced by gemcitabine [35] and G2-M checkpoint induced by topoisomerase I inhibitors such as irinotecan [36]. Nguyen et al showed 5–22 fold enhancement of paclitaxel cytotoxicity when combined with Hsp90 inhibitors [37]. Breast cancer cell lines exposed to Hsp90 inhibitors were sensitized to the apoptotic effects of paclitaxel by activation of caspase-3 and -9 [38]. Cells with intact retinoblastoma gene (RB) that were exposed to 17-AAG followed by paclitaxel underwent G1 arrest and enhanced apoptosis. This schedule dependence was not noted in cells harboring a mutated RB. Akt inactivation is another mechanism by which Hsp90 inhibitors sensitize tumor cells to induction of apoptosis by paclitaxel [39]. These studies formed the rationale for combination phase I trials of 17-AAG with paclitaxel [40], docetaxel [41], cisplatin [42], gemcitabine-cisplatin [43] and irinotecan [44].
Similarly, Hsp90 inhibitors are thought to enhance the radiosensitivity of many tumor cell lines. Raf-1, Akt and HER2 are radioresponse proteins that protect against radiation induced cell death. Hsp90 inhibition causes degradation of the radioresponse proteins, increases apoptosis and enhances G2 arrest [45]. Lastly, in an attempt to show synergistic activity by simultaneously inhibiting the Raf-signaling pathway, a phase I trial evaluated the combination of 17-AAG with Sorafenib and showed clinical activity in renal cell carcinoma and melanoma patients [46].
Alternatively, agents such as cisplatin may enhance the activity of Hsp90 inhibitors [47]. Under non-stress conditions, heat shock factor 1 (HSF1) binds with Hsp90, but when an Hsp90 inhibitor blocks the target, HSF1 is released, thereby causing a heat shock response that potentially limits the effect of Hsp90 inhibition. Cisplatin blocks the HSF1 mediated heat shock response by blocking HSF1 binding to the promoter region of the transcription factor [48].
Despite, these early encouraging results, the development of 17-AAG was halted in July 2008. However, more recently, 17-AAG was successfully converted to nanoparticle albumin-bound, nab-17AAG/ABI-1010 to improve the safety and efficacy over their surfactant and solvent based counterparts [49]. This agent is currently being evaluated in a phase I trial in combination with Abraxane in patients with advanced solid tumors. Patients will be administered ABI-1010 intravenously weekly for 3 weeks of a 28-day cycle; results of this trial are awaited [50].
2.1.2. 17-DMAG (17-desmethoxy-17-N,N-dimethylaminoethylaminogeldanamycin)
Substitution of the C-17 methoxy group of GM with N, N-dimethylethylamine resulted in 17-DMAG (Table 1, entry 2). The presence of an ionizable amino group resulted in increased water solubility, better oral bioavailability and equal or greater anti-tumor activity compared to 17-AAG [51, 52]. 17-DMAG was developed by the NCI and Kosan and entered phase I clinical trials in 2005. Various schedules were evaluated including twice weekly [53], daily × 3 or 5 days every 3 weeks (1.5–46 mg/m2) [54], weekly (2.5–80 mg/m2) [55], twice weekly × 2 weeks and every 3 weeks (8–32 mg/m2)[56]. Toxicity in these trials included peripheral neuropathy [53], renal dysfunction [53, 55], fatigue [55], cardiotoxicity [56], ocular adverse events comprised of blurred vision, keratitis, dry eyes [55], pneumonitis with dyspnea and thrombocytopenia [56]. Efficacy assessment included 1 CR (castrate refractory prostate cancer) and 1 PR (melanoma) [55] and 3 CR in acute myeloid leukemia [56]. There was also one phase I trial of orally administered 17-DMAG where in patients were administered 17-DMAG either daily or every other day for 4 out of 6 weeks [57]. In the 28 patients treated, no DLT was observed and common drug-related toxicities included fatigue, anorexia, proteinuria and peripheral edema. Bioavailability in 14 patients was 51% and 49% on day 1 and 21 respectively and was not dose-dependent. Early signs of activity included SD in hemangioendothelioma, melanoma and renal cell carcinoma.
17-DMAG (60–100 mg/m2), administered weekly has also been investigated in combination with trastuzumab (phase I study) in 28 patients with metastatic breast cancer and metastatic ovarian cancer. DLT was grade 3 keratitis, which was reversible, and 1 patient had a decline in left ventricular ejection fraction. Anti-tumor activity was seen in patients with HER2+ metastatic breast cancer (1 PR, 1 MR and 5 patients with SD). Tumor regression including CR was also noted in 1 patient with HER2+ metastatic breast cancer and another patient with ovarian cancer (HER2 status unknown) [58].
Despite the CR with 17-DMAG in patients with acute myeloid leukemia, Kosan discontinued its development in 2008 to “commit resources to the development of 17-AAG for the treatment of breast cancer as a result of a comparative analysis with 17-AAG based on several factors, including clinical experience to date, strength of intellectual property protection and risk, and time to commercialization” [59]
2.1.3. IPI-504 [17-allylamino-17-demethoxygeldanamycin hydroquinone hydrochloride]
In a different approach, Infinity Pharmaceuticals developed IPI-504 (retaspimycin) (Table 1, entry 3), a new GM analog, which is the water-soluble hydroquinone hydrochloride salt derivative of 17-AAG [60]. IPI-504 can be prepared by reduction of 17-AAG with sodium dithionite followed by conversion to its hydrochloride salt. IPI-504 and 17-AAG actually exist in a redox equilibrium in vivo between the hydroquinone (as in IPI-504) and quinone (as in 17-AAG) forms through the action of oxidoreductases. The hydroquinone form in IPI-504 is a more potent inhibitor of Hsp90 and is likely the more relevant form in vivo. Additionally, since the quinone ring has been identified as a primary cause for the hepatotoxicity of GM, its reduction to a hydroquinone has predictably resulted in diminished toxicity. For a number of reasons, including improved PK and toxicity properties, IPI-504 appears to be the most promising inhibitor derived from the ansamycin class [61]. Various phase I-III trials have been conducted in patients with multiple myeloma, non-small cell lung cancer, castrate resistant prostate cancer, breast cancer and gastrointestinal stromal tumor.
In the phase I trial in patients with relapsed and refractory multiple myeloma, IPI-504 was infused on days 1, 4, 8, 11 of a 21-day cycle. The primary objective was MTD which was determined to be 400 mg/m2 [62].
A phase I/II trial was conducted in 12 patients with non-small cell lung cancer after they had progressed on epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKI). Patients were administered with IPI-504 twice weekly on a 4 week cycle at doses (150 to 300 mg/m2). DLT was increased liver enzymes and fatigue. Of the 9 patients who were evaluable, 7 had best response of SD. Of the 4 patients who had evaluable disease by 18F-flurodeoxyglucose (FDG) positron emission tomography (PET), 2 had SD and 2 had PR [63]. This promising activity led to the phase II portion of the study which evaluated 76 patients at 400 mg/m2 administered twice weekly on a 21 day cycle [64]. The primary objective was overall RR; secondary aims included safety, PFS and analysis by molecular subtypes. The most common side effects were fatigue, diarrhea and nausea. Grade 3 or higher liver function abnormalities were observed in 9 patients. Overall RR was 7% (5 PR) with 10% in patients who were EGFR-wild type and 4% in those who had EGFR mutations. Although the target overall RR was 20% for the two EGFR groups, 2 of the 3 patients with oncogenic rearrangement of the anaplastic lymphoma kinase (ALK) gene also had PR and the third had SD for 7.2 months with 24% reduction in tumor size. This was the first trial that showed that Hsp90 inhibitors have clinical activity in non-small cell lung cancer, particularly among those with ALK rearrangements [64].
A phase Ib trial evaluated IPI-504 in combination with docetaxel in 16 patients with advanced solid tumors of which 6 were non-small cell lung cancer. Patients were treated with 300–550 mg/m2 of IPI-504 in combination with 75 mg/m2 of docetaxel on a once every 3 weeks schedule. DLT included fatigue, neutropenia, asymptomatic bradycardia, increase in liver enzymes with acute respiratory distress syndrome; all of which were reversible on trial. MTD was identified as 450 mg/m2 [65]. In the expansion phase, 23 patients with non-small cell lung cancer were treated with 300 mg/m2 of IPI-504 in combination with 75 mg/m2 of docetaxel weekly in a 21-day cycle. Among the 23 patients, there was no drug-related hepatotoxicity. Gastrointestinal toxicity was low grade and easily manageable. The overall RR was 26% (6 PR, 7SD). Higher response rates were seen in heavy smokers and those with squamous cell histology. Based on these data, an adaptive randomized phase II study is underway comparing IPI-504 and docetaxel versus placebo + docetaxel in pretreated patients with non-small cell lung cancer and a smoking history. This trial is designed to identify and validate potential predictive biomarkers of the activity of combination treatment [66].
A single agent phase II trial evaluated the safety and efficacy of IPI-504 in two groups of men with castrate resistant prostate cancer. Group A were chemotherapy-naïve patients and group B included patients who had disease progression on previous docetaxel-based therapy. IPI-504 was administered at 400 mg/m2 twice weekly on a 21-day cycle. Of the 19 patients enrolled in this study, 15 had prior docetaxel-based therapies. There was no prostate specific antigen (PSA) or RECIST responses although one patient remained on study for 9 cycles with 48% PSA decline. Serious adverse events were noted in group B which included grade 5 hepatic failure and grade 5 ketoacidosis and the study concluded that further evaluation in this population was not warranted [67].
Recently, the results of a phase II trial of IPI-504 in combination with trastuzumab in patients with metastatic HER2+ metastatic breast cancer were presented at the 2011 Annual ASCO scientific meeting. 26 patients were treated with 300 mg/m2 of IPI-504 weekly in combination with trastuzumab 6 mg/kg once every 3 weeks. Endpoints of this study included overall RR and tolerability of IPI-504. The trial used a Simon two-stage design with prespecified criteria; if the DLT rate was <25% and at least 2 responses were seen, then the trial would be expanded. No DLTs were reported. The most common adverse events noted were fatigue, nausea and diarrhea. Among the 20 patients evaluable for efficacy, 1 PR was observed and 1 patient had SD for 6 months. Thus, there was modest clinical activity but the trial did not meet the prespecified criteria for trial expansion [68].
Patients with TKI- resistant gastrointestinal stromal tumor were treated with IPI-504 in a phase I dose-escalation trial. The initial trial design included only 45 patients with metastatic and unresectable gastrointestinal stromal tumor but later, 18 patients with soft tissue sarcomas were also included. IPI-504 was evaluated in five dose levels administered either twice weekly for 2 weeks followed by 1 week off or twice weekly for 3 weeks in continuous 21 day cycles. The objectives of this trial were to determine the MTD, find the dose and schedule for subsequent trials and to assess the biologic activity of IPI-504 with anatomic and FDG-PET imaging. FDG-PET imaging was done at baseline, day 11 and 21. IPI-504 was well tolerated up to 400 mg/m2 twice weekly followed by 1 week off. DLT was headache and myalgia. Other common adverse events included fatigue, diarrhea and elevated liver enzymes. Of the 36 patients evaluable for efficacy on this schedule, 67% had SD and 3 % had a PR. For the 18 patients evaluable by FDG-PET, 22% had a PET PR and 66% had SD. Median PFS was 12 weeks [69].
The RING (retaspimycin in GIST) trial was an international phase III, double blinded, placebo controlled trial of IPI-504 in patients with refractory gastrointestinal stromal tumor that had failed previous therapies with either imatinib or sunitinib. There was no limit on previous therapies. Target enrollment was 200 patients, however the study was terminated after 46 patients were enrolled as a higher mortality rate was observed for patients in the treatment arm. Review of the patients’ demographics revealed that the higher mortality rate was observed in those enrolled in the treatment arm who had received three or more therapies since their initial diagnosis and had more advanced disease compared to patients enrolled in the earlier IPI-504 trials [70]. Due to the safety concerns and the increased mortality rate in this population, and because 17-AAG and IPI-504 are interconvertible in vivo, the phase III trial of 17-AAG was also terminated [61].
2.1.4. IPI-493 (17-desmethoxy-17-amino geldanamycin)
IPI-493, the primary active, long-lived metabolite of 17-AAG was developed by Infinity Pharmaceuticals as an oral formulation (Table 1, entry 4). However, IPI-493 suffers from poor pharmaceutical properties including low solubility and is difficult to administer in pharmacologically relevant doses. Because drug exposure of IPI-504 was superior to IPI-493 Infinity has halted the development of this agent to focus exclusively on retaspimycin [71].
2.2. Purine and Purine-like Analogues
The rational design of Hsp90 inhibitors became possible following the availability of x-ray crystal structures of Hsp90 bound to ATP/ADP as well as to GM and RD. Hsp90 has a unique fold among most other ATPases resulting in a distinctive conformation adopted by bound nucleotide that has enabled for its selective inhibition by small-molecules over other ATP/ADP binding proteins [72]. Taking advantage of this unique fold and using a structure-based approach, Chiosis et al. designed the first reported synthetic Hsp90 inhibitor, PU3 (Fig. 1), based on the purine-scaffold by incorporating features that enabled the molecule to adopt a bent conformation [73]. As the prototype for the purine-scaffold class, PU3 has been optimized by numerous groups through a variety of strategies to result in potent and selective inhibitors with improved pharmaceutical properties. Those that have advanced to clinical trials include the purines CNF2024/BIIB021, MPC-3100, and PU-H71 as well as the purine-like Debio 0932 (CUDC-305) (Table 1, entry 5–8). Each of these molecules have in common an −NH2 group attached to a purine or purine-like core and an aryl moiety separated by approximately 5 Å as critical elements for binding to Hsp90. The aryl moiety is attached to the purine or purine-like core by a one atom linker [74].
2.2.1. CNF 2024/BIIB021
CNF 2024/BIIB021 was discovered and first developed by Conforma Therapeutics and later by Biogen Idec (Table 1, entry 5) [75–77]. CNF2024/BIIB021 is unique amongst the other members in this class in that the aryl moiety is attached to the 9-position of the purine. In order to maintain the critical distance of 5 Å, the amino group was moved to the 2-position.
This agent has been evaluated in phase I and II clinical trials. A phase I clinical trial evaluated BIIB021 administered orally daily for 3 weeks in a 28-day cycle in patients with chronic lymphocytic leukemia versus orally twice weekly for 3 weeks in a 28-day cycle in patients with advanced solid tumors. MTD was determined at 800 mg twice weekly and DLT was syncope and dizziness in patients with advanced solid tumors. Other grade 3 or 4 toxicities included fatigue, hyponatremia and hypoglycemia. There was one incidence of grade 3 abnormal liver enzymes in a patient with chronic lymphocytic leukemia. Efficacy assessment revealed that one patient with chronic lymphocytic leukemia had a 39% reduction in the lymph node size at 25 mg dose and 11 of 16 evaluable patients with solid tumors had SD [78].
Another phase I dose-escalation trial evaluated BIIB021 in combination with trastuzumab in patients with HER2+ metastatic breast cancer. Primary endpoint was to evaluate the safety and tolerability of BIIB021 when administered orally twice weekly in combination with 6 mg/kg trastuzumab every 3 weeks. MTD was determined at 600 mg twice weekly. DLT were diarrhea and partial seizure that presented as aphasia. Other frequent adverse events included fatigue, nausea, dizziness, headache and rash among others. Of the 30 patients enrolled, 2 patients had confirmed PR by RECIST and 10 had SD. Three patients had metabolic PR (>25% decline in the SUV max) and 16 had SD on FDG-PET imaging [79].
Based on these data, phase II studies with this compound in combination with Aromasin were planned [80], however, BIIB021 is no longer listed on the Biogen Idec pipeline. Apparently, Biogen Idec no longer will develop BIIB021 as they are shifting focus away from oncology for strategic purposes, and plan to outlicense its further development [81].
2.2.2. MPC-3100
MPC-3100 (Table 1, entry 6) is being developed by Myrexis, a company under the umbrella of Myriad Pharmaceuticals [82]. A phase I, modified Fibonacci, dose-escalation trial evaluated oral daily dose of MPC-3100 for 21 days in a 28-day cycle in patients with refractory or relapsed cancer. Single patient cohorts were studied for the first three dose levels (50, 100, 165 mg/m2). The first patient treated at 245 mg/m2 developed grade 2 prerenal azotemia. Two additional patients were therefore treated in this cohort. One of these patients experienced a DLT of supraventricular tachycardia and respiratory failure after day 21 dose. Patients were enrolled at the 245 mg/m2 cohort which was expanded after the development of the first DLT [83]. MPC-3100 recently completed the Phase 1 study and results are awaited. Due to the poor solubility and bioavailability of MPC-3100, Myrexis announced the planned introduction of a pro-drug, MPC-0767. The expected filing of an IND for the pro-drug candidate MPC-0767 is in the first quarter of 2012 [84].
2.2.3. Debio 0932 (CUDC-305)
Scientists at Curis replaced the N3 of the purine (Fig. 1, depicted in red on PU3) with a carbon to result in a purine-like derivative, Debio 0932 (CUDC-305) [85], which is being developed by Debiopharm (Table 1, entry 7) [86]. In April 2010, Debiopharm initiated the phase I trial that is designed to evaluate the MTD and safety of Debio 0932 in patients with advanced solid tumors or lymphoma. The first part of this study (phase Ia) is an open-label, dose-escalation, phase trial where the study drug will be administered orally (25–100 mg) in both daily and every other day regimens. A total of 80 patients are expected to be treated in the phase Ia portion. The phase Ib portion is an expansion cohort of certain solid tumors and /or lymphoma patients which will further assess the safety, PK and PD of this agent at a potential phase II dose level to make a preliminary assessment of anti-tumor activity. About 40 patients are expected to be treated on this phase Ib portion. The trial is currently ongoing and results are awaited [87].
2.2.4. PU-H71
The Chiosis group at Memorial Sloan-Kettering has discovered the Hsp90 inhibitor PUH71 [88–90], which is being evaluated in a phase I clinical trial in patients with advanced solid tumors, lymphoma and myeloproliferative disorders under license to Samus Therapeutics(Table 1, entry 8) The primary objective is to assess the safety, tolerability, MTD, and PK of PU-H71 in patients with advanced solid tumors and lymphoma when administered intravenously three times weekly for two weeks in a 21-day cycle. Secondary objectives are to assess anti-tumor activity as defined by response rate, CR, partial response (PR), stable disease, duration of response, and progression free survival (PFS), and to evaluate 124I-PU-H71 as a non-invasive means to determine tumor PK and intratumor concentration in a selected group of patients with advanced malignancies. Exploratory objectives are to evaluate 124I-PU-H71 as a non-invasive means to predict tumor dose-response relationships in a selected group of patients with advanced malignancies, to evaluate changes in tumor client proteins using optional tumor biopsies performed pre- and post-treatment, to evaluate ex vivo the sensitivity of the tumor to PU-H71 by performing optional tumor biopsies pre- treatment and to evaluate changes in client proteins in the circulating tumor cells pre- and post-treatment. This phase I trial began patient accrual in early August 2011.
This agent is also being studied at a different dosing schedule at the NCI in patients with advanced solid tumors and low-grade non-hodgkins lymphoma. Patients will receive PU-H71 intravenously weekly × 2 weeks of 21-day cycle. This study will follow a modified accelerated titration design; the accelerated phase will end when one patient experiences a DLT or two patients experience grade 2 drug-related toxicities during the first cycle, after which the study will follow a standard 3 + 3 design. The primary objectives are to establish the safety, tolerability, MTD, recommended phase 2 dose, and PK of PU-H71 when administered on the above mentioned schedule. Secondary objectives are to perform pharmacodynamic (PD) studies to ascertain the effect of PU-H71 on Hsp90 client proteins in tumor tissue at the MTD, and on Hsp70 in tumor tissue, serum, and peripheral blood mononuclear cells at the MTD. PK and PD studies will be conducted during cycle 1. Up to 10 additional patients will be entered at the MTD to further define toxicity and perform PD studies at this dose. Planned accrual is up to 50 patients. This study is actively recruiting patients since June 2011 [91].
2.3. Resorcinol derivatives
RD is a macrocyclic lactone antibiotic first isolated from the fungus Monosporium bonorden in 1953 (Fig. 1) [92]. In addition to a resorcinol moiety (depicted in red in Fig. 1), RD also contains reactive epoxide and α,β,γ,δ-unsaturated carbonyl groups. RD is not stable in serum and is devoid of in vivo activity, however, the resorcinol core is maintained in a number of molecules entering clinical trials including NVP-AUY922, KW-2478, and AT13387 as well as STA-9090 (Table 1, entry 9–13). While these molecules were not discovered through direct modification of RD they clearly resemble it by maintaining the resorcinol core as a critical element for binding.
2.3.1. Triazole derivatives STA-9090 (Ganetespib)
STA-9090 (Table 1, entry 9) is an unspecified novel resorcinol-containing triazole compound developed by Synta Pharmaceuticals and has been tested in multiple clinical trials in both advanced solid tumors and hematological malignancies.
Goldman et al evaluated STA-9090 in a phase I trial in 35 patients with advanced solid tumors wherein they received this agent intravenously weekly for 3 weeks in a 28-day cycle. MTD was determined at 216 mg/m2. DLT was fatigue, diarrhea and elevated amylase. Other common adverse events included abdominal pain, anemia, nausea, constipation and dyspnea. There was 1 PR in a patient with rectal cancer and several SD were noted in patients with gastrointestinal stromal tumor, non-small cell lung cancer and renal cell carcinoma [93]. Another phase I dose-escalation trial is evaluating twice weekly dosing (2–144 mg/m2) for 3 weeks in a 28-day cycle. Forty-nine patients have been treated so far. DLT was elevated liver enzymes. Of the evaluable patients, 1 PR was noted in a patient with metastatic melanoma and there were 2 SD in patients with non-small cell lung cancer. STA-9090 was well tolerated up to 120 mg/m2; the 144 mg/m2 dose cohort has been expanded and dose escalation is ongoing [94].
A safety and efficacy phase I/II study in patients with acute myeloid leukemia and other hematological malignancies evaluated weekly infusions of STA-9090 for 4 consecutive weeks per cycle at 3 dose levels (120–200 mg/m2). One patient had a DLT of elevated liver enzymes. There were no formal responses noted but one patient with refractory acute myeloid leukemia had SD and bone marrow blast reduction lasting 10 weeks. The recommended phase II dose has not been recommended and accrual is ongoing [95]. Another multicenter, phase I trial evaluated STA-9090 administered intravenously twice weekly at doses ranging 14–110 mg/m2 for 4 weeks. DLT included hyponatremia, hyperbilirubinemia, elevated liver enzymes and prolonged QTc. MTD has not been determined. Clinical responses included hematological responses in 2 patients with chronic myeloid leukemia (CML) and acute myeloid leukemia respectively. In addition, 4 patients with myelofibrosis had SD; 1 of these responses lasted 7 months and the patient went on to receive allogenic stem cell transplant [96].
A phase II trial of STA-9090 was conducted in patients with non-small cell lung cancer. Patients received weekly infusions of STA-9090 at 200 mg/m2 for 3 weeks in a 28-day cycle. Patients were enrolled based on their mutation status; cohort A: EGFR, cohort B: KRAS, cohort C: EGFR and KRAS wild type (WT) and cohort D: EGFR and KRAS WT with adenocarcinoma histology. Additional mutational analysis of BRAF, PIK3CA, ERBB2 and MET, as well as FISH analysis for ALK translocation (EML4-ALK gene-rearrangement), were performed in a subset of patients. Study design also included cohort E which would allow patients with clinical benefit in cohorts A through D to receive weekly docetaxel therapy in addition to ganetespib after progression of disease. Primary endpoint was PFS at 16 weeks with expansion for that particular cohort. Common adverse events reported were fatigue, diarrhea, nausea, anorexia and dyspnea which were all manageable and easily reversible. Expansion criteria were achieved for cohort C, including 1 PR and 7 patients with SD > 16 weeks [97]. Updated results were presented at the 14th world Conference on lung cancer in July, 2011 in Amsterdam. It was noted that 8 patients in cohort C/D harbored the EML4-ALK-rearrangement gene (ALK+tumors). Six out of 8 (75%) patients were crizotinib naïve and had tumor shrinkage in the target lesions. Additionally, another patient with ALK+ tumor with crizotinib-refractory disease had impressive tumor regressions in the lung and responded to single agent ganetespib. Eight of thirteen (62%) patients with KRAS mutation also had tumor shrinkage. One patient with mixed response (16% target lesion shrinkage and development of new lesions) rolled over to cohort E at 8 months. A phase IIb/III trial of ganetespib and docetaxel in second line advanced non-small cell lung cancer patients has been initiated [98].
Similar to the patients with non-small cell lung cancer, patients with gastrointestinal stromal tumor were also treated with STA-9090 at 200 mg/m2 weekly for 3 weeks in a 28-day cycle. In this Simon’s two stage study design, if > 4/23 patients in stage 1 had a CBR > 16 weeks, then enrollment would continue to stage 2. Hsp90 client protein levels were analyzed in pre- and post-treatment tumor biopsies. Common adverse events noted were fatigue, diarrhea, nausea, vomiting and increased alkaline phosphatase. 12/23 evaluable patients had SD meeting the formal criteria to enroll to stage 2. However, paired tumor biopsies in 4 patients did not show prolonged inhibition of activated KIT or its downstream pathways. Accrual has been limited to patients with PDGFRA mutations to allow alternative schedules and combinations [99].
An open label phase II trial of STA-9090 is currently ongoing in patients with unselected metastatic breast cancer. STA-9090 is administered at the same dose and schedule used in patients with non-small cell lung cancer and gastrointestinal stromal tumor. Primary endpoint is to determine overall RR using RECIST 1.1 [100].
2.3.2. Isoxazole derivatives NVP-AUY922/VER52296
Workman and colleagues from the Cancer Research UK Centre for Cancer Therapeutics first identified the resorcinol containing pyrazole CCT018159 from a HTS of a library of 56,000 compounds capable of inhibiting the ATPase activity of yeast Hsp90 [101]. Using a structure-based approach, scientists at Vernalis and the Cancer Research UK Centre for Cancer Therapeutics optimized this hit into the isoxazole NVP-AUY922/VER52296 (Table 1, entry 10) [102, 103]. This compound is now being developed by Novartis and is currently in phaseI/II clinical trials in patients with relapsed or refractory multiple myeloma and HER2+ and ER+ metastatic breast cancer.
Single agent NVP-AUY922 was administered intravenously weekly in 28-day cycle to patients with advanced solid tumors in a phase I study. A total of 96 patients were treated at doses ranging 2–70 mg/m2. Endpoints also included PK and PD analyses. DLT was atrial flutter, diarrhea, fatigue, darkening of vision and anorexia. Other common adverse events included nausea (35%), vomiting (18%), and night blindness (20%). MTD was 70mg/m2. SD was reported in 16 patients and 9 patients had a partial metabolic response on FDG-PET [104]. This led to the phase II expansion in patients with HER2+ and ER+ metastatic breast cancer. Preliminary imaging and biomarker data from this phase II trial was reported in the 2011 Annual ASCO Scientific meeting. Patients with HER2+ and ER+ disease underwent FDG-PET with trastuzumab and bevacizumab respectively at baseline and during treatment. Two HER2+ patients had partial metabolic response and 1 of these patients had confirmed PR by RECIST [105].
A phase I/II study to determine the MTD of NVP-AUY922 alone and in combination with bortezomib, with or without dexamethasone, in patients with relapsed or refractory multiple myeloma has been completed and results are awaited [106].
Although the chemical structure of HSP990, an oral Hsp90 inhibitor, has not yet been reported, it is claimed to be a follow-up compound of NVP-AUY922. A phase I trial is evaluating HSP990 when administered either weekly or twice weekly in patient with advanced solid tumors. Endpoints include MTD, DLT, efficacy, PK and PD. The study is currently actively recruiting patients [107]. Another phase I trial is assessing the once weekly dose in adult Japanese and Korean patients with advanced solid tumors [108].
2.3.3. Other Resorcinol derivatives
2.3.3.1. KW-2478
The novel resorcinol analog KW-2478 was discovered by Kyowa Hakko Kirin Pharma through “a unique lead optimization strategy, including microbial screening, x-ray crystallography, cell-based screening, and in vivo models” (Table 1, entry 12) [109]. A phase I safety, PK and PD study of KW-2478 was recently completed in patients with relapsed/refractory multiple myeloma, chronic lymphocytic leukemia or B-cell non-hodgkins lymphoma. KW-2478 (14–99 mg/m2) was administered intravenously daily on days 1–5 of a 14-day cycle. There were no DLT’s observed up to 99 mg/m2. Drug related toxicities included grade 1/2 hypertension in one patient and grade 3 QTc prolongation in another. The study is ongoing at a dose of 132 mg/m2 and further dose-escalation is planned [110]. Another phase I/II trial is evaluating this agent in combination with bortezomib when administered on days 1, 4, 8, 11 of a 21-day cycle in patients with relapsed and refractory multiple myeloma [111].
2.3.3.2. AT-13387
AT13387 was discovered by scientists at Astex following optimization of a resorcinol-containing lead generated through a fragment-based drug discovery approach (Table 1, entry 13) [112, 113]. Three phase I trials are evaluating this agent in various dosing schedules including weekly or twice weekly intravenous infusions for 3 weeks in a 28-day cycle [114] and twice a week (2 days in a row) for the first three weeks in a 28-day cycle [115, 116] in patients with advanced solid tumors. It is also being evaluated with or without Imatinib in a multicenter, randomized phase II study in patients with advanced gastrointestinal stromal tumor who have progressed following treatment with TKI’s. AT-13387 will be administered IV weekly for 3 weeks of a 28-day cycle. Primary endpoint of this trial is to assess reduction/stabilization of tumor at 4 months using RECIST 1.1 criteria [117].
2.4. Dihydroindazolone derivatives
SNX-5422 is a pyrazole-containing Hsp90 inhibitor that was discovered by scientists at Serenex following optimization of a hit identified using an ATP-affinity column (Table 1, entry 14) [118]. This column was used to first load ATP-binding proteins, then was challenged with a library of structurally diverse 8,000 compounds to identify molecules capable of displacing unspecified proteins from the column. Hsp90 was identified as the target of an initial hit compound. SNX-5422 is actually the glycine prodrug of SNX-2112 (Fig. 1). In May 2007, Serenex Inc started a phase I trial for SNX-5422 in patients with solid tumor and lymphoma. SNX-5422 (4–21.28 mg/m2) was administered orally every other day daily for 21 days in a 28-day cycle. Objectives were to determine MTD and PK of this agent. There were no DLT’s observed among the 11 patients treated. Grade 1 toxicities included nausea and fatigue. The trial was enrolling at 21.28 mg/m2 [119]. In March 2008, Pfizer Inc announced that it has entered into an agreement to acquire Serenex Inc [120]. Later, the development of SNX-5422 was discontinued based on reports of ocular toxicity and the potential for irreversible retinal damage [121].
2.5. Others
Other Hsp90 inhibitors include DS-2248 (Daiichi Sankyo, Inc; undergoing phase I evaluation) [122] and XL-888 (Exelixis; sponsor has decided to terminate the safety study of this agent) [123]. The structures for these two inhibitors have not been disclosed.
3. Assays for monitoring response to Hsp90 inhibition in clinic
Several clinical assays have been developed to assess Hsp90-target inhibition, mainly by measuring the levels of a number of Hsp90 onco-client proteins from samples obtained pre- and post-Hsp90 inhibitor administration. As mentioned, Hsp90 inhibition causes the proteasomal degradation of such client proteins [1, 2, 12]. Hsp70 induction, another read-out of Hsp90 inhibition established in several cancer models, was also evaluated.
Most such PD assays to date have been performed on peripheral blood mononuclear cells (PBMCs). Several potential biomarkers, such as CDK4, Raf-1, c-KIT reduction and Hsp70 induction were measured in these studies. While PD monitoring of PBMCs has provided a readily accessible and reproducible index of in vivo biologic activity of Hsp90 inhibitors in clinical trials, the current consensus from these studies is that drug effects in these cells do not predict tumor-specific activity [18, 53, 54].
This is not surprising in light of a body of evidence showing a higher sensitivity of cancer cells to Hsp90 inhibition when compared to normal cells, as well as preferential accumulation of these inhibitors in tumor cells rather than normal cells [124–126]. As a means of explanation, Kamal et al proposed that Hsp90 in tumors exists entirely in multi-chaperone complexes of high affinity for certain small molecule inhibitors, whereas Hsp90 from normal tissues is in a latent, uncomplexed state of low inhibitor binding potential [123]. Recent work by Moulick et al substantiates but also significantly extends this view by proposing that Hsp90 forms biochemically distinct complexes in cancer cells [125]. In this view, a major fraction of cancer cell Hsp90 retains “house keeping” chaperone functions similar to normal cells, whereas a functionally distinct Hsp90 pool enriched or expanded in cancer cells specifically interacts with oncogenic proteins required to maintain tumor cell survival. This Hsp90 fraction perhaps represents a cell stress specific form of chaperone complex, that is expanded and constitutively maintained in the tumor cell context, and that may execute functions necessary to maintain the malignant phenotype. Such roles are to regulate the folding of overexpressed (i.e. HER2), mutated (i.e. mB-Raf) or chimeric proteins (i.e. Bcr-Abl) [1–3], but also to facilitate scaffolding and complex formation of molecules involved in aberrantly activated signaling complexes (i.e. STAT5, BCL6) [88, 125]. The abundance of this tumor Hsp90 species, which is not dictated by Hsp90 expression alone, is potentially predictive of the cell's sensitivity to Hsp90 inhibition [125].
Preclinical data also suggest that the levels of soluble IGFBP2 and HER2 extracellular domain levels [127] or serum Hsp70 might be a useful PD marker of drug response [128], however, the utility of these potential biomarkers has yet to be validated in clinical settings [129].
The use of biopsies to measure PD changes has also been attempted in an effort to assay target modulation [16], but remains limited because of the logistical and ethical issues associated with repeated invasive procedures. Studies in melanoma patients noted changes in biomarkers in patients that experienced prolonged stable disease when treated with 17-AAG [16] at the dose levels 320 and 450 mg/m2/week. When 17-AAG was administered i.v. once weekly × 6 weeks at 450 mg/m2 dose and schedule of 17-AAG, the effects of 17-AAG on Raf-1 kinase expression were short-lived, and no objective anti-melanoma responses were seen [130], suggesting collectively that at this dose and schedule, the tumor Hsp90 target was not optimally inhibited.
As an alternative to tumor biopsies, changes in the levels of tumor HER2 and VEGF levels are now being investigated non-invasively by PET using gallium-68 or zirconium-89 labeled antibodies [131–133]. This sensitive assay is able to monitor tumor receptor expression in real time following systemic Hsp90 inhibitor administration, and is currently incorporated in several ongoing clinical studies with Hsp90 inhibitors [133–135]. While results of these studies are awaited, it is evident that the use of these assays remains limited to tumors that express these markers.
PET with [18F]-FDG was also tested as an assay to measure early response to Hsp90 therapy. Preclinical studies in HER2 tumors suggested no changes in FDG-PET at 24 hours post Hsp90 inhibitor administration, in contrast to HER2 PET [136]. When incorporated into a Phase I trial of IPI-504 in patients with metastatic TKI-resistant gastrointestinal stromal tumor who received the Hsp90 inhibitor on days 1, 4, 8 and 11 of a 21 day cycle, serial monitoring with 18FDG-PET/CT imaging at baseline, day 11, and day 21, showed a reduced FDG-PET signal. Reactivation of tumor FDG uptake correlated with planned breaks in drug administration, and the signal decreased again when treatment resumed [137]. On this trial only SD was noted and it remains unclear whether a complete FDG uptake inhibition rather than reduction in uptake is needed to result in CR and PR. Metabolic PR and SD have been reported with BIIB021 and NVP-AUY922 using FDG-PET imaging [79, 104]. These data suggest that, at least in highly glycolytic tumors, FDG-PET may provide a useful assay to measure Hsp90 tumor inhibition over a longer drug administration time.
The Hsp90 inhibitor PU-H71 has an endogenous iodine atom (127I), which can be conveniently replaced with the PET radionuclide 124I to result in the imaging agent 124I-PU-H71. Since the PET imaging agent is virtually identical to PU-H71, the use of this radioligand allows for assessing drug distribution by non-invasive PET imaging. A phase 0 trial of 124I-PU-H71 which evaluates the radiation dosimetry, metabolism and biodistribution of this PET agent is currently ongoing [138]. The purpose of this study is to see how PU-H71 accumulates and how long it lasts inside tumors. The results of the phase 0 trial will help plan how to use PU-H71 in the phase I setting; specifically it will allow optimization of the PET agent to assess tumor pharmacokinetics of PU-H71 in the phase I study. The PET assay will also be investigated in the phase I to evaluate tumor dose-response correlations by measuring PU-H71 tumor concentrations for up to 48 hours post co-infusion of PU-H71 with 124I-PU-H71 [139]. Together, this may provide useful information on identifying tumors more likely to respond to PU-H71 therapy and in designing most optimal dose and schedules for the administration of the Hsp90 inhibitor.
3. Conclusion
Given their potential to degrade a number of different oncoproteins and affect multiple signaling pathways, Hsp90 inhibitors have been hypothesized to be active in a wide variety of cancers. To date, the greatest clinical activity as evidenced by objective tumor regressions, has been observed with the geldanamycin analogue tanespimycin when given in combination with trastuzumab to patients with HER2+ metastatic breast cancer. Pre-clinically, HER2 is among the most sensitive client proteins to Hsp90 inhibition and this may account for the activity observed in this setting. Recently, objective tumor responses were reported with ganetespib in all tumors that harbored the ALK rearrangement. This further emphasizes the targeted approach of Hsp90 inhibition, in that responses are perhaps dependent upon addiction of the tumors to Hsp90 client oncoproteins. While these results validate Hsp90 inhibition as a relevant anticancer strategy, the full potential of these inhibitors to be active in a broader spectrum of tumor types has yet to be achieved. This differential response could be because we are unable to select patients who might best benefit from therapy and perhaps have not successfully optimized the dosing and scheduling of this class of agents.
As a class, Hsp90 inhibitors demonstrate rapid clearance from normal tissues and the blood compartment with prolonged retention in tumors, hence traditional serum pharmacokinetics may be insufficient to guide dosing and scheduling [4, 88, 89, 90, 103, 125, 140]. Additionally, the current drug development paradigm based on identifying maximum tolerated dose may not be applicable in the case of these inhibitors where target modulation is critical to producing their anti-tumor effects. In order to optimally deliver Hsp90 inhibitor therapy, evaluation of the effects of these agents at the level of the tumor (including client protein modulation) is essential. To this end, tumor biopsies and novel targeted imaging studies must be incorporated into future clinical trials of these drugs and efforts at identifying robust biomarkers of response and resistance will be crucial next steps in rationalizing the clinical development of the growing list of Hsp90 inhibitors. Ultimately this investment into understanding the fundamentals of drug delivery and patient selection for these unique inhibitors will allow the next generation of Hsp90 targeted therapies to realize their full potential and promise as anti-cancer therapeutics.
Highlights.
we summarize the current status of Hsp90 inhibitors based on their chemical classification
we present the current stage of clinical development for Hsp90 inhibitors in cancers
we discuss the assays and biomarkers currently implemented in clinic for Hsp90 inhibitors
we present strategies aimed at enhancing the clinical effectiveness of Hsp90 inhibitors in cancer
Acknowledgement
G Chiosis is funded in part by Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research" and "The Experimental Therapeutics Center of Memorial Sloan-Kettering Cancer Center", the Geoffrey Beene Cancer Research Center of MSKCC, Leukemia and Lymphoma Society, Breast Cancer Research Fund, the SPORE Pilot Award and Research &Therapeutics Program in Prostate Cancer, the Hirshberg Foundation for Pancreatic Cancer, the Byrne Fund, National Institutes of Health (1U01 AG032969-01A1, 1R01 CA155226-01, 1R21AI090501-01, 3P30CA008748), Department of Defense (R03-BC085588), Susan G Komen for the Cure and the Institute for the Study of Aging and The Association for Frontotemporal Dementias (Grant #281207 AFTD). T Taldone discloses a grant support from the Department of Defense (PDF-BC093421), K Jhaveri from Conquer Cancer Foundation: Young Investigator Award and S Modi from ASCO Cancer Foundation/ Breast Cancer Research Foundation: Advanced Clinical Research Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zuehlke A, Johnson JL. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers. 2010;93:211–217. doi: 10.1002/bip.21292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 3.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel HJ, Modi S, Chiosis G, Taldone T. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opinion on Drug Discovery. 2011;6:559–587. doi: 10.1517/17460441.2011.563296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taldone T, Sun W, Chiosis G. Discovery and development of heat shock protein 90 inhibitors. Bioorg Med Chem. 2009;17:2225–2235. doi: 10.1016/j.bmc.2008.10.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janin YL. ATPase inhibitors of heat-shock protein 90, second season. Drug Discov Today. 2010;15:342–353. doi: 10.1016/j.drudis.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 7.DeBoer C, Meulman PA, Wnuk RJ, Peterson DH. Geldanamycin, a new antibiotic. Journal of Antibiotics. 1970;23:442–447. doi: 10.7164/antibiotics.23.442. [DOI] [PubMed] [Google Scholar]
- 8.Whitesell L, Shifrin SD, Schwab G, Neckers LM. Benzoquinonoid ansamycins possess selective tumoricidal activity unrelated to src kinase inhibition. Cancer Research. 1992;52:1721–1728. [PubMed] [Google Scholar]
- 9.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 11.Grenert JP, Sullivan WP, Fadden P, Haystead TA, Clark J, Mimnaugh E, Krutzsch H, Ochel HJ, Schulte TW, Sausville E, Neckers LM, Toft DO. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem. 1997;272:23843–23850. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- 12.Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem J. 2008;410:439–453. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 13.Neckers L. Chaperoning oncogenes: Hsp90 as a target of geldanamycin. Handb Exp Pharmacol. 2006:259–277. doi: 10.1007/3-540-29717-0_11. [DOI] [PubMed] [Google Scholar]
- 14.Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol. 1995;36:305–315. doi: 10.1007/BF00689048. [DOI] [PubMed] [Google Scholar]
- 15.Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol. 1998;42:273–279. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- 16.Banerji U, O'Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y, Simmons L, Maloney A, Raynaud F, Campbell M, Walton M, Lakhani S, Kaye S, Workman P, Judson I. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol. 2005;23:4152–4161. doi: 10.1200/JCO.2005.00.612. [DOI] [PubMed] [Google Scholar]
- 17.Goetz MP, Toft D, Reid J, Ames M, Stensgard B, Safgren S, Adjei AA, Sloan J, Atherton P, Vasile V, Salazaar S, Adjei A, Croghan G, Erlichman C. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol. 2005;23:1078–1087. doi: 10.1200/JCO.2005.09.119. [DOI] [PubMed] [Google Scholar]
- 18.Grem JL, Morrison G, Guo XD, Agnew E, Takimoto CH, Thomas R, Szabo E, Grochow L, Grollman F, Hamilton JM, Neckers L, Wilson RH. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J Clin Oncol. 2005;23:1885–1893. doi: 10.1200/JCO.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 19.Ramanathan RK, Trump DL, Eiseman JL, Belani CP, Agarwala SS, Zuhowski EG, Lan J, Potter DM, Ivy SP, Ramalingam S, Brufsky AM, Wong MK, Tutchko S, Egorin MJ. Phase I pharmacokinetic-pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin (17AAG, NSC 330507), a novel inhibitor of heat shock protein 90, in patients with refractory advanced cancers. Clin Cancer Res. 2005;11:3385–3391. doi: 10.1158/1078-0432.CCR-04-2322. [DOI] [PubMed] [Google Scholar]
- 20.Solit DB, Ivy SP, Kopil C, Sikorski R, Morris MJ, Slovin SF, Kelly WK, DeLaCruz A, Curley T, Heller G, Larson S, Schwartz L, Egorin MJ, Rosen N, Scher HI. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. Clin Cancer Res. 2007;13:1775–1782. doi: 10.1158/1078-0432.CCR-06-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nowakowski GS, McCollum AK, Ames MM, Mandrekar SJ, Reid JM, Adjei AA, Toft DO, Safgren SL, Erlichman C. A phase I trial of twice-weekly 17-allylamino-demethoxy-geldanamycin in patients with advanced cancer. Clin Cancer Res. 2006;12:6087–6093. doi: 10.1158/1078-0432.CCR-06-1015. [DOI] [PubMed] [Google Scholar]
- 22.Weigel BJ, Blaney SM, Reid JM, Safgren SL, Bagatell R, Kersey J, Neglia JP, Ivy SP, Ingle AM, Whitesell L, Gilbertson RJ, Krailo M, Ames M, Adamson PC. A phase I study of 17-allylaminogeldanamycin in relapsed/refractory pediatric patients with solid tumors: a Children's Oncology Group study. Clin Cancer Res. 2007;13:1789–1793. doi: 10.1158/1078-0432.CCR-06-2270. [DOI] [PubMed] [Google Scholar]
- 23.Bagatell R, Gore L, Egorin MJ, Ho R, Heller G, Boucher N, Zuhowski EG, Whitlock JA, Hunger SP, Narendran A, Katzenstein HM, Arceci RJ, Boklan J, Herzog CE, Whitesell L, Ivy SP, Trippett TM. Phase I pharmacokinetic and pharmacodynamic study of 17-N-allylamino-17-demethoxygeldanamycin in pediatric patients with recurrent or refractory solid tumors: a pediatric oncology experimental therapeutics investigators consortium study. Clin Cancer Res. 2007;13:1783–1788. doi: 10.1158/1078-0432.CCR-06-1892. [DOI] [PubMed] [Google Scholar]
- 24.Heath EI, Hillman DW, Vaishampayan U, Sheng S, Sarkar F, Harper F, Gaskins M, Pitot HC, Tan W, Ivy SP, Pili R, Carducci MA, Erlichman C, Liu G. A Phase II Trial of 17-Allylamino-17-Demethoxygeldanamycin in Patients with Hormone-Refractory Metastatic Prostate Cancer. Clinical Cancer Research. 2008;14:7940–7946. doi: 10.1158/1078-0432.CCR-08-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solit D, Osman I, Polsky D, Panageas K, Daud A, Goydos J, Teitcher J, Wolchok J, Germino J, Krown S, Coit D, Rosen N, Chapman P. Phase II Trial of 17-Allylamino-17-Demethoxygeldanamycin in Patients with Metastatic Melanoma. Clinical Cancer Research. 2008;14:8302–8307. doi: 10.1158/1078-0432.CCR-08-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ronnen E, Kondagunta GV, Ishill N, Sweeney SM, Deluca JK, Schwartz L, Bacik J, Motzer RJ. A phase II trial of 17-(Allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest New Drugs. 2006;26:543–546. doi: 10.1007/s10637-006-9208-z. [DOI] [PubMed] [Google Scholar]
- 27.Richardson PG, Chanan-Khan AA, Alsina M, Albitar M, Berman D, Messina M, Mitsiades CS, Anderson KC. Tanespimycin monotherapy in relapsed multiple myeloma: Results of a phase 1 dose-escalation study. British Journal of Haematology. 2010;150:438–445. doi: 10.1111/j.1365-2141.2010.08265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Bagatell R, Ma W, Wheler J, Rosen N, Norton L, Cropp GF, Johnson RG, Hannah AL, Hudis CA. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J Clin Oncol. 2007;25:5410–5417. doi: 10.1200/JCO.2007.11.7960. [DOI] [PubMed] [Google Scholar]
- 29.Modi S, Stopeck AT, Linden HM, Solit DB, Chandarlapaty S, Rosen N, D'Andrea G, Dickler MN, Moynahan ME, Sugarman S, Ma W, Patil S, Norton L, Hannah AL, Hudis C. HSP90 Inhibition is Effective in Breast Cancer: A Phase 2 Trial of Tanespimycin (17AAG) plus Trastuzumab in Patients with HER2-Positive Metastatic Breast Cancer Progressing on Trastuzumab. Clin Cancer Res. 2011 doi: 10.1158/1078-0432.CCR-11-0072. [DOI] [PubMed] [Google Scholar]
- 30.Chandarlapaty S, Scaltriti M, Angelini P, Ye Q, Guzman M, Hudis CA, Norton L, Solit DB, Arribas J, Baselga J, Rosen N. Inhibitors of HSP90 block p95-HER2 signaling in Trastuzumab-resistant tumors and suppress their growth. Oncogene. 2010;29:325–334. doi: 10.1038/onc.2009.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richardson P, Chanan-Khan A, Lonial S, Krishnan AY, Carroll M, Alsina M, Albitar M, Berman D, Messina M, Anderson K. Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: results of a phase 1/2 study. British Journal of Haematology. 2011;153:729–740. doi: 10.1111/j.1365-2141.2011.08664.x. [DOI] [PubMed] [Google Scholar]
- 32.Richardson PG, Badros AZ, Jagannath S, Tarantolo S, Wolf JL, Albitar M, Berman D, Messina M, Anderson KC. Tanespimycin with bortezomib: activity in relapsed/refractory patients with multiple myeloma. Br J Haematol. 2010;150:428–437. doi: 10.1111/j.1365-2141.2010.08264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong Z, Simmons J, Timmermans P. Prevention and treatment of bortezemib-induced peripheral neuropathy by the Hsp90 inhibitor tanespimycin (KOS-953) in the rat. European Journal of cancer Supplements. 2008;6:23. [Google Scholar]
- 34.PressRelease, Bristol-Myers Squibb Halts Development of Tanespimycin. 2008 available from: http://www.myelomabeacon.com/news/2010/07/22/tanespimycin-development-halted/, in,
- 35.Arlander SJ, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM. Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. J Biol Chem. 2003;278:52572–52577. doi: 10.1074/jbc.M309054200. [DOI] [PubMed] [Google Scholar]
- 36.Flatten K, Dai NT, Vroman BT, Loegering D, Erlichman C, Karnitz LM, Kaufmann SH. The role of checkpoint kinase 1 in sensitivity to topoisomerase I poisons. J Biol Chem. 2005;280:14349–14355. doi: 10.1074/jbc.M411890200. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen DM, Lorang D, Chen GA, Stewart JHt, Tabibi E, Schrump DS. Enhancement of paclitaxel-mediated cytotoxicity in lung cancer cells by 17-allylamino geldanamycin: in vitro and in vivo analysis. The Annals of thoracic surgery. 2001;72:371–378. doi: 10.1016/s0003-4975(01)02787-4. discussion 378–379. [DOI] [PubMed] [Google Scholar]
- 38.Ramalingam SS, Egorin MJ, Ramanathan RK, Remick SC, Sikorski RP, Lagattuta TF, Chatta GS, Friedland DM, Stoller RG, Potter DM, Ivy SP, Belani CP. A phase I study of 17-Allylamino-17-demethoxygeldanamycin combined with paclitaxel in patients with advanced solid malignancies. Clinical Cancer Research. 2008;14:3456–3461. doi: 10.1158/1078-0432.CCR-07-5088. [DOI] [PubMed] [Google Scholar]
- 39.Solit DB, Basso AD, Olshen AB, Scher HI, Rosen N. Inhibition of heat shock protein 90 function down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer Res. 2003;63:2139–2144. [PubMed] [Google Scholar]
- 40.Ramalingam SS, Egorin MJ, Ramanathan RK, Remick SC, Sikorski RP, Lagattuta TF, Chatta GS, Friedland DM, Stoller RG, Potter DM, Ivy SP, Belani CP. A phase I study of 17-allylamino-17-demethoxygeldanamycin combined with paclitaxel in patients with advanced solid malignancies. Clin Cancer Res. 2008;14:3456–3461. doi: 10.1158/1078-0432.CCR-07-5088. [DOI] [PubMed] [Google Scholar]
- 41.Solit DB, Rosen N. Hsp90: A novel target for cancer therapy. Current Topics in Medicinal Chemistry. 2006;6:1205–1214. doi: 10.2174/156802606777812068. [DOI] [PubMed] [Google Scholar]
- 42.McCollum AK, Lukasiewicz KB, TenEyck CJ, Lingle WL, Toft DO, Erlichman C. Cisplatin abrogates the geldanamycin-induced heat shock response. Molecular Cancer Therapeutics. 2008;7:3256–3264. doi: 10.1158/1535-7163.MCT-08-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hubbard J, Erlichman C, Toft DO, Qin R, Stensgard BA, Felten S, Ten Eyck C, Batzel G, Ivy SP, Haluska P. Phase I study of 17-allylamino-17 demethoxygeldanamycin, gemcitabine and/or cisplatin in patients with refractory solid tumors. Investigational New Drugs. 2011;29:473–480. doi: 10.1007/s10637-009-9381-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tse AN, Klimstra DS, Gonen M, Shah M, Sheikh T, Sikorski R, Carvajal R, Mui J, Tipian C, O'Reilly E, Chung K, Maki R, Lefkowitz R, Brown K, Manova-Todorova K, Wu N, Egorin MJ, Kelsen D, Schwartz GK. A phase 1 dose-escalation study of irinotecan in combination with 17-allylamino-17-demethoxygeldanamycin in patients with solid tumors. Clinical Cancer Research. 2008;14:6704–6711. doi: 10.1158/1078-0432.CCR-08-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yin X, Zhang H, Lundgren K, Wilson L, Burrows F, Shores CG. BIIB021, a novel Hsp90 inhibitor, sensitizes head and neck squamous cell carcinoma to radiotherapy. Int J Cancer. 2010;126:1216–1225. doi: 10.1002/ijc.24815. [DOI] [PubMed] [Google Scholar]
- 46.Vaishampayan UN, Burger AM, Sausville EA, Heilbrun LK, Li J, Horiba MN, Egorin MJ, Ivy P, Pacey S, LoRusso PM. Safety, efficacy, pharmacokinetics, and pharmacodynamics of the combination of sorafenib and tanespimycin. Clinical Cancer Research. 2010;16:3795–3804. doi: 10.1158/1078-0432.CCR-10-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bagatell R, Paine-Murrieta GD, Taylor CW, Pulcini EJ, Akinaga S, Benjamin IJ, Whitesell L. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res. 2000;6:3312–3318. [PubMed] [Google Scholar]
- 48.McCollum AK, Lukasiewicz KB, Teneyck CJ, Lingle WL, Toft DO, Erlichman C. Cisplatin abrogates the geldanamycin-induced heat shock response. Mol Cancer Ther. 2008;7:3256–3264. doi: 10.1158/1535-7163.MCT-08-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tao C, Yu C, De TK, Everett N, Frankel T, Ci S, Trieu Voon-Shiong P, Desai N. Preparation of nanoparticle albumin bound 17AAG (nab-17AAG) suitable for intravenous administration. Proc Amer Assoc Cancer Res. 2005;46 Abstract 1435. [Google Scholar]
- 50.Pitot HC. [Updated July 14, 2011];A Trial of ABI-010 & ABI-007 in Patients With Advanced Non-Hematologic Malignancies. available from: http://clinicaltrials.gov/ct2/show/NCT00820768?term=ABI-010&rank=1, in,
- 51.Messaoudi S, Peyrat JF, Brion JD, Alami M. Recent advances in Hsp90 inhibitors as antitumor agents. Anticancer Agents Med Chem. 2008;8:761–782. doi: 10.2174/187152008785914824. [DOI] [PubMed] [Google Scholar]
- 52.Hollingshead M, Alley M, Burger AM, Borgel S, Pacula-Cox C, Fiebig HH, Sausville EA. In vivo antitumor efficacy of 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride), a water-soluble geldanamycin derivative. Cancer Chemother Pharmacol. 2005;56:115–125. doi: 10.1007/s00280-004-0939-2. [DOI] [PubMed] [Google Scholar]
- 53.Kummar S, Gutierrez ME, Gardner ER, Chen X, Figg WD, Zajac-Kaye M, Chen M, Steinberg SM, Muir CA, Yancey MA, Horneffer YR, Juwara L, Melillo G, Ivy SP, Merino M, Neckers L, Steeg PS, Conley BA, Giaccone G, Doroshow JH, Murgo AJ. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. European Journal of Cancer. 2010;46:340–347. doi: 10.1016/j.ejca.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramanathan RK, Egorin MJ, Erlichman C, Remick SC, Ramalingam SS, Naret C, Holleran JL, TenEyck CJ, Ivy SP, Belani CP. Phase I pharmacokinetic and pharmacodynamic study of 17-dimethylaminoethylamino-17-demethoxygeldanamycin, an inhibitor of heat-shock protein 90, in patients with advanced solid tumors. Journal of Clinical Oncology. 2010;28:1520–1526. doi: 10.1200/JCO.2009.25.0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pacey S, Wilson RH, Walton M, Eatock MM, Hardcastle A, Zetterlund A, Arkenau HT, Moreno-Farre J, Banerji U, Roels B, Peachey H, Aherne W, De Bono JS, Raynaud F, Workman P, Judson I. A phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clinical Cancer Research. 2011;17:1561–1570. doi: 10.1158/1078-0432.CCR-10-1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lancet JE, Gojo I, Burton M, Quinn M, Tighe SM, Kersey K, Zhong Z, Albitar MX, Bhalla K, Hannah AL, Baer MR. Phase i study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia. 2010;24:699–705. doi: 10.1038/leu.2009.292. [DOI] [PubMed] [Google Scholar]
- 57.Flaherty K, Gore L, Avadhani A, Leong S, Harlacker K, Zhong Z. Phase 1, pharmacokinetic (PK) and pharmacodynamic (PD) study of oral alvespimycin (A; KOS-1022; 17-DMAG): two different schedules in patients with advanced malignancies. J Clin Oncol. 2007;25 [Google Scholar]
- 58.Miller K, Rosen LS, Modi S, Schneider B, Roy J, Chap L, Paulsen M, Kersey K, Hannah AL, Hudis C. Phase I trial of alvespimycin (KOS-1022; 17-DMAG) and trastuzumab (T) J Clin Oncol. 2007;25:1115. [Google Scholar]
- 59.F. Press Release, Kosan Announces Senior Management Changes and Clinical Portfolio Priorities. 2008 available from: http://phx.corporate-ir.net/phoenix.zhtml?c=121014&p=irolnewsArticle&ID=1113760&highlight=, in,
- 60.Sydor JR, Normant E, Pien CS, Porter JR, Ge J, Grenier L, Pak RH, Ali JA, Dembski MS, Hudak J, Patterson J, Penders C, Pink M, Read MA, Sang J, Woodward C, Zhang Y, Grayzel DS, Wright J, Barrett JA, Palombella VJ, Adams J, Tong JK. Development of 17-allylamino-17-demethoxygeldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proc Natl Acad Sci U S A. 2006;103:17408–17413. doi: 10.1073/pnas.0608372103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sydor J, Normant E, Pien C, Porter J, Ge J, Grenier L, Pak R, Ali J, Dembski M, Hudak J, Patterson J, Penders C, Pink M, Read M, Sang J, Woodward C, Zhang Y, Grayzel D, Wright J, Barrett J, Palombella V, Adams J, Tong J. Development of 17-allylamino-17-demethoxygeldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proceedings of the National Academy of Sciences. 2006;103:17408–17413. doi: 10.1073/pnas.0608372103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siegel D, Jaggannath S, Mazumder A. Update on Phase I clinical trial of IPI-504, a novel, water-soluble Hsp90 inhibitor, in patients with relapsed/refractory multiple myeloma (MM) Blood. 2006;108 abstract 3579. [Google Scholar]
- 63.Sequist L, Janne P, Sweeney J. Phase 1/2 Trial of the Novel Hsp90 Inhibitor IPI-504, in Patients with Relapsed and/or Refractory Stage IIIb or Stage IV Non-Small Cell Lung Cancer (NSCLC) Stratified by EGFR Mutation Status. AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics. 2007 [Google Scholar]
- 64.Sequist LV, Gettinger S, Senzer NN, Martins RG, Jänne PA, Lilenbaum R, Gray JE, Iafrate AJ, Katayama R, Hafeez N, Sweeney J, Walker JR, Fritz C, Ross RW, Grayzel D, Engelman JA, Borger DR, Paez G, Natale R. Activity of IPI-504, a Novel Heat-Shock Protein 90 Inhibitor, in Patients With Molecularly Defined Non-Small-Cell Lung Cancer. Journal of Clinical Oncology. 2010;28:4953–4960. doi: 10.1200/JCO.2010.30.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Riely G, Stoller R, Egorin M, Solit D, Dunbar J, Savage A, Walker J, Grayzel D, Ross R, Weiss GJ. A phase Ib trial of IPI-504 (retaspimycin hydrochloride), a novel Hsp90 inhibitor, in combination with docetaxel. J Clin Oncol. 2009;27 abstract 3547. [Google Scholar]
- 66.Riely G, Gettinger SN, Stoller RG, Gabrail NY, Dy GK, Weiss GJ, Tunkey C, Skiliris G, Strychor S, Dunbar J, DeLucia D, Ross RW, Gray JE. safety and Activity of IPI-504 (restaspimycin hydrochloride) and Docetaxel in Pretreated Patients with Metastatic Non-Small Cell Lung Cancer (NSCLC) J Clin Oncol. 2011;29 Abstract 7516. [Google Scholar]
- 67.Oh W, Stadler WM, Srinivas S, Chu F, Bubley G, Quigley M, Goddard J, Dunbar J, Grayzel D, Ross RW. A single arm phase II trial of IPI-504 in patients with castration resistant prostate cancer (CRPC) Genitourinary Cancers Symposium. 2009 Abstract 219. [Google Scholar]
- 68.Modi S, Saura C, Henderson CA, Lin NU, Mahtani RL, Goddard J, Rodenas E, O'Shaughnessy J, Baselga J. Efficacy and safety of retaspimycin hydrochloride (IPI-504) in combination with trastuzumab in patients (pts) with pretreated, locally advanced or metastatic HER2-positive breast cancer. J Clin Oncol. 2011;29 abstract 590. [Google Scholar]
- 69.Wagner A, Morgan JA, Chugh R, Rosen LS, George S, Gordon MS, Devine CM, Van Den Abbeele AD, Grayzel D, Demetri GD. Inhibition of heat shock protein 90 (Hsp90) with the novel agent IPI-504 in metastatic GIST following failure of tyrosine kinase inhibitors (TKIs) or other sarcomas: Clinical results from phase I trial. J Clin Oncol. 2008;26 Abstract 10503. [Google Scholar]
- 70.Johnston A, Allaire M. Infinity Halts RING Trial in Advanced Gastrointestinal Stromal Tumors. 2009 Available from: http://investor.ipi.com/releasedetail.cfm?ReleaseID=377328,
- 71.Infinity. [Updated June 10, 2011];A Phase 1 Dose Escalation Study of IPI-493. available from: http://clinicaltrials.gov/ct2/show/NCT00724425?term=IPI-493&rank=1, in.
- 72.Chene P. ATPases as drug targets: learning from their structure. Nat Rev Drug Discov. 2002;1:665–673. doi: 10.1038/nrd894. [DOI] [PubMed] [Google Scholar]
- 73.Chiosis G, Timaul MN, Lucas B, Munster PN, Zheng FF, Sepp-Lorenzino L, Rosen N. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol. 2001;8:289–299. doi: 10.1016/s1074-5521(01)00015-1. [DOI] [PubMed] [Google Scholar]
- 74.Taldone T, Chiosis G. Purine-scaffold Hsp90 inhibitors. Curr Top Med Chem. 2009;9:1436–1446. doi: 10.2174/156802609789895737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang H, Neely L, Lundgren K, Yang YC, Lough R, Timple N, Burrows F. BIIB021, a synthetic Hsp90 inhibitor, has broad application against tumors with acquired multidrug resistance. Int J Cancer. 2010;126:1226–1234. doi: 10.1002/ijc.24825. [DOI] [PubMed] [Google Scholar]
- 76.Lundgren K, Zhang H, Brekken J, Huser N, Powell RE, Timple N, Busch DJ, Neely L, Sensintaffar JL, Yang YC, McKenzie A, Friedman J, Scannevin R, Kamal A, Hong K, Kasibhatla SR, Boehm MF, Burrows FJ. BIIB021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein Hsp90. Mol Cancer Ther. 2009;8:921–929. doi: 10.1158/1535-7163.MCT-08-0758. [DOI] [PubMed] [Google Scholar]
- 77.Kasibhatla S, Hong K, Biamonte MA, Busch DJ, Karjian PL, Sensintaffar JL, Kamal A, Lough RE, Brekken J, Lundgren K, Grecko R, Timony GA, Ran Y, Mansfield R, Fritz LC, Ulm E, Burrows FJ, Boehm MF. Rationally Designed High-Affinity 2-Amino-6-halopurine Heat Shock Protein 90 Inhibitors That Exhibit Potent Antitumor Activity. Journal of Medicinal Chemistry. 2007;50:2767–2778. doi: 10.1021/jm050752+. [DOI] [PubMed] [Google Scholar]
- 78.Elfiky A, Saif MW, Beeram M, O'Brien S, Lammanna N, Castro JE, Woodworth J, Perea R, Storgard C, Von Hoff DD. BIIB021, an oral, synthetic non-ansamycin Hsp90 inhibitor: Phase I experience. J Clin Oncol. 2008;26 abstract 2503. [Google Scholar]
- 79.Modi S, Ismail-Khan R, Munster P, Lucas M, Galluppi GR, Tangri S, Chen Y, Yamashita M, Storgard CM, Moulder SX. Phase 1 Dose-Escalation Study of the Heat Shock Protein 90 Inhibitor BIIB021 with Trastuzumab in HER2+ Metastatic Breast Cancer. CTRC-AACR San Antonio Breast Cancer Symposium.2010. [Google Scholar]
- 80.Biogen-Idec. [Updated June 16, 2011];Hormone Receptor Positive Metastatic Breast Cancer (HR+ mBC) BIIB021 Plus Aromasin Schedule Finding. available from: http://clinicaltrials.gov/ct2/show/NCT01004081?term=BIIB021&rank=2, in,
- 81.Mitchell P. Biogen Idec restructures, sharpens neurology focus. Nature Biotechnology. 2011;29(number 1):7–8. doi: 10.1038/nbt0111-7. [DOI] [PubMed] [Google Scholar]
- 82. www.myrexis.com/pipeline/mpc-3100, in.
- 83.Yu M, Samlowski WE, Baichwal V, Brown B, Evans BA, Woodland D, Mather G, Patnaik A, Tolcher AW, Papadopoulos K. MPC-3100, a fully synthetic, orally bioavailable Hsp90 inhibitor, in cancer patients. J Clin Oncol. 2010;28 abstract e13112. [Google Scholar]
- 84.Myrexis. http://www.thepharmaletter.com/file/107269/myrexis-to-drop-development-of-azixa-4th-qtr-loss-widens-names-new-ceo.html, in.
- 85.Bao R, Lai CJ, Wang DG, Qu H, Yin L, Zifcak B, Tao X, Wang J, Atoyan R, Samson M, Forrester J, Xu GX, DellaRocca S, Borek M, Zhai HX, Cai X, Qian C. Targeting heat shock protein 90 with CUDC-305 overcomes erlotinib resistance in non-small cell lung cancer. Mol Cancer Ther. 2009;8:3296–3306. doi: 10.1158/1535-7163.MCT-09-0538. [DOI] [PubMed] [Google Scholar]
- 86.Debiopharm. http://www.debiopharm.com/our-business/pipeline.html, in.
- 87.van Ingen H, Sanlaville Y. Study of Debio 0932 in Patients With Advanced Solid Tumours or Lymphoma. available from: http://clinicaltrials.gov/ct2/show/NCT01168752?term=debio&rank=3, in, Verified February, 2011.
- 88.Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, Moulick K, Taldone T, Gozman A, Guo Y, Wu N, de Stanchina E, White J, Gross SS, Ma Y, Varticovski L, Melnick A, Chiosis G. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci U S A. 2009;106:8368–8373. doi: 10.1073/pnas.0903392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cerchietti LC, Lopes EC, Yang SN, Hatzi K, Bunting KL, Tsikitas LA, Mallik A, Robles AI, Walling J, Varticovski L, Shaknovich R, Bhalla KN, Chiosis G, Melnick A. A purine scaffold Hsp90 inhibitor destabilizes BCL-6 and has specific antitumor activity in BCL-6-dependent B cell lymphomas. Nat Med. 2009;15:1369–1376. doi: 10.1038/nm.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marubayashi S, Koppikar P, Taldone T, Abdel-Wahab O, West N, Bhagwat N, Caldas-Lopes E, Ross KN, Gonen M, Gozman A, Ahn JH, Rodina A, Ouerfelli O, Yang G, Hedvat C, Bradner JE, Chiosis G, Levine RL. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010;120:3578–3593. doi: 10.1172/JCI42442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kummar S. [Updated June 22, 2011];A Phase I Study of the Hsp90 Inhibitor, PU-H71, in Patients With Refractory Solid Tumors and Low-Grade Non-Hodgkin's Lymphoma. available from: http://bethesdatrials.cancer.gov/clinical-research/search_detail.aspx?ProtocolID=NCI-11-C-0150, in,
- 92.Delmotte P, Delmotte-Plaque J. A new antifungal substance of fungal origin. Nature. 1953;171:344. doi: 10.1038/171344a0. [DOI] [PubMed] [Google Scholar]
- 93.Goldman J, Raju RN, Gordon GA, Vukovic VM, Bradley R, Rosen LS. A phase I dose-escalation study of the Hsp90 inhibitor STA-9090 administered once weekly in patients with solid tumors. J Clin Oncol. 2010;28 abstract 2529. [Google Scholar]
- 94.Cho D, Heath EI, Cleary JM, Kwak EL, Gandhi L, Lawrence DP, Zack C, Teofilovici F, Bradley R, Karol MD, Shapiro G, LoRusso P. A phase I dose-escalation study of the Hsp90 inhibitor ganetespib (STA-9090) administered twice weekly in patients with solid tumors: Updated report. J Clin Oncol. 2011;29 Abstract 3051. [Google Scholar]
- 95.Lancet J, Smith D, Bradley R, Komrokji RS, Teofilovici F, Rizzieri D. A Phase I/II Trial of the Potent Hsp90 Inhibitor STA-9090 Administered Once Weekly In Patients with Advanced Hematologic Malignancies. Blood (ASH Annual Meeting Abstracts) 2010;116 Abstract 3294. [Google Scholar]
- 96.Padmanabhan S, Kelly K, Heaney M, Hodges S, Chanel S, Frattini M, Swords R, Bradley R, Teoflivici F, DeAngelo DJ. A Phase I Study of the Potent Hsp90 Inhibitor STA-9090 Administered Twice Weekly In Subjects with Hematologic Malignancies. Blood (ASH Annual Meeting Abstracts) 2010;116 Abstract 2898. [Google Scholar]
- 97.Wong K, Koczywas M, Goldman JW, Paschold EH, Horn L, Lufkin JM, Blackman RK, Teofilovici F, Shapiro G, Socinski MA. An open-label phase II study of the Hsp90 inhibitor ganetespib (STA-9090) as monotherapy in patients with advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2011;29 Abstract 7500. [Google Scholar]
- 98.S.P. Corp. Study of Ganetespib (STA-9090) + Docetaxel in Advanced Non Small Cell Lung Cancer. available from: http://clinicaltrials.gov/ct2/show/NCT01348126?term=sta9090+and+docetaxel&rank=3, in, Verified September, 2011.
- 99.Demetri G, Heinrich MC, Chmielowski B, Morgan JA, George S, Bradley R, Blackman RK, Teofilovici F, Fletcher JA, Tap WD, von Mehrn M. An open-label phase II study of the Hsp90 inhibitor ganetespib (STA-9090) in patients (pts) with metastatic and/or unresectable GIST. J Clin Oncol. 2011;29 Abstract 10011. [Google Scholar]
- 100.Modi S. [Updated May, 2011];Open-label Study of STA-9090 for Patients With Metastatic Breast Cancer. available from: http://clinicaltrials.gov/ct2/show/NCT01273896?term=STA-9090+and+breast&rank=1, in,
- 101.Cheung KMJ, Matthews TP, James K, Rowlands MG, Boxall KJ, Sharp SY, Maloney A, Roe SM, Prodromou C, Pearl LH, Aherne GW, McDonald E, Workman P. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorganic and Medicinal Chemistry Letters. 2005;15:3338–3343. doi: 10.1016/j.bmcl.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 102.Gaspar N, Sharp SY, Eccles SA, Gowan S, Popov S, Jones C, Pearson A, Vassal G, Workman P. Mechanistic evaluation of the novel HSP90 inhibitor NVP-AUY922 in adult and pediatric glioblastoma. Mol Cancer Ther. 2010;9:1219–1233. doi: 10.1158/1535-7163.MCT-09-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall F, Aherne W, Rowlands M, Hayes A, Martins V, Urban F, Boxall K, Prodromou C, Pearl L, James K, Matthews TP, Cheung KM, Kalusa A, Jones K, McDonald E, Barril X, Brough PA, Cansfield JE, Dymock B, Drysdale MJ, Finch H, Howes R, Hubbard RE, Surgenor A, Webb P, Wood M, Wright L, Workman P. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68:2850–2860. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- 104.Samuel T, Sessa C, Britten C, Milligan KS, Mita MM, Banerji U, Pluard P, Quadt C, Shapiro G. AUY922, a novel HSP90 inhibitor: Final results of a first-in-human study in patients with advanced solid malignancies. J Clin Oncol. 2010;28 Abstract 2528. [Google Scholar]
- 105.Schroder C, Pederson JV, Chua S, Swanton C, Akimov M, Ide S, Fernandez-Ibarra C, Jurasz-Dzik A, DeVries E, Gaykema SB, Banerji U. USe of Biomarkers and imaging to evaluate the treatment effect of AUY922, an Hsp90 inhibitor, in patients with HER2+ or ER+ metastatic breast cancer. J Clin Oncol. 2011;29:e11024. [Google Scholar]
- 106.Novartis. [Updated August 10, 2011];A Phase I-Ib/II Study to Determine the Maximum Tolerated Dose (MTD) of AUY922 Alone and in Combination With Bortezomib, With or Without Dexamethasone, in Patients With Relapsed or Refractory Multiple Myeloma. available from: http://clinicaltrials.gov/ct2/show/NCT00708292?term=AUY922&rank=2, in,
- 107.Novartis. A Study of HSP990 Administered by Mouth in Adult Patients With Advanced Solid Tumors. available from: http://clinicaltrials.gov/ct2/show/NCT00879905?term=HSP990&rank=1, in, Verified March, 2011.
- 108.Novartis. Dose Escalation of HSP990 in Japan/Korea. available from: http://clinicaltrials.gov/ct2/show/NCT01064089?term=HSP990&rank=2, in, Verified Januray, 2011.
- 109.Nakashima T, Ishii T, Tagaya H, Seike T, Nakagawa H, Kanda Y, Akinaga S, Soga S, Shiotsu Y. New molecular and biological mechanism of antitumor activities of KW-2478, a novel nonansamycin heat shock protein 90 inhibitor, in multiple myeloma cells. Clin Cancer Res. 2010;16:2792–2802. doi: 10.1158/1078-0432.CCR-09-3112. [DOI] [PubMed] [Google Scholar]
- 110.Cavenagh J, Yong K, Byrne J, Cavet J, Johnson P, Morgan G, Williams C, Akinaga S, Francis G, Kilborn J. The Safety, Pharmacokinetics and Pharmacodynamics of KW-2478, a Novel Hsp90 Antagonist, in Patients with B-Cell Malignancies: A First-in-Man, Phase I, Multicentre, Open-Label. Dose Escalation Study 50th ASH Annual Meeting and Exposition; 2008. pp. 958–959. [Google Scholar]
- 111.I. Kyowa Hakko Kirin Pharma. A Study of KW-2478 in Combination With Bortezomib in Subjects With Relapsed and/or Refractory Multiple Myeloma. available from: http://clinicaltrials.gov/ct2/show/NCT01063907, in, Verified July, 2011.
- 112.Woodhead AJ, Angove H, Carr MG, Chessari G, Congreve M, Coyle JE, Cosme J, Graham B, Day PJ, Downham R, Fazal L, Feltell R, Figueroa E, Frederickson M, Lewis J, McMenamin R, Murray CW, O'Brien MA, Parra L, Patel S, Phillips T, Rees DC, Rich S, Smith DM, Trewartha G, Vinkovic M, Williams B, Woolford AJA. Discovery of (2,4-Dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1- ylmethyl)-1,3-dihydroisoindol-2-yl]methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design. Journal of Medicinal Chemistry. 2010;53:5956–5969. doi: 10.1021/jm100060b. [DOI] [PubMed] [Google Scholar]
- 113.Murray CW, Carr MG, Callaghan O, Chessari G, Congreve M, Cowan S, Coyle JE, Downham R, Figueroa E, Frederickson M, Graham B, McMenamin R, O'Brien MA, Patel S, Phillips TR, Williams G, Woodhead AJ, Woolford AJA. Fragment-based drug discovery applied to Hsp90. Discovery of two lead series with high ligand efficiency. Journal of Medicinal Chemistry. 2010;53:5942–5955. doi: 10.1021/jm100059d. [DOI] [PubMed] [Google Scholar]
- 114.Wolanski A. [Updated June 10, 2011];Study to Assess the Safety of Escalating Doses of AT13387 in Patients With Metastatic Solid Tumors. available from: http://clinicaltrials.gov/ct2/show/NCT00878423?term=AT-13387&rank=4, in,
- 115.NCI. AT13387 in Adults With Refractory Solid Tumors. available from: http://clinicaltrials.gov/ct2/show/NCT01246102?term=AT-13387&rank=3, in, Verified November 20, 2010.
- 116.Kummar S. AT13387 in Treating Patients With Refractory Solid Tumors. available from: http://clinicaltrials.gov/ct2/show/NCT01245218?term=AT-13387&rank=2, in, Verified November, 2010.
- 117.Demetri G. A Study to Investigate the Safety and Efficacy of AT13387, Alone or in Combination With Imatinib, in Patients With GIST. available from: http://clinicaltrials.gov/ct2/show/NCT01294202?term=AT-13387&rank=1, in, Verified June, 2011.
- 118.Fadden P, Huang KH, Veal JM, Steed PM, Barabasz AF, Foley B, Hu M, Partridge JM, Rice J, Scott A, Dubois LG, Freed TA, Silinski MAR, Barta TE, Hughes PF, Ommen A, Ma W, Smith ED, Spangenberg AW, Eaves J, Hanson GJ, Hinkley L, Jenks M, Lewis M, Otto J, Pronk GJ, Verleysen K, Haystead TA, Hall SE. Application of chemoproteomics to drug discovery: Identification of a clinical candidate targeting Hsp90. Chemistry and Biology. 2010;17:686–694. doi: 10.1016/j.chembiol.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 119.Bryson J, Infante JR, Ramanathan RK, Jones SF, Von Hoff DD, Burris HA. A Phase 1 dose-escalation study of the safety and pharmacokinetics (PK) of the oral Hsp90 inhibitor SNX-5422. J Clin Oncol. 2008;26 abstract 14613. [Google Scholar]
- 120.Pfizer. Pfizer Annual report. available from: http://www.pfizer.com/files/annualreport/2008/financial/financial_highlights2008.pdf, in, 2008.
- 121.Rajan A, Kelly RJ, Trepel JB, Kim YS, Alarcon SV, Kummar S, Gutierrez M, Crandon S, Zein WM, Jain L, Mannargudi B, Figg WD, Houk BE, Shnaidman M, Brega N, Giaccone G. A Phase 1 study of PF-04929113 (SNX-5422), an orally bioavailable heat shock protein 90 inhibitor in patients with refractory solid tumor malignancies and lymphomas. Clinical Cancer Research. 2011 doi: 10.1158/1078-0432.CCR-11-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.DaiichiSankyo-Inc. A Study of DS-2248, in Subjects With Advanced Solid Tumors. available from: http://clinicaltrials.gov/ct2/show/NCT01288430?term=DS-2248&rank=1, in, Verified April, 2011.
- 123.Exelixis. [Updated March 31, 2011];Safety Study of Pharmacokinetics of XL888 in Adults With Solid Tumors. available from: http://clinicaltrials.gov/ct2/show/NCT00796484?term=XL-888&rank=1, in, Verified April, 2011.
- 124.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 125.Vilenchik M, Solit D, Basso A, Huezo H, Lucas B, He H, Rosen N, Spampinato C, Modrich P, Chiosis G. Targeting wide-range oncogenic transformation via PU24FCl, a specific inhibitor of tumor Hsp90. Chem Biol. 2004;11:787–797. doi: 10.1016/j.chembiol.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 126.Moulick K, Ahn J, Zong H, Rodina A, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7:818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang H, Chung D, Yang YC, Neely L, Tsurumoto S, Fan J, Zhang L, Biamonte M, Brekken J, Lundgren K, Burrows F. Identification of new biomarkers for clinical trials of Hsp90 inhibitors. Mol Cancer Ther. 2006;5:1256–1264. doi: 10.1158/1535-7163.MCT-05-0537. [DOI] [PubMed] [Google Scholar]
- 128.Dakappagari N, Neely L, Tangri S, Lundgren K, Hipolito L, Estrellado A, Burrows F, Zhang H. An investigation into the potential use of serum Hsp70 as a novel tumour biomarker for Hsp90 inhibitors. Biomarkers. 2010;15:31–38. doi: 10.3109/13547500903261347. [DOI] [PubMed] [Google Scholar]
- 129.Eiseman JL, Guo J, Ramanathan RK, Belani CP, Solit DB, Scher HI, Ivy SP, Zuhowski EG, Egorin MJ. Evaluation of plasma insulin-like growth factor binding protein 2 and Her-2 extracellular domainas biomarkers for 17-allylamino-17-demethoxygeldanamycin treatment of adult patients with advanced solid tumors. Clin Cancer Res. 2007;13:2121–2127. doi: 10.1158/1078-0432.CCR-06-2286. [DOI] [PubMed] [Google Scholar]
- 130.Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, Teitcher J, Wolchok JD, Germino FJ, Krown SE, Coit D, Rosen N, Chapman PB. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res. 2008;14:8302–8307. doi: 10.1158/1078-0432.CCR-08-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Holland JP, Caldas-Lopes E, Divilov V, Longo VA, Taldone T, Zatorska D, Chiosis G, Lewis JS. Measuring the pharmacodynamic effects of a novel Hsp90 inhibitor on HER2/neu expression in mice using Zr-DFO-trastuzumab. PLoS One. 2010;5:e8859. doi: 10.1371/journal.pone.0008859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nagengast WB, de Korte MA, Oude Munnink TH, Timmer-Bosscha H, den Dunnen WF, Hollema H, de Jong JR, Jensen MR, Quadt C, Garcia-Echeverria C, van Dongen GA, Lub-de Hooge MN, Schroder CP, de Vries EG. 89Zr-bevacizumab PET of early antiangiogenic tumor response to treatment with HSP90 inhibitor NVP-AUY922. J Nucl Med. 2010;51:761–767. doi: 10.2967/jnumed.109.071043. [DOI] [PubMed] [Google Scholar]
- 133.Akhurst T, Morris P, Modi S, Carraquillo J, Solit D, Hudis C, Larson S, Smith-Jones P. Positron emission tomography (PET) with radiolabeled F(ab')2-trastuzumab fragments in patients (pts) with HER2-positive metastatic breast cancer (MBC): Initial feasibility results. Breast Cancer Symposium. 2008 [Google Scholar]
- 134.Schroder C, Pederson JV, Chua S, Swanton C, Akimov M, Ide S, Fernandez-Ibarra C, Jurasz-Dzik A, DeVries E, Gaykema SB, Banerji U. Use of biomarkers and imaging to evaluate the treatment effect of AUY922, an HSP90 inhibitor, in patients with HER2+ or ER+ metastatic breast cancer. J Clin Oncol. 2011;29 [Google Scholar]
- 135.Schroder C, DeVries E. [Updated March 12,2010];Imaging HSP90 Inhibitor AUY922 on VEGF-89ZR-bevacizumab Positron Emission Tomography (PET) available from: http://www.clinicaltrials.gov/ct2/show/NCT01081613?term=AUY922&rank=14, in,
- 136.Smith-Jones PM, Solit D, Afroze F, Rosen N, Larson SM. Early tumor response to Hsp90 therapy using HER2 PET: comparison with 18F-FDG PET. J Nucl Med. 2006;47:793–796. [PMC free article] [PubMed] [Google Scholar]
- 137.Demetri G, Sledge G, Morgan JA, Wagner A, Quigley MT, Polson K, Pokela J, van den Abbeele A, Adams J, Grayzel D. Inhibition of the Heat Shock Protein 90 (Hsp90) chaperone with the novel agent IPI-504 to overcome resistance to tyrosine kinase inhibitors (TKIs) in metastatic GIST: Updated results of a phase I trial. J Clin Oncol, ASCO Annual Meetings Proceedings Part I, 25, N. 2007;18S:10024. [Google Scholar]
- 138.Dunphy M. PET Imaging of Cancer Patients Using 124I-PUH71: A Pilot Study. available from: http://clinicaltrials.gov/ct2/show/NCT01269593?term=PET+imaging+of+cancer+patients&rank=1, in, Verified June, 2011.
- 139.Gerecitano J. [Updated July 12, 2011];The First-in-human Phase I Trial of PU-H71 in Patients With Advanced Malignancies. available from: http://clinicaltrials.gov/ct2/show/NCT01393509?term=PU-H71&rank=1, in,
- 140.Chandarlapaty S, Sawai A, Ye Q, Scott A, Silinski M, Huang K, Fadden P, Partdrige J, Hall S, Steed P, Norton L, Rosen N, Solit DB. SNX2112, a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER kinase-dependent cancers. Clin Cancer Res. 2008;14:240–248. doi: 10.1158/1078-0432.CCR-07-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]