

Abstract
DNA breaks can be repaired with high-fidelity by homologous recombination. A ubiquitous protein that is essential for this DNA template-directed repair is RecA1. After resection of broken DNA to produce single-stranded DNA (ssDNA), RecA assembles on this ssDNA into a filament with the unique capacity to search and find DNA sequences in double-stranded DNA (dsDNA) that are homologous to the ssDNA. This homology search is vital to recombinational DNA repair, and results in homologous pairing and exchange of DNA strands. Homologous pairing involves DNA sequence-specific target location by the RecA-ssDNA complex. Despite decades of study, the mechanism of this enigmatic search process remains unknown. RecA is a DNA-dependent ATPase, but ATP hydrolysis is not required for DNA pairing and strand exchange2,3, eliminating active search processes. Using dual optical trapping to manipulate DNA, and single-molecule fluorescence microscopy to image DNA pairing, we demonstrate that both the three-dimensional conformational state of the dsDNA target and the length of the homologous RecA-ssDNA filament play important roles in the homology search. We discovered that as the end-to-end distance of the target dsDNA molecule is increased, constraining its available 3-dimensional conformations, the rate of homologous pairing decreases. Conversely, when the length of the ssDNA in the nucleoprotein filament is increased, homology is found faster. We propose a model for the DNA homology search process termed “intersegmental contact sampling”, wherein the intrinsic multivalent nature of the RecA nucleoprotein filament is employed to search DNA sequence space within 3-dimensional domains of DNA, exploiting multiple weak contacts to rapidly search for homology. Our findings highlight the importance of the 3-dimensional conformational dynamics of DNA, reveal a previously unknown facet of the homology search, and provide insight into the mechanism of DNA target location by this member of a universal family of proteins.
The mechanism by which the RecA-family of DNA strand exchange proteins (which include T4 UvsX, archaeal RadA, and eukaryotic Rad51) locate DNA sequence identity is unknown. Ensemble studies have constrained possible mechanisms by establishing that neither ATP hydrolysis is needed3,4 nor 1-dimensional sliding is operative5. Consequently, the manner by which the RecA nucleoprotein filament promotes the efficient, rapid, and accurate search for homology has remained undefined for decades6. Single-molecule methods have the potential to provide new insight into this long-standing question. In fact, magnetic tweezer experiments demonstrated that the endpoint of homologous pairing can be detected as a change in the length of a single dsDNA target molecule7,8. However, the mechanism by which homology was found and DNA pairing occurred was not revealed. Therefore, we sought to directly observe the manner by which RecA nucleoprotein filaments locate their homologous target in dsDNA.
Initially we attempted to directly observe fluorescent RecA nucleoprotein filaments interacting with bacteriophage λ dsDNA in real-time by using total internal reflected fluorescence microscopy (TIRFM)9. Fully homologous fluorescent ssDNA that was complementary to three different loci of λ DNA (Fig. 1a) was generated by incorporation of 5-(3-aminoallyl) dUTP into ssDNA comprising a region of λ DNA using PCR, followed with covalent attachment of ATTO565 (see Methods section). RecA nucleoprotein filaments were assembled on these fluorescent ssDNA substrates in ensemble reactions containing ssDNA-binding protein (SSB) and the non-hydrolysable ATP analog, ATPγS (5’-O-3’- thiotriphosphate)4. ATPγS was used to maintain the filament in its active form, eliminate filament disassembly, and prevent dissociation of DNA pairing products7,10-12. Using biochemical assays, we confirmed that the fluorescent ssDNA generated by this procedure was functional for RecA-mediated DNA pairing (Supplementary Fig. 1). The λ dsDNA, biotinylated at each end, was attached under flow to the interior surface of a single-channel microfluidic device (flowcell) (Fig. 1b). Due to sequential attachment of each end to the streptavidin-coated surface, most DNA molecules were extended to nearly (~80%) B-form length, and extension could be maintained in the absence of flow (Figure 1b, i and ii).
Figure 1. DNA pairing by RecA, imaged using single-molecule TIRFM, suggests that the three-dimensional conformation of target dsDNA is important in the homology search.
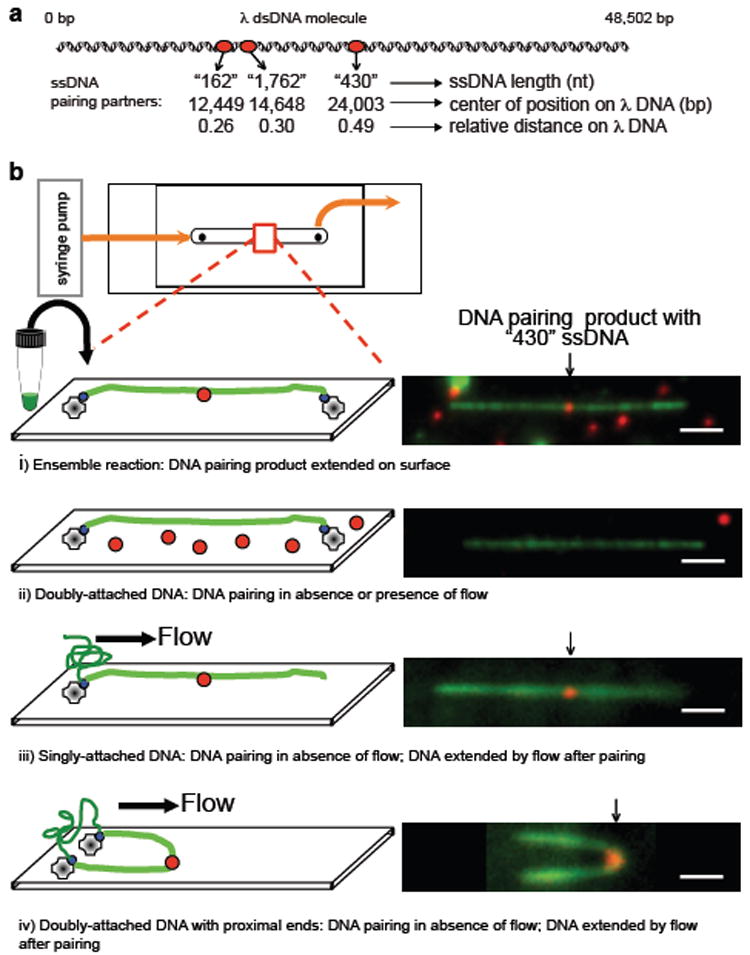
a, DNA substrates. b, DNA pairing between λ DNA (green) and RecA filament assembled on 430 nt ssDNA (red): i) ensemble reaction examined by TIRFM; ii-iv) in situ reactions, dsDNA attached prior to pairing: ii) doubly-attached extended DNA, iii) singly-attached DNA, and iv) doubly-attached DNA with ends in proximity. Homologously paired products were observed in iii and iv when DNA was relaxed by stopping flow, and then flow-extended for visualization. White bar = 2.4 μm.
To confirm DNA pairing at the homologous λ DNA target site, reactions were conducted under ensemble conditions, and products extended on the surface of a flowcell for analysis by single-molecule, two-color TIRFM; dsDNA was imaged by YOYO-1 binding (green) and ssDNA via ATTO565 (red). DNA pairing products were observed; the sites of interaction coincided with the region of homology within the λ DNA molecule (Fig. 1b, i). For the 430 nt ssDNA, all bound fluorescent ssDNA-RecA filaments were at the homologous locus (observed fractional distance 0.51±0.02; n= 21; Supplementary Fig. 2).
Next, we attempted to detect homologous pairing in real-time using single-molecule TIRFM. Preformed RecA nucleoprotein filaments were introduced into a flowcell to which λ DNA molecules were tethered; buffer flow was stopped; and the reaction monitored in real-time (Fig. 1b, ii). Although the dsDNA was readily visible, we failed to observe any interaction between the fluorescent nucleoprotein filaments and extended λ DNA, even for reaction periods longer than 1 hour. However, we noticed that in addition to the desired doubly-tethered extended λ DNA molecules, some DNA molecules were attached only via one end (Fig. 1b, iii). When flow was stopped to score pairing with the doubly-tethered λ DNA molecules, these singly-tethered molecules relaxed to a randomly coiled state. Unexpectedly, when these unconstrained DNA molecules were subsequently re-extended by buffer flow, 80% (n=20) revealed a stable pairing product (Fig. 1b, iii). This finding suggested that either a free DNA end or random coiled DNA was needed for pairing. In the same field of view, there were also λ DNA molecules that had both ends attached, but at a relatively close end-to-end distance (Fig, 1b, iv). When the flow was stopped, we observed that these molecules also participated in homologous pairing during the time that flow was off, demonstrating that a free DNA end was not required. These unanticipated results revealed that DNA pairing did not occur on DNA that was extended to near its entropic elastic limit, and suggested that the DNA homology search required the 3-dimensional states that are accessible in randomly coiled DNA. Collectively, they suggested that a coiled conformation of the target dsDNA is crucial.
To address this possibility, we developed an alternative single-molecule imaging strategy that permitted reproducible measurement of the effects of dsDNA conformational structure, unperturbed by flow, on the DNA homology search process. This method utilizes a specialized flowcell (Fig. 2a), two optical laser traps operated in position-clamp mode, epifluorescent detection, fluorescent ssDNA-RecA filaments and a λ DNA-dumbbell (a single λ DNA molecule with a 1 μm polystyrene bead attached at each end (see Methods section13)). The DNA pairing assay was performed in situ using the dsDNA-dumbbell target, and the dual optical trap configuration was utilized to reliably vary the end-to-end distance of the dsDNA. The flowcell has 4 channels and a flow-free reservoir. Movement of DNA-dumbbells between channels of the flowcell was accomplished via stage translation; manipulation of optical traps relative to one another was accomplished using a steering mirror controlling one of the traps. Each experiment (Fig. 2b; Supplementary Movie 1) consisted of the following steps: 1) In channel 1, a streptavidin-coated bead was trapped in each of the two optical traps. 2) The beads were moved to channel 2 to capture a λ dsDNA molecule (biotinylated on both ends, and stained with YOYO-1) on one bead. 3) The beads were moved into channel 3, and by independent steering of a trap, the distal end of the DNA was attached to the second bead. 4) The DNA-dumbbell was moved to the dye-free channel for de-staining, and the end-to-end distance was fixed. 5) The DNA-dumbbell was moved to the flow-free reservoir containing the fluorescent ssDNA-RecA filaments. 6) After a defined incubation time, the DNA-dumbbell was moved back to channel 4, which is free of nucleoprotein filaments, extended to its contour length (~16 μm), and examined for DNA pairing products.
Figure 2. Visualization of RecA-promoted DNA pairing with an individual optically-trapped DNA-dumbbell, imaged by epifluorescence.

a, Four channel flowcell with a flow-free reservoir. b, DNA-dumbbell assembly and RecA-pairing reaction: 1) trap two beads (yellow); 2) capture λ DNA molecule (green) on one bead; 3) capture free DNA end with second bead using steerable optical trap; 4) set center-to-center bead distance, and remove YOYO-1; 5) incubate DNA-dumbbell in reservoir with RecA nucleoprotein filaments (red); and 6) extend DNA to visualize products. c, Images of pairing products with 430 and 1,762 nt nucleoprotein filaments.
Shown in Fig. 2c are representative products of reactions where the DNA-dumbbells were initially held at a center-to-center bead distance of 2 μm, and incubated for 2 minutes in the reservoir that contained RecA nucleoprotein filaments. For the two homologous ssDNA nucleoprotein filaments shown (430 nt and 1,762 nt), the pairing is clearly at the homologous locus. For a 2 minute incubation with dsDNA at a bead-to-bead distance of 2 μm and the 430 nt substrate, 90% of the dsDNA molecules (n=29) contained a nucleoprotein filament stably bound to the expected region of homology (Fig. 3a). To determine the effect of end-to-end distance (i.e., three-dimensional conformation) on the RecA-mediated DNA pairing reaction, the reactions were performed at increasing bead separations (Fig. 3a). As the bead distance was increased from 2 to 8 μm, the efficiency of DNA pairing period decreased to near zero, extrapolating to zero at ~9 μm; for comparison, in the TIRFM experiments where no DNA pairing was detected in situ, the DNA end-to-end distance was ~13 μm.
Figure 3. DNA three-dimensional conformation and nucleoprotein filament length contribute to homology search.
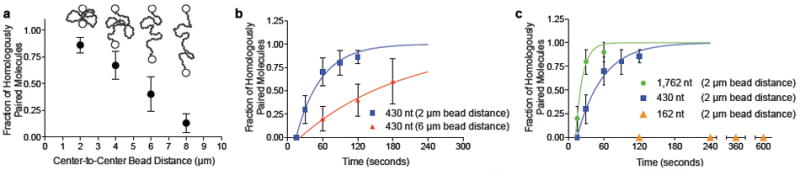
a, Effect of DNA end-to-end distance; 430 nt substrate (2 min); error bars: standard error of mean (SEM) from multiple experiments, n =10 to 29). b, Time course; 430 nt substrate: 2 μm (squares) and 6 μm (triangles) separation; respective pairing rates, 0.023 (±0.002) s-1 (n≥10) and 0.0056 (±0.0006) s-1 (n≥5). c, Effect of ssDNA length; 162 nt (triangles; n=5, 6, 4, and 2 at times indicated), 430 nt (squares; same data as Figure 3b; n≥10), and 1,762 nt (circles; n≥10); error bars, SEM; 2 μm separation; respective rates: zero, 0.023 (±0.002) s-1, and 0.086 (±0.026) s-1.
We compared the time-course of homologous pairing for fixed center-to-center bead distances of 2 μm and 6 μm (Fig. 3b), to determine the effect of decreasing DNA conformational states on the rate of the reaction. For the 2 μm separation, the rate of DNA pairing increased with half-time of ~30 seconds, and approached a yield of 100%. When the separation was increased to 6 μm, the rate slowed 4-fold to a half-time of ~125 seconds, but nonetheless approached 100% (Fig. 3b). To establish the kinetic reaction order, we conducted single-molecule DNA pairing assays as a function of RecA nucleoprotein filament concentration (Supplementary Fig. S3). The reaction rate was independent of nucleoprotein filament concentration, showing that DNA pairing under these conditions is not diffusion-limited, but rather, limited by a rate-determining unimolecular step, as in the ensemble studies14, but that was dependent on dsDNA conformation and not the pairing step itself.
To understand the nature of the complex that limits the rate of DNA pairing, we varied the length of RecA nucleoprotein filaments. Shown in Fig. 3c is a comparison of the time-courses for 162, 430, and 1,762 nt nucleoprotein filaments. Increasing the ssDNA length ~4-fold, from 430 to 1762 nt, increased the observed rate of pairing ~3.8-fold. However, when the length of the ssDNA was decreased to 162 nt, we did not observe any stably bound homologously paired products after incubations for 10 minutes at the closest bead-to-bead distance possible (2 μm), even though this substrate was active in ensemble DNA pairing reactions (Supplementary Fig. 2). We conclude that that the length of the RecA nucleoprotein filament is crucial factor in rate-limiting step of homologous pairing.
In addition to the anticipated stable, homologously paired end products, short-lived non-homologous interactions were observed (Fig. 4a). These events, which occurred outside of the homologous regions, were relatively unstable and dissociated during the movement of the molecule from the reservoir to the observation channel, during the separation of beads, or after the λ DNA molecule was extended (Supplementary Movie 2). At most, these heterologous events lasted for a few tens of seconds and never persisted on the minute timescale. When the molecules from the 2 μm dataset were analyzed, 22% of the reactions with the 430 nt ssDNA and 40% of reactions with the 1,762 nt ssDNA had these unstable heterologously paired intermediates (Fig. 4b); for the 162 nt ssDNA, only 1 heterologously bound filament was seen out of 28 molecules.
Figure 4. RecA nucleoprotein filaments exhibit transient non-homologous interactions and loop-release events.
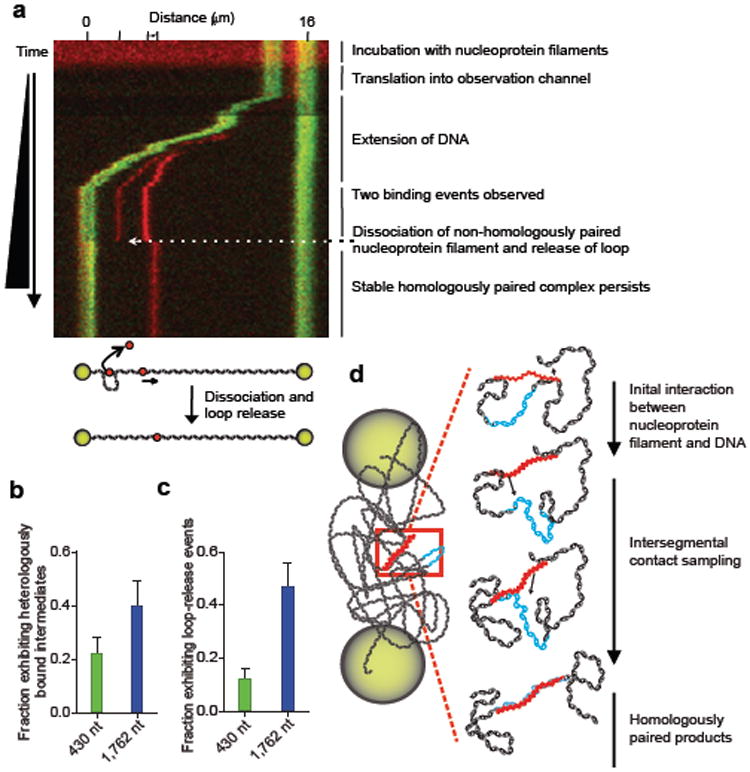
a, Kymograph of DNA-dumbbell during bead separation (Figure 2b, step 6). Distance scale (top) and tick marks show positions of beads (green) and nucleoprotein filaments (red); illustration depicts dissociation of heterologously bound filament. Fraction of dsDNA-dumbbells with, b, non-homologously bound intermediates and, c, loop release events; 430 nt (blue) and 1,762 nt (green) filaments; n = 50 and 30, respectively; error bars indicate SEM. d, Model for RecA homology search by intersegmental contact sampling; for simplicity, only two simultaneous points of interaction are depicted.
Some intermediates of the pairing process had a second filament bound non-specifically to spatially-separated regions of the λ DNA molecule. For such a heterologously-bound nucleoprotein filament, when the relaxed DNA molecule was moved into the observation channel and the beads were separated for observation, the existence of a loop could be inferred from sudden recoil of the homologously paired spot. As the beads were separated, the weaker of the two heterologous interactions was released, and there was a simultaneous movement (“jump”) of the fluorescence at the homologous pairing locus (Fig. 4a; Supplementary Movie 3) resulting from the release of DNA that was constrained in the loop. Approximately ~12% (n = 50) of the DNA-dumbbells displayed loop release events for the 430 nt nucleoprotein filament and, consistent with expectations, when the length of the nucleoprotein filament was increased to 1,762 nt, the number of molecules with transient loop structures increased to 47% (n = 30) (Fig. 4c).
Our results clearly establish that both the 3-dimensional conformation of dsDNA and the length of the nucleoprotein filament are important determinants of the rate for DNA homologous pairing. These findings lead us to propose a model termed “intersegmental contact sampling” to describe the search for homology by a RecA nucleoprotein filament (Fig. 4d). One of the key features of the model is that the RecA nucleoprotein filament has a polyvalent interaction surface capable of binding simultaneously and non-specifically, but weakly, with non-contiguous segments of dsDNA. The second related feature of this model is that 3-dimensional conformational entropy of the dsDNA greatly enhances the probability that DNA sequence homology will be found through iterated homology sampling, via multiple weak contacts, by this polyvalent filament. This model is compatible both with our key experimental findings, which we expect would apply to the search in the presence of ATP as well, and with the involvement of heterologously-bound intermediates that have been inferred from biochemical studies15,16. Our data show that dsDNA extended to near contour length fails to produce homologously paired products. This observation provides an explanation for the observation that the formation of stable DNA pairing products in single-molecule studies utilizing magnetic tweezers required negative plectonemic supercoils in the DNA target7,8. In contrast, when a ssDNA-RecA filament was extended to near its contour length, homologous pairing with fully homologous coiled dsDNA occurred 7, which is compatible with our finding that the coiled structure of dsDNA is essential to the homology search. Here we established that as the end-to-end distance of the dsDNA was decreased, allowing it to assume a more random coil-like 3-dimensional conformation, the rate of DNA pairing increased because the local DNA concentration increases, and the likelihood that DNA segments will be in close proximity also greatly increases. The increased local DNA concentration results in a greater statistical probability that a single nucleoprotein filament can simultaneously interact with and sample multiple regions of the same DNA molecule. This, in turn, is manifest as a kinetically more efficient homology sampling process. In further support of the intersegmental contact sampling model, when the length of the ssDNA in the nucleoprotein filament is increased the observed rate of pairing is increased, as well as the number of nucleoprotein filaments with multiple, transient, heterologous intersegmental interactions. This shows that longer nucleoprotein filaments can simultaneously and independently sample more segments of the target dsDNA than shorter nucleoprotein filaments. Kinetically, our findings are consistent with the following two-step scheme:
where Khet is the equilibrium constant for the binding of a RecA nucleoprotein filament (NPF) to heterologous dsDNA (the kinetic steps comprising Khet are rapid relative to ks) and ks is the rate-limiting unimolecular rate constant for intersegmental homology searching step within the dsDNA molecule or domain. In general, this kinetic formalism predicts a hyperbolic dependence of homologous pairing on the component concentrations unless the equilibrium constant for formation of the heterologous complex is large; when this is case, the observed rate is defined by the first-order rate constant, ks. Given that the rate of target location is independent of nucleoprotein filament concentration, this implies that the heterologously-bound complex is saturated at a filament concentration of 100 pM (Supplementary Fig. 3), placing a limit on the equilibrium dissociation constant of <10 pM (i.e., Khet > 1011 M-1). In the context of this kinetic model, values for ks are defined by the experiments in Fig. 3b & c, which show that the rate of the intersegmental homology search decreases 4-fold when the DNA end-to-end distance increases from 1 μm to 5 μm, and increases ~4-fold when the ssDNA length increases ~4-fold. The latter is suggests that the intradomainal search is enhanced proportionately by the increase in either heterologous contacts or the reach of the longer ssDNA. In many regards, the homology search by RecA has parallels to target location by sequence-specific DNA binding proteins, with the notable exception that the specificity of the RecA filament is determined by the sequence of the associated ssDNA. Seminal work on the DNA target selection by transcriptional regulatory proteins identified sliding, hopping, and intersegmental transfer as potentially facilitating mechanisms17,18. Here we have established intersegmental transfer as the operative pathway used by RecA to find DNA sequence homology; this behavior is distinct from the sliding and hopping used to enhance the rate of target location by most regulatory proteins, which are typically univalent or bivalent with regard to site binding18. Our approach now provides a framework for future studies on the previously mysterious homology search by recombination proteins. It is applicable to studies of more complex systems such as eukaryotic Rad51, where it can provide insight into the function of the many accessory proteins that enhance DNA pairing9. Finally, more broadly, the imaging strategy and flow-free cell design can easily be adapted to visualize target location and mechanism of processes as diverse as DNA replication and repair, RNA interference, transcription, and protein translation, where the 3-dimenensional conformations of nucleic acids are undoubtedly important.
Methods Summary
RecA and SSB were purified as described19,20. Fluorescent ssDNA was prepared as detailed in the Methods section. Nucleoprotein filaments were formed as described4 in SM buffer (25 mM TrisOAc (pH 7.5), 1 mM DTT, and 4 mM Mg(OAc)2), SSB (at a ratio of 1 SSB monomer:11 nt), 2 nM molecules fluorescent ssDNA, and 1 mM ATPγS were incubated for 10 min at 37°C; RecA was added at 1 monomer per 1.7 nt, and incubated 1 hour. Nucleoprotein filaments were diluted to 0.2 nM prior to use.
For DNA pairing using TIRFM, biotinylated λ DNA (1 pM, molecules) in SM2 (SM and 50 mM NaCl) was bound to the flowcell and then washed to remove free DNA, and to attach the second DNA end. Reactions were started by addition of 0.2 nM nucleoprotein filaments. For ensemble experiments visualized by TIRFM, nucleoprotein filaments and λ DNA were incubated either1 hour (162 nt substrate) or 30 minutes (430 nt substrate) at 37°C.
Visualization of RecA-mediated pairing with individual DNA-dumbbells was performed at 37°C. The flowcell was treated for 1 h with BSA (1 mg/ml) in SM3 (50 mM TrisOAc (pH 8.2), 50 mM DTT, 1 mM Mg(OAc)2, and 15% sucrose). Biotinylated λ DNA and buffers were pumped into the flowcell at a linear flow rate of ~100 μm s-1. Channels contained (see Figure 2): 1) SM3, 18 fM streptavidin-coated polystyrene beads (1 μm, Bangs Laboratories), and 5 nM YOYO-1 (Invitrogen); 2) SM3, 100 nM YOYO-1, and 10 pM (molecules) biotinylated λ DNA; 3) SM3; 4) SM and 15% sucrose. The reaction reservoir contained 0.2 nM nucleoprotein filaments in SM with 15% sucrose and 0.5 mM ATPγS.
Methods
Microscope
An Eclipse TE2000-U, upright microscope with a total internal reflected fluorescence (TIRF) attachment (Nikon), using a CFI Plan Apo TIRF 100x, 1.45 N.A., oil-immersed objective was the basis of the instrument developed. Infrared trapping capability, operated in position-clamp mode, was achieved essentially as previously described21 with the addition of a polarizer (Newport) to split the beam and generate two traps and a steering mirror (Newport) to control the x-y position of one of the beams. Laser excitation of the sample in TIRF mode was achieved using a Cyan 488 nm laser (Picarro) or 561 nm laser (Cobolt). Epifluorescence illumination was achieved with an X-Cite 120W mercury vapor lamp (Lumen Dynamics). The fluorescence emission was directed through a polychroic mirror (515/30 nm and 600/40 nm, Chroma). Light was guided into a Dual-View apparatus (Optical Insights) where the green and red components were spatially separated (dichroic 565dxcr, emission HQ515/30 nm and HQ600/40 nm Chroma). Movies were captured on a DU-897E iXon CCD camera (Andor, 100 ms exposure), and processed using IQ imaging software (Andor).
Biotinylated λ Duplex DNA
Multiple biotin moieties were incorporated into both ends of Bacteriophage λ DNA (NEB) by an end-filling reaction. A 30 μl reaction contained 1X NEB buffer #2, 33μM each of dATP, dTTP, dCTP and biotin-11-dGTP (Perkin Elmer), 5 μg λ DNA, and 5 units of Klenow exo- (NEB). The reaction was incubated for 15 min at 25°C then terminated by the addition EDTA to a final concentration of 10 mM and heat inactivation of Klenow at 75°C for 20 min. The reaction was then diluted to a 100 μl final volume with Nanopure water (Millipore) and passed through an S-400 spin column (GE Healthcare) equilibrated with TE buffer (10 mM Tris HCl (pH 7.5) and 1 mM EDTA).
Fluorescent ssDNA Substrates
Primers sequences utilized to generate dsDNA products by PCR that were used to make the fluorescent ssDNA substrates:
87 bp product for D-loop assay with pUC19 supercoiled DNA: forward primer 5′-biotin-CGACGGCCAGTGAATTCCCCGA-3′, reverse primer 5′-TTACGCCAAGCTTACTCGGGAAACAT-3′;
162 bp product (identical to λ DNA between base pairs 12,368-12,529): forward primer 5′-biotin-TAACGTCATGTCAGAGCAGAAAAAG-3′, reverse primer 5′-GCAATACCATCAAAGGTCTGCGTG-3′;
430 bp product (identical to λ DNA between base pairs 23,788-24,217): forward primer 5′-biotin-ACTGTTCTTGCGGTTTGGAGG-3′, reverse primer 5′-CTATCGGAAGTTCACCAGCCAG-3′;
1,762 bp product (identical to λ DNA between base pairs 13,767-15,528): forward primer 5′-biotin-GGATGCGGTGAACTTCGTCAAC-3′, reverse primer 5′-CCCCTTACTGCTTCCTTTACCC-3′.
PCR reactions contained 1X ThermoPol buffer (NEB), 0.2 mM dATP, 0.2 mM dCTP, 0.2 mM dGTP, 0.1 mM dTTP, 0.2 mM 5-(3-aminoallyl) dUTP (Fermentas), 0.25 ng/μl lambda DNA (NEB) (pUC19 for 87 nt substrate), 0.5 μM each primer and 0.05 U/μl Vent exo- polymerase (NEB). The thermocycler (iCycler, Bio-Rad) program involved initial denaturation at 95°C 2 min, 30 cycles of a denaturation phase at 95°C 30 sec, an annealing phase at 60.6/63°C/62.2°C/59.4°C 30 sec for 87, 162, 430 or 1762 nt products, respectively, and an extension phase at 72°C 0.25/0.25/1/5 min for 87, 162, 430 or 1762 nt products, respectively. The final PCR step was extension at 72°C for 5 min. The reactions were then processed with a QIAquick PCR purification kit (Qiagen). Following purification, the DNA was ethanol precipitated at -20°C. To fluorescently label the PCR products, a 20 μl reaction containing 10-20 μg of PCR generated DNA containing amine-modified nucleotides, 200 mM sodium bicarbonate (pH 9.0), and 5 mM ATTO565 NHS-ester (ATTO-TEC GmbH) was incubated for 1-2 hr at 25°C protected from light. Alexa Fluor 488 succinimidyl ester (Invitrogen) was used to label the 87 nt substrate used in the D-loop assay. Following incubation, 180 μl Nanopure water was added and a QIAquick PCR purification kit (Qiagen) was used to remove free label. Purified labeled DNA was stored at 4°C until the strand-separation step. Alkali denaturation in combination with the single 5′-biotin incorporated from the forward primer in the PCR reaction was utilized to produce ssDNA from the fluorescently labeled duplex PCR product as follows: 800 μl avidin-agarose (400 μl settled gel; Thermo Scientific) was prepared in 1.5 mL Eppendorf tube using centrifugation to pellet agarose. All centrifugation steps were performed using a bench top centrifuge at 4,524 × g for 1 min. The resin was pelleted and washed three times with 1 mL binding/wash buffer (10 mM Tris HCl (pH 7.5), 1 mM EDTA, and 150 mM NaCl). Fluorescently labeled biotinylated dsDNA (~10-20 μg, from the PCR reaction above) was diluted to 1 mL with binding/wash buffer. The diluted DNA was added to the prepared avidin-agarose, and mixed end-over-end for 1 h protected from light. The agarose and bound DNA were pelleted by centrifugation and washed three times with 1 mL wash/binding buffer to remove unbound DNA. The ssDNA was eluted by alkali denaturation of the dsDNA, by addition of 200 μl of 0.15 M NaOH to the pelleted agarose and mixing end-over-end 10 min to release the non-biotinylated strand. The slurry was transferred to an empty microspin column (Bio-Rad) and centrifuged at 4700 × g to recover the eluted ssDNA. A Microspin S-400 column (GE Healthcare) was used to exchange the ssDNA into TE. Samples of each fraction were analyzed by polyacrylamide or agarose gel electrophoresis. Fractions containing ssDNA were pooled, purified and concentrated with QIAquick PCR purification kit (Qiagen). The DNA concentration was determined using an extinction coefficient of 8,919 M-1 cm-1 at 260 nm, taking into account a correction factor of 0.34 for absorbance at 260 nm by the dye. The dye concentration was determined using an extinction coefficient of 120,000 M-1 cm-1 at 563 nm.
Flowcell Fabrication
Channels and holes were CO2 laser etched into glass slides (Fisher Scientific 25×75×1 mm) covered with an adhesive abrasive blasting mask (Epilog) using a 30 Watt Mini-24 Laser Engraver (Epilog Lasers). Following the engraving step, the slides were blasted using 220 grit silicon carbide (Electro Abrasives) to remove residual laser-ablated glass from the channels. A cover glass (Corning No. 1, 24×60 mm) was attached with UV Optical adhesive #74 (Norland Products) applied through capillary action. The adhesive was cured by placing the flowcell 30 cm from a 100 Watt HBO lamp (Zeiss, Inc.) for 20 minutes followed by a final heat curing at 70°C for 12 h. PEEK tubing with 0.5 mm inner diameter (Upchurch Scientific) was inserted into each of the etched holes to create inlet and outlet connection ports using 5 min Epoxy (Devcon).
Surface Preparation of Single-Channel Flowcell for TIRFM Experiments
The entire surface modification procedure was done at 25°C. The flowcells were cleaned with 1 M NaOH for 30-60 minutes, then washed two times with 1 mL Nanopure water, and followed by 1 mL of buffer (25 mM TrisOAc (pH 7.5), 50 mM NaCl). 1 mg/mL biotinylated BSA (Thermo Scientific) in the above buffer was then incubated in the flowcell for 5 minutes followed by a wash with 1 mL of buffer. Then, 0.1 mg/mL streptavidin (Promega) in buffer was incubated in the flowcell for 5 minutes, followed by a wash with 1 mL of buffer. Finally, the flowcell was blocked with 1.5 mg/mL Roche Blocking Reagent (Roche) in buffer for 30-60 minutes, and washed with 1 mL buffer. The prepared flowcell was then mounted on the microscope and attached to the syringe pump (KD Scientific).
D-loop Assay
RecA and SSB were purified as previously described19,20. The AlexaFluor 488-labeled 87 nt ssDNA substrate prepared as described above. A 10 μl reaction containing 25 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mM DTT, 2 mM ATPγS, 100 μg/ml BSA, 4.5 μM RecA, and 105 nM fluorescently labeled 87 nt ssDNA was incubated for 8 minutes at 37°C. The reaction was started with addition of 35 nM supercoiled DNA (pUC19) and incubated at 37°C for 20 minutes. The reaction was stopped by mixing with 5μl of stop solution (4.8% SDS, 7 mg/ml proteinase K) and incubating for 10 minutes at 37°C. Products were resolve by electrophoresis in a 1% ultrapure agarose gel (Invitrogen) using TAE (40 mM Tris, 20 mM acetic acid, and 1 mM EDTA) at 100 volts until the bromophenol blue had migrated 4 cm. The gel was imaged and analyzed with a STORM scanner and Image Quant software (Molecular Dynamics). The efficiency of the reaction was calculated as the fraction of ssDNA that formed D-loops multiplied by 3 to correct for the 3-fold molar excess of ssDNA relative to supercoiled pUC19 in the reaction.
Single-molecule DNA Pairing Experiments
Nucleoprotein filaments were formed essentially as described previously4 in SM buffer (25 mM TrisOAc (pH 7.5), 1 mM DTT, and 4 mM Mg(OAc)2), SSB (at a ratio of 1 SSB monomer:11 nt), 2 nM molecules fluorescent ssDNA, and 1 mM ATPγS were incubated for 10 min at 37°C; RecA was added at a ratio of 1 monomer:1.7 nt, and incubated for an hour. Nucleoprotein filaments were then diluted 10-fold to a final concentration of 0.2 nM in buffer prior to introduction into the flowcell. In the DNA pairing experiments using TIRFM, biotinylated λ DNA (1 pM, molecules) in SM2 buffer (SM and 50 mM NaCl) was introduced into the flowcell and allowed to bind for several minutes. The flowcell was then washed with 500 μl SM2 buffer to remove free DNA as well as to extend and attach the second end of the λ DNA molecules. The reaction was started by the addition of 0.2 nM nucleoprotein filaments in SM2 buffer. For ensemble experiments visualized by TIRFM, the nucleoprotein filaments and lambda DNA were incubated for 1 hour (162 nt substrate) or 30 minutes (430 nt substrate) at 37°C prior to visualization in a single-channel flowcell
Visualization of RecA-mediated pairing with individual DNA-dumbbells was performed at 37°C. The flowcell surface was treated for 1 h with BSA (1 mg/ml) in single-molecule (SM3) buffer (50 mM TrisOAc (pH 8.2), 50 mM DTT, 1 mM Mg(OAc)2, and 15% sucrose). Biotinylated λ DNA and buffers were pumped at a linear flow rate of ~100 μm s-1 into the flowcell. The channels contained (see Figure 2): 1) SM3 buffer, 18 fM streptavidin-coated polystyrene beads (1 μm; Bangs Laboratories), and 5 nM YOYO-1 (Invitrogen). 2) SM3 buffer, 100 nM YOYO-1, and 10 pM (molecules) biotinylated λ DNA. 3) SM3 buffer. 4) SM buffer and 15% sucrose. The reaction reservoir contained 0.2 nM nucleoprotein filaments in SM with 15% sucrose and 0.5 mM ATPγS.
Data Analysis
Data were analyzed using GraphPad Prism v5.04. The kinetic data were fit to a single exponential function (Y=Y0 + (Plateau-Y0)*(1-exp(-k*x)). In Fig. 4 b and c, none of the time courses pass through the origin. We are not certain whether this is an intrinsic characteristic of the homology search or limitation of the experimental procedure: e.g., the time for the DNA to relax from flow-induced stretching after movement into the reservoir. We note that the half-time for the relaxation of extended λ DNA is ~6 seconds22; during this time, the dsDNA is not in its equilibrium coiled configuration and initial interaction with the RecA nucleoprotein filament would be limited by the DNA polymer dynamics.
Supplementary Material
Acknowledgments
We are grateful to member of the laboratory for their comments on this work. A.L.F. was funded by an American Cancer Society Postdoctoral Fellowship (PF-08–046–01-GMC) and S.C.K.by the National Institutes of Health (GM-62653 and GM-64745).
Footnotes
Author Contributions A.L.F. and S.C.K. conceived the general ideas, designed the experiments, and interpreted the data. A.L.F. performed experiments. A.L.F. and S.C.K. wrote the manuscript.
The authors declare no competing financial interests.
References
- 1.Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Menetski JP, Kowalczykowski SC. Interaction of recA protein with single-stranded DNA. Quantitative aspects of binding affinity modulation by nucleotide cofactors. J Mol Biol. 1985;181:281–295. doi: 10.1016/0022-2836(85)90092-0. [DOI] [PubMed] [Google Scholar]
- 3.Kowalczykowski SC, Krupp RA. DNA-strand exchange promoted by RecA protein in the absence of ATP: implications for the mechanism of energy transduction in protein-promoted nucleic acid transactions. Proc Natl Acad Sci U S A. 1995;92:3478–3482. doi: 10.1073/pnas.92.8.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menetski JP, Bear DG, Kowalczykowski SC. Stable DNA heteroduplex formation catalyzed by the Escherichia coli RecA protein in the absence of ATP hydrolysis. Proc Natl Acad Sci U S A. 1990;87:21–25. doi: 10.1073/pnas.87.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adzuma K. No sliding during homology search by RecA protein. J Biol Chem. 1998;273:31565–31573. doi: 10.1074/jbc.273.47.31565. [DOI] [PubMed] [Google Scholar]
- 6.Kowalczykowski SC. Biochemistry of genetic recombination: Energetics and mechanism of DNA strand exchange. Annu Rev Biophys Biophys Chem. 1991;20:539–575. doi: 10.1146/annurev.bb.20.060191.002543. [DOI] [PubMed] [Google Scholar]
- 7.Fulconis R, Miné J, Bancaud A, Dutreix M, Viovy JL. Mechanism of RecA-mediated homologous recombination revisited by single molecule nanomanipulation. EMBO J. 2006;25:4293–4304. doi: 10.1038/sj.emboj.7601260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Heijden T, et al. Homologous recombination in real time: DNA strand exchange by RecA. Mol Cell. 2008;30:530–538. doi: 10.1016/j.molcel.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 9.Forget AL, Kowalczykowski SC. Single-molecule imaging brings Rad51 nucleoprotein filaments into focus. Trends Cell Biol. 2010;20:269–276. doi: 10.1016/j.tcb.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McEntee K, Weinstock GM, Lehman IR. Binding of the recA protein of Escherichia coli to single- and double-stranded DNA. J Biol Chem. 1981;256:8835–8844. [PubMed] [Google Scholar]
- 11.Honigberg SM, Gonda DK, Flory J, Radding CM. The pairing activity of stable nucleoprotein filaments made from recA protein, single-stranded DNA, and adenosine 5’-(gamma-thio)triphosphate. J Biol Chem. 1985;260:11845–11851. [PubMed] [Google Scholar]
- 12.Galletto R, Amitani I, Baskin RJ, Kowalczykowski SC. Direct observation of individual RecA filaments assembling on single DNA molecules. Nature. 2006;443:875–878. doi: 10.1038/nature05197. [DOI] [PubMed] [Google Scholar]
- 13.van Mameren J, et al. Counting RAD51 proteins disassembling from nucleoprotein filaments under tension. Nature. 2009;457:745–748. doi: 10.1038/nature07581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Julin DA, Riddles PW, Lehman IR. On the mechanism of pairing of single- and double-stranded DNA molecules by the recA and single-stranded DNA-binding proteins of Escherichia coli. J Biol Chem. 1986;261:1025–1030. [PubMed] [Google Scholar]
- 15.Gonda DK, Radding CM. By searching processively recA protein pairs DNA molecules that share a limited stretch of homology. Cell. 1983;34:647–654. doi: 10.1016/0092-8674(83)90397-5. [DOI] [PubMed] [Google Scholar]
- 16.Tsang SS, Chow SA, Radding CM. Networks of DNA and recA protein are intermediates in homologous pairing. Biochemistry. 1985;24:3226–3232. doi: 10.1021/bi00334a023. [DOI] [PubMed] [Google Scholar]
- 17.Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 18.Berg OG. In: The Biology of Nonspecific DNA Protein Interactions. Revzin A, editor. CRC Press; 1990. pp. 71–85. [Google Scholar]
- 19.Mirshad JK, Kowalczykowski SC. Biochemical characterization of a mutant RecA protein altered in DNA-binding loop 1. Biochemistry. 2003;42:5945–5954. doi: 10.1021/bi027233i. [DOI] [PubMed] [Google Scholar]
- 20.Harmon FG, Kowalczykowski SC. RecQ helicase, in concert with RecA and SSB proteins, initiates and disrupts DNA recombination. Genes Dev. 1998;12:1134–1144. doi: 10.1101/gad.12.8.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bianco PR, et al. Processive translocation and DNA unwinding by individual RecBCD enzyme molecules. Nature. 2001;409:374–378. doi: 10.1038/35053131. [DOI] [PubMed] [Google Scholar]
- 22.Perkins TT, Quake SR, Smith DE, Chu S. Relaxation of a single DNA molecule observed by optical microscopy. Science. 1994;264:822–826. doi: 10.1126/science.8171336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.