

Abstract
Laccases are copper-containing oxidases that are involved in sclerotization of the cuticle of mosquitoes and other insects. Oxidation of exogenous compounds by insect laccases may have the potential to produce reactive species toxic to insects. We investigated two classes of substituted phenolic compounds, halogenated di- and trihydroxybenzenes and substituted di-tert-butylphenols, on redox potential, oxidation by laccase and effects on mosquito larval growth. An inverse correlation between the oxidation potentials and laccase activity of halogenated hydroxybenzenes was found. Substituted di-tert-butylphenols however were found to impact mosquito larval growth and survival. In particular, 2,4-di-tert-butyl-6-(3-methyl-2-butenyl)phenol (15) caused greater than 98% mortality of Anopheles gambiae larvae in a concentration of 180 nM, whereas 2-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-methylpropanal oxime (13) and 6,8-di-tert-butyl-2,2-dimethyl-3,4-dihydro-2H-chromene (33) caused 93% and 92% mortalities in concentrations of 3.4 and 3.7 μM, respectively. Larvae treated with di-tert-butylphenolic compounds died just before pupation.
Keywords: Anopheles gambiae, anti-larval activity, halogenated di- and trihydroxybenzenes, laccases, mosquito larvicides, redox potential, substituted di-tert-butylphenols
I. Introduction
Mosquitoes, the most important arthropods affecting human health, are vectors of human diseases. The control of mosquitoes with insecticides is currently in jeopardy, since there have been no new public health insecticides developed for mainstream vector control in the past 30 years. Laccases, copper-containing oxidases, are present in fungi, plants, bacteria, and insects and have been implicated in a variety of physiological processes and various applications including bioremediation, bio-fuel cells, pigmentation, cell wall synthesis, detoxification, wound healing, and biosensors [1]. Substituted o-, m-, and p-phenols, polyphenols, and aromatic amines and N-hydroxamates are oxidized by laccases with a concomitant reduction of oxygen to water [2–7]. The involvement of laccases in cuticle sclerotization or tanning is essential to insect survival [8,9]. Laccase inhibitors are limited and few useful organic compounds have been found [10–12]. Our studies aimed at identifying laccase substrates that would be oxidized to produce compounds potentially toxic to mosquito larvae, through covalent modification of laccase [13,14] or other molecular targets within the insect, pursued two approaches: (1) production of methine quinone intermediates for covalent linkage to nucleophilic protein moieties, represented by class 1 compounds in Figure 1, and (2) formation of phenoxy free radical intermediates in the laccase active site, represented by class 2 compounds in Figure 1. These two approaches led us to synthesize these two classes of substituted phenolic compounds (Figure 1) and to study their oxidation by laccase, redox potentials and antilarval activities. The phenolic compounds are laccase substrates and do not appear to inhibit laccase, however, several class 2 compounds possess potent antilarval activity. The results may shed light onto future design of environmentally compatible laccase substrates and mosquito larvicides.
Figure 1.

Synthesized halogenated polyphenols, aminophenols and di-tert-butyl substituted phenols.
II. Results and Discussion
Our initial plan was to position a leaving group such as bromine or chlorine into phenolic molecules. Upon oxidation by laccase, the resulting cyclic haloenone is a reactive molecule that undergoes addition-elimination reaction with a nucleophile such as the amino function of lysine or hydroxyl function of serine or tyrosine of laccase or some other nearby insect proteins. If such covalent linkage reaction could occur, halogenated phenols may lead to an inhibition of protein function and result in toxicity. Figure 2 depicted a proposed oxidation of p-dihydroquinone 1 to p-quinone 16 followed by an addition-elimination reaction with a nucleophile of protein. To test this notion, we treated p-quinone 16 (vide infra; for synthesis) with L-alanine benzyl ester (17; as a p-toluenesulfonic acid salt) and 2 equivalents of triethylamine in dichloromethane at 25°C (Figure 2). The amino function of L-alanine serves as a nucleophile. As expected, addition product 18 was isolated in 65% yield. The quinone structure of 18 was revealed by its IR spectrum in which the quinone C=O and C=C absorptions appear at 1663 and 1580 cm−1, respectively. This encouraging result prompted us to synthesize class 1 molecules containing halogenated phenols and aminophenols such as compounds 1 – 8 (Figure 1). Oxidation of 3-chloro-4-hydroxy-5-methoxybenzaldehyde (2) by laccase may provide a hydroxymethine quinone, which should have similar reactivity to that of the quinone derived from 1. Compounds 3 ~ 5 will afford either reactive ortho- or para-quinones. 4-Bromo-5-chloro-6-methoxybenzene-1,3-diol (6) possessing meta-dihydroxy functions should not produce quinone. o-Aminophenols, such as compounds 7 and 8, were investigated since the resulting iminoquinones could be more reactive than the quinone analogs and may complex with the copper in the laccase binding site (T1 site) [13,14].
Figure 2.
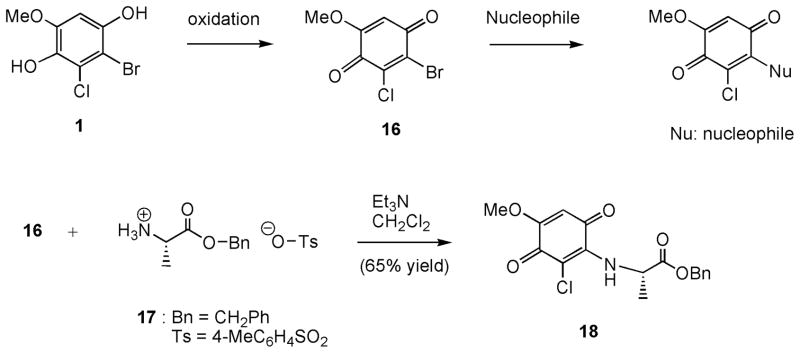
Proposed covalent linkage of para-quinone 2 with nucleophile and reaction of quinone 2 with L-alanine.
Since substituted di-tert-butyl phenols including MON-0585 (Figure 1) were implicated as insect growth regulators [15–17], and they can be oxidized by Co(II)-Schiff base complex and oxygen to produce 4-substituted 2,6-di-tert-butyl-6-hydroperoxy-2,4-cyclohexadienones [18,19], we therefore envision that substituted di-tert-butylphenols may undergo oxidation with laccase catalyzed by coppers and oxygen in the active site to provide di-tert-butyl-hydroperoxy-2,4-cyclohexadienones that are toxic to insects through their reaction with endogenous nucleophiles. Hence, class 2 compounds including di-tert-butylphenols 9 – 15 were synthesized and their redox potentials, oxidation by laccase, and anti-larval activities determined. MON-0585, 2,6-di-tert-butyl-4-(α,α-dimethylbenzyl)phenol, reported by Monsanto Co. [16], was prepared and used as a control.
II.1. Synthesis
In our reported total synthesis of (+)-chloropuupehenone [20], chlorinated phenols such as compounds 2 and 3 were prepared from vanillin. Utilizing these two chlorinated phenolic compounds, class 1 compounds, 1 and 4 – 6, were synthesized via a sequence of protection, Baeyer-Villiger oxidation, regioselective bromination, and deprotection reactions. Compounds 7 and 8 were readily available from the reduction of 4-hydroxy-3-nitrobenzaldehyde (31). p-Dihydroxybenzene 1 was synthesized from 3-chlorovanillin (2) by a sequence of reactions depicted in Scheme 1: (i) protection of C4-hydroxyl function with t-butyldimethylsilyl chloride; (ii) Baeyer-Villiger oxidation [21] of the aldehyde function with m-chloroperbenzoic acid (MCPBA); (iii) basic methanolysis of the resulting formate moiety with K2CO3 in methanol; (iv) regioselective bromination with N-bromosuccinimide in N,N-dimethylformamide (DMF); (v) deprotection of the silyl ether moiety; and (iv) reduction of the quinone moiety with hydrogen and palladium/carbon in ethanol. In the bromination reaction of phenol 20 with N-bromosuccinimide (NBS) in DMF, compounds 21 and 16 were isolated in 31% and 48% yield, respectively. Regioselective bromination onto the less hindered C2 of phenol 20 has been reported previously [20]. Compound 16 likely derived from a hydrolytic cleavage of the silyl ether function of 21 from HBr generated through NBS and the phenol and/or a trace amount of water presence in the reaction mixture followed by oxidation to the quinone. The infrared spectrum of quinone 16 does not display hydroxyl absorption, but quinone stretch at 1683 cm−1, and the spectrum of dihydroquinone 1 shows strong hydroxyl absorption at 3303 cm−1. Desilylation of compound 21 with tetra-n-butylammonium fluoride gave quinone 16. Oxidation of dihydroquinone to benzoquinone under the NBS reaction conditions has been reported [22], however, oxidation under tetra-n-butylammonium fluoride reaction conditions is unexpected.
Scheme 1.

Synthesis of compounds 1 and 4.
The t-butyldimethylsilyl and two benzyl protecting groups of compound 22 [20] were readily removed by a sequential treatment with tetra-n-butylammonium fluoride and hydrogen-palladium/carbon to give triol 4 in a 72% overall yield. Triol 4 is soluble in water and readily oxidized in air to give the corresponding quinone, which gradually decomposes to produce black solids. Hence, the compound is stored in a dry box under nitrogen atmosphere.
o-Dihydroxybenzene 5 was readily synthesized from compound 24 [20] by methylation of the phenolic hydroxyl moiety with trimethyloxonium tetrafluoroborate followed by desilylation with tetra-n-butylammonium fluoride (Scheme 2). Compound 5 does not undergo oxidation to give the corresponding o-quinone as that of p-dihydroxybenzene 1 (vide supra), which evidence from its IR spectrum showing strong hydroxyl absorption bands at ν 3436 and 3219 cm−1. Since the less hindered C5-hydroxyl moiety of 3-chloro-4,5-dihydroxybenzaldehyde (3) [20] should react faster with an electrophile than the more hindered C4-hydroxy, protection of C5-OH followed by methylation of C4-OH, and subsequent functional group manipulation would lead to m-dihydroxybenzene analog 6 (Scheme 2). Thus, silylation of 3 with t-butyldimethylsilyl chloride, 4-dimethylaminopyridine (DMAP), and triethylamine gave a 7:1 ratio of silyl ether 26 and the C4-silyl ether byproduct, which were separated by silica gel column chromatography. Methylation of C4-hydroxy moiety of 26 with trimethyloxonium tetrafluoroborate, followed by Baeyer-Villiger oxidation of the aldehyde function with MCPBA, and basic methanolysis of the resulting formate afforded phenol 29. Regio-selective bromination of 29 with NBS followed by removal of the silyl ether protecting group produced m-dihydroxybenzene 6.
Scheme 2.

Synthesis of compounds 5 and 6.
o-Aminophenols 7 [23] and 8 [24] were readily prepared from the reduction of both nitro and aldehyde functions of 4-hydroxy-3-nitrobenzaldehyde (31) using a catalytic amount of palladium under 30 psi of hydrogen in ethanol (Scheme 3). The two products (1:1) were separated by silica gel column chromatography. Apparently, compound 7 derived from a reduction of the benzylic hydroxyl moiety of compound 8 under the hydrogenation reaction conditions.
Scheme 3.
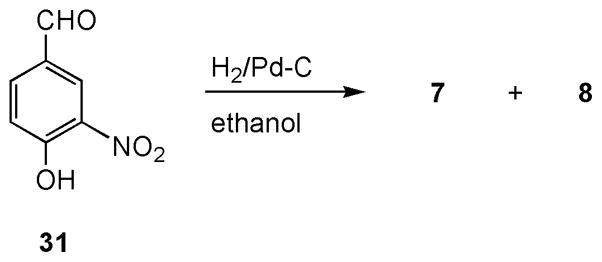
Synthesis of compounds 7 and 8.
Various halogenated polyphenols and substituted aminophenols were synthesized starting from 3-chlorovanilin (2) and 4-hydroxy-3-nitrobenzaldehyde (31), respectively. The reaction sequences are simple and should be applicable to the synthesis of various substituted polyphenols.
The synthesis of class 2 compounds mainly utilized Friedel-Crafts reaction of di-tert-butylphenols. Following the reported procedure [25], compounds 9 and 10 were prepared by an acylation reaction of 2,6-di-t-butylphenol with isobutyryl chloride and aluminum chloride followed by bromination with cupric bromide in dichloromethane and ethyl acetate (to give compound 9), and reduction with lithium aluminum hydride in ether (to produce compound 10). Oxidation of alcohol 10 with o-iodoxybenzoic acid (IBX) in DMSO afforded aldehyde 11, further oxidation with silver nitrate and sodium hydroxide gave acid 12 [19] (Scheme 4). Treatment of aldehyde 11 with hydroxylamine produced oxime 13, and a reductive amination reaction of 11 with benzylamine followed by sodium cyanoborohydride furnished amine 14. Compound 15 was synthesized in 46% yield from a Friedel-Crafts alkylation reaction of 2,4-di-tert-butylphenol (32) with 2-methyl-3-buten-2-ol and BF3•ether in dichloromethane at 25°C. A 10% yield of benzopyran 33 was also isolated, which likely derived from an acid-catalyzed ring closing reaction of 15. MON-0585 or 2,6-di-tert-butyl-4-cumylphenol was prepared by a Friedel-Crafts alkylation reaction of 4-cumylphenol and isobutylene in dichloromethane in the presence of a catalytic amount of sulfuric acid in a sealed tube at 80°C for 6 hours [26].
Scheme 4.
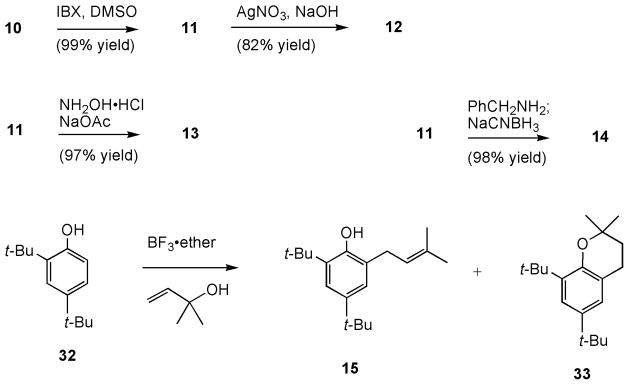
Synthesis of compounds 11 – 15, and 33.
II.2. Redox potentials and laccase activities
Dopamine, N-acetyldopamine (NADA) and related catecholamines are oxidized by laccase in insects during sclerotization and pigmentation of cuticle [27,28]. Insects with decreased laccase function develop serious cuticle defects and die;29 therefore, an inhibitor of laccases might function as a novel type of insecticide. Alternatively, oxidation of phenolic compounds by laccase may produce compounds that are toxic to insects through their reaction with nucleophilic groups in proteins. As vertebrate animals possess tyrosinase (or monophenol monooxygenase) [30,31], compounds that are selectively oxidized by laccase but not tyrosinase may serve as pro-insecticides. Selective laccase inhibitors or substrates generating toxic oxidized products might have useful properties of toxicity to insects but not vertebrates. An electrochemical study based on cyclic voltammetry (CV) was carried out to understand the inherent redox properties of the substrates. We tested compounds 1 – 8, 34, and 35 for evidence of irreversible inhibition of fungal laccase but did not observe inhibition (data not shown). However, these compounds were oxidized by laccase (vide infra).
Various known laccase substrates such as hydroquinone, catechol, 2-aminophenol, 1,2-phenylenediamine, 2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), 3-amino-4-hydroxybenzoic acid (34), 4-amino-3-hydroxybenzoic acid (35), and our synthesized materials, compounds 1 – 15, 18, 33, and MON-0585 were studied for their redox potentials (Table S1) and ability to be oxidized by fungal laccase (Table 1).
Table 1.
Kinetic constants of fungal laccase with known laccase substrates and substituted phenols.
| Substrate | kcat (min−1) | Km (mM) | kcat/Km (min−1mM−1) | ε (M−1cm−1)a | Wavelength (nm) |
|---|---|---|---|---|---|
| Hydroquinone | 7,130 | 0.02 | 35,650 | 17,252 [34] | 248 |
| catechol | 2,010 | 0.60 | 3,350 | 2,211 [34] | 450 |
| 2-aminophenol | 2,250 | 0.19 | 11,840 | 17,865 [35] | 434 |
| 1,2-phenylenediamine | 1,040 | 0.43 | 2,420 | 13,000 [36] | 440 |
| ABTS | 8,360 | 0.09 | 92,890 | 36,000 [34] | 414 |
| Compound 1 | 1,100 | 0.07 | 15,710 | 2,590 | 400 |
| Compound 2 | 6,310 | 2.20 | 2,870 | 762 | 410 |
| Compound 3 | 1,900 | 0.04 | 47,500 | 3,600 | 440 |
| Compound 4 | 8,680 | 0.05 | 173,600 | 1,700 | 500 |
| Compound 5 | 5,380 | 0.04 | 134,500 | 876 | 450 |
| Compound 6 | 5,760 | 0.07 | 82,290 | 2,280 | 350 |
| Compound 7 | 7,870 | 0.11 | 71,550 | 5,180 | 390 |
| Compound 8 | 3,780 | 0.08 | 47,250 | 9,840 | 390 |
| Compound 34 | 2,930 | 0.07 | 41,860 | 14,480 | 440 |
| Compound 35 | 2,550 | 0.17 | 15,000 | 13,760 | 440 |
Most compounds present some redox properties as listed in Table S1. Interestingly, even though the CV features varied dramatically, the main oxidation potential of the substrates showed a good correlation with the laccase activity. Besides the oxidation potential, these compounds also showed different electrochemical oxidation/reduction reversibility which is indicated by the separation between the peak potentials of the oxidation and reduction waves in CV, i.e. ΔEp [32]. In general, we divided the compounds into four groups shown in Figures S1 – S4. The first group of compounds 4, 5, 7, 8, 9, 34, and 35 all present at least one pair of redox waves with a small peak separation (ΔEp) ranging from ~60 mV to 100 mV, indicating that they are electrochemically active with nearly reversible redox properties. The second group consists of ABTS, compounds 1 and 3, and MON-0585, which show a peak separation between 140 mV to 290 mV. These compounds are not as active as the first group but still show quasi-reversible redox properties. The third group includes hydroquinone, catechol, 2-aminophenol, 1,2-phenylenediamine and compounds 6, and which show nearly irreversible redox waves as indicated by the large peak separation between the redox waves (with ΔEp = 370 − 420 mV) or complete absence of the reduction waves corresponding to the oxidation waves at electropotential above 0.0 V. The fourth group either only shows one of the redox waves or has an enormous peak separation (> 540 mV), including compounds 2, 10–15, 18, and 33. Interestingly, compounds 4 and 5 have very similar structures and thus both show reversible redox properties but the redox potentials shift. Compounds 2 and 3 also have very close structures but compound 2 is much more irreversible. Compound 33 did not show measureable redox signal besides the large peak due to electrolysis of water at 2.0 V, which is likely due to the low solubility.
It is noted that compounds 9 – 15, 18, and 33 are the most water-insoluble materials. This causes unreliable CV measurements in aqueous PBS buffer solution containing 20% ethanol as reported in this study. They either present high oxidation potentials (> 0.68 V) or do not show oxidation waves within the applicable potential range in aqueous solution (with an upper limit at ~ + 0.8 to 1.2 V in most experiments). These compounds likely absorb into larvae and produce larvae-killing activity (vide infra).
To determine how well fungal laccase can oxidize the water soluble compounds, we determined the kinetic constants of fungal laccase using compounds 1 – 8, 34, and 35 along with known laccase substrates such as hydroquinone, catechol, 2-aminophenol, 1,2-phenylenediamine, and ABTS [33]. Results are summarized in Table 1. The known laccase substrates serve as positive controls and for comparison. A relationship between catalytic efficiency (kcat/Km) and redox potential is depicted in Figure 3.
Figure 3.
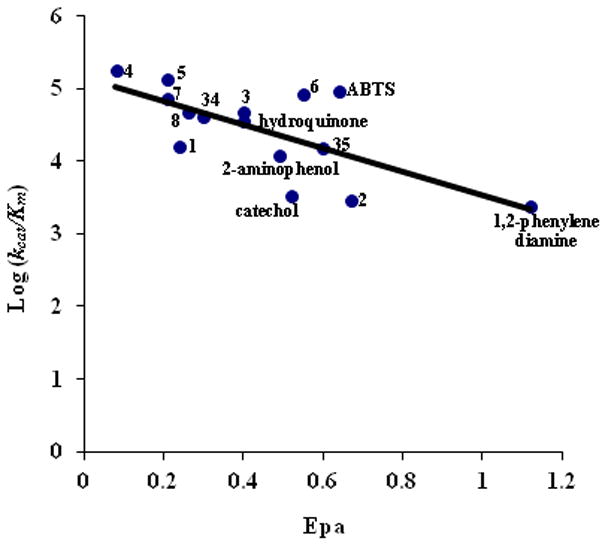
Inversion correlation of laccase catalytic efficiency and oxidation potential of Compounds 1 – 8, 34, 35, ABTS, 2-aminophenol, catechol, hydroquinone, and 1,2-phenylenediamine.
As shown in Figure 3, there is an inverse correlation between laccase catalytic efficiency and oxidation potential of the water-soluble compounds 1 – 8, 34, 35, ABTS, 2-aminophenol, catechol, hydroquinone, and 1,2-phenylenediamine. These results are consistent with previous studies [37–39]. These compounds show either nearly reversible or quasi-reversible redox properties except 1,2-phenylenediamine. The lower the peak potential for oxidation (i.e. Epa1 in Table S1), the higher the laccase activity is. 1,2-Phenylenediamine shows a very high oxidation potential and thus gives the lowest laccase activity. However, no clear correlation was found between the electrochemical reaction rate and reversibility of the compounds, both of which are reflected by the peak separation redox waves (i.e. ΔEp = Epa − Epc). Compounds 9 – 15, 33, and MON-0585 are insoluble in water, and their oxidation by laccase could not be detected.
II.3. Anti-mosquito larval activities
Since laccases oxidize phenolic compounds in their function in insect cuticle sclerotization [9], we investigated potential anti-mosquito larval activities of laccase substrates such as compound 4 and dihydroquinone, di-t-butylphenols such as compounds 9 – 15, compound 18, and 33. MON-0585 was used as a positive control. Table 2 summarizes toxicities of substituted phenols to third-instar larvae of Anopheles gambiae in a three-day bioassay. The water-soluble samples such as dihydroquinone and triol 4 were dissolved in water and used, while other water-insoluble materials were dissolved in DMSO and used for the treatment of mosquito larvae in aqueous solution. Laccase substrates such as dihydroquinone and compound 4 and quinone 18 were not toxic at the concentrations tested, however, di-t-butylphenols including benzopyrane 33 (without phenolic OH function) produced significant mortality at 1,000 μg/mL concentration. Most strikingly, compound 15 possesses similar anti-larval activity to that of MON-0585 at 50 μg/mL concentration. The trifluoroacetic acid salt of amine 14 and sodium salt of carboxylic acid 12 are water-soluble and they do not possess anti-larval activity. Hence, the water-soluble compounds may not have been absorbed by larvae and consequently lacked anti-larval activity. Water-insoluble materials such as compound 15 likely absorb into larvae and produce larvae-killing activity. Compound 15 appears to be much more toxic to mosquito larvae than methoprene, which caused only about 60% mortality at 100 μg/L in 3 days against the third-instar larvae of Culex molestus [40]. Larvae treated with di-tert-butylphenolic compounds died just before pupation. Because the larval mortality occurred over the course of 3 days, it suggests that the target does not involve the neurological system typical of most insecticides. We examined microscopic sections of larvae from the control and compound 15 treated groups, and found that the cuticle was thin in compound 15 treated larvae when compared to the control larvae (Figure 4). The treated larvae appeared to start molting by separating the larval cuticle, but failed to remove the old cuticle. There was very little apparent synthesis of new pupal cuticle in the treated larvae. This was observed not only in the head, but also in the thorax and abdomen of the insects killed by compound 15. Thus, it appears that these compounds affect the development of the cuticle, a target of the mosquito that is different from the neurological targets of currently existing insecticides. However, the exact target and mechanism of action of these compounds remain to be studied.
Table 2.
Toxicities of substituted phenols to third-instar larvae of Anopheles gambiae in a three-day bioassay. Results are mean ± standard errors (SE) of four replicates (n = 4); each with 12–15 third-instar larvae. Means followed by the same letter in the same column are not significantly different (P > 0.05) according to Fisher’s LSD multiple comparison test after arcsine square root transformation of the percentage date (ProStat; Poly Software International, 2002).
| Compound | % Mortality (mean ± SE) | |
|---|---|---|
| 50 μg/L | 1000 μg/L | |
| Control | 0 ± 0 b | 3.50 ± 3.50 d |
| Dihydroquinone | 1.25 ± 1.25 b | 5.38 ± 1.80 d |
| Compound 4 | 0 ± 0 b | 11.4 ± 3.85 d |
| Compound 9 | 0 ± 0 b | 60.0 ± 11.5 c |
| Compound 10 | 0 ± 0 b | 46.7 ± 3.25 c |
| Compound 11 | 0 ± 0 b | 65.0 ± 5.00 c |
| Compound 12 | 5.50 ± 3.45 b | 12.3 ± 1.35 d |
| Compound 13 | 5.41 ± 3.28 b | 93.1 ± 3.99 ab |
| Compound 14 | 3.71 ± 2.14 b | 7.70 ± 3.14 d |
| Compound 15 | 98.1 ± 1.93 a | 98.3 ± 1.68 ab |
| Compound 18 | 5.00 ± 3.18 b | 6.66 ± 2.71 d |
| Compound 33 | 5.77 ± 3.69 b | 91.7 ± 6.31 b |
| MON-0585 | 93.3 ± 4.71 a | 100 ± 0 a |
Figure 4.
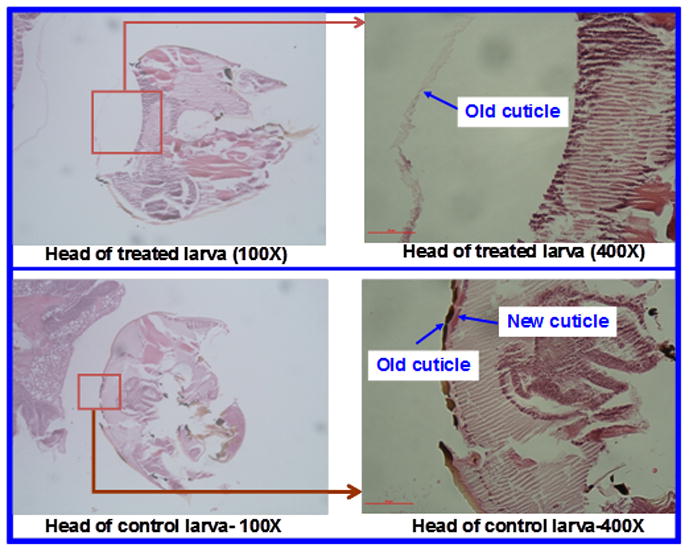
Microscopic comparisons of An. gambiae larvae in the control and killed by compound 15.
III. Conclusion
Oxidation of phenols by laccases led us to synthesize various phenolic compounds containing hydroxyl, halide, aldehyde, amino, and di-tert-butyl moieties. The redox potential, laccase activity, and antilarval activity were investigated. Phenolic compounds containing hydroxyl, halide, aldehyde, and amino functions were found to be laccase substrates but not inhibitors. The redox potentials and laccase activities of various phenolic compounds were correlated. It was found that a decrease of oxidation potential leads to higher laccase activity. A number of di-tert-butylphenolic compounds such as compounds 13, 15 and 33 were found to have potent anti-larval activity. Their mechanism of action appears to be different from that of MON-0585 and remains to be investigated.
4. Experimental Section
General Methods
Nuclear magnetic resonance spectra were obtained at 400 MHz for 1H and 100 MHz for 13C in deuteriochloroform, unless otherwise indicated, and reported in ppm. Infrared spectra were taken from a Nicolet 380 FT-IR instrument (Thermo Scientific) in solid forms and are reported in wave numbers (cm−1). Low-resolution mass spectra were taken from an API 2000-triple quadrupole ESI-MS/MS mass spectrometer (from Applied Biosystems). High-resolution Mass spectra were obtained from a LCT Premier (Waters Corp., Milford MA) time of flight mass spectrometer. The instrument was operated at 10,000 resolution (W mode) with dynamic range enhancement that attenuates large intensity signals. Mass correction for exact mass determinations was made automatically with the lock mass feature in the MassLynx data system. A reference compound in an auxiliary sprayer is sampled every third cycle by toggling a “shutter” between the analysis and reference needles. The reference mass is used for a linear mass correction of the analytical cycles. Cyclic voltammetric experiments were performed on a CHI400A potentiostat (CH Instruments, TX) with a three-electrode setup consisting of a 3-mm diameter glassy carbon disk electrode embedded in epoxy as a working electrode, a Ag/AgCl (saturated KCl) as the reference electrode, and a coiled Pt wire as the counter electrode. 4-Hydroxy-3-nitrobenzaldehyde, 3-amino-4-hydroxybenzoic acid, 4-amino-3-hydroxybenzoic acid, and 2-methyl-3-buten-2-ol were purchased from Aldrich Co., and L-alanine benzyl ester p-toluenesulfonic acid salt was from Advanced CHEMTECH. Trametes verisolor (fungal) laccase was purchased from Sigma (product # 53739). The compounds, 1 – 15, 33, and MON-0585 used in biological studies are ~98% pure as indicated by HPLC analysis (see Supplemental Data for HPLC graphs).
O-benzyl N-(6-Chloro-2,5-dioxo-4-methoxy-1,4-cyclohexadienyl)-L-alanine (18)
A solution of 50 mg (0.20 mmol) of quinone 16, 42 mg (0.2 mmol) of L-alanine benzyl ester p-toluenesulfonic acid salt, and 40 mg (0.40 mmol) of triethylamine in 5 mL of dichloromethane under argon was stirred at 25°C for 12 h. The reaction solution was diluted with dichloromethane and washed with water and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a gradient mixture of hexane and ethyl acetate as eluant to give 45 mg (65% yield) of compound 18. IR (neat) ν 3300, 3100 (w), 2939, 1735, 1663, 1580, 1500, 1445, 1381, 1258, 1190, 1123, 1004, 829, 694; 1H NMR δ 7.37 (s, 5 H, Ph), 5.78 (s, 1 H, =CH), 5.21 (s, 2 H, CH2Ph), 5.20 (pentet, J = 8 Hz, 1 H, CHN), 3.85 (s, 3 H, OMe), 1.56 (d, J = 8 Hz, 3 H, Me); 13C NMR δ 185.2, 180.9, 172.3 (2 C), 161.3, 141.8, 135.1, 128.9 (2 C), 128.8 (2 C), 128.5, 103.2, 67.8, 57.1, 52.0, 20.4; MS m/z 372.2 (M+Na+), 350.2 (M+1); HRMS calcd for C17H16ClNNaO5+ (M+Na+) 372.0609, found 372.0614.
4-(tert-Butyldimethylsilyloxy)-3-chloro-5-methoxybenzaldehyde (19)
To a solution of 0.30 g (1.61 mmol) of aldehyde 2 [20] in 5 mL of dichloromethane at 0°C under argon, were added 0.27 ml (1.93 mmol) of triethylamine, 39 mg of 4-dimethylaminopyridine (0.32 mmol), and 0.29 g (1.93 mmol) of t-butyldimethylsilyl chloride. The solution was stirred at 25°C for 8 hours, diluted with 50 mL of diethyl ether, washed with water and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a mixture of hexane and diethyl ether (4:1) as eluant to give 0.47 g (97% yield) of compound 19. 1H NMR δ 9.76 (s, 1 H), 7.45 (d, J = 2.0 Hz, 1 H), 7.27 (d, J = 2.0 Hz, 1 H), 3.85 (s, 3 H), 1.01 (s, 9 H), 0.21 (s, 6 H); 13C NMR δ 190.0, 151.9, 147.7, 130.0, 126.6, 126.3, 108.4, 55.6, 25.8 (3 C), 19.0, −3.8 (2 C); HRMS calcd for C14H22ClO3Si (M+H+) 301.1021, found 301.1028.
4-(tert-Butyldimethylsilyloxy)-3-chloro-5-methoxyphenol (20)
To a solution of 0.20 g (0.66 mmol) of compound 19 in 10 mL of dichloromethane under argon at 25°C, was added 0.25 g (1.0 mmol) of m-chloroperbenzoic acid (70% purity), and the solution was heated to reflux for 8 hours. The reaction solution was cooled to 25°C, diluted with aqueous sodium thiosulfate, and extracted three times with diethyl ether. The organic layers were combined, washed with saturated aqueous NaHCO3, water, and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a mixture of hexane and diethyl ether (2:1) as eluant to give 0.21 g (98% yield) of 4-(tert-butyldimethylsilyloxy)-3-chloro-5-methoxyphenyl formate (20A). 1H NMR δ 8.26 (s, 1H, OCHO), 6.78 (d, J = 2.9 Hz, 1 H), 6.58 (d, J = 2.9 Hz, 1 H), 3.80 (s, 3 H), 1.03 (s, 9 H), 0.20 (s, 6 H); 13C NMR δ 159.3, 151.8, 143.2, 140.4, 125.9, 114.4, 104.1, 55.7, 26.0, 19.0, −3.9; HRMS calcd for C14H22ClO4Si (M+H+) 317.0976, found 317.0968.
A solution of 0.21 g (0.65 mmol) of formate 20A and 0.45 g (3.2 mmol) of K2CO3 in 10 mL of methanol was stirred at 25°C for 4 h., and the solution was diluted with 10 mL of aqueous NH4Cl. The solution was extracted with ethyl acetate twice, and the combined extract was washed with brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a mixture of hexane and diethyl ether (2:1) as eluant to give 0.14 g (72% yield) of phenol 20: 1H NMR δ 6.43 (d, J = 2.8 Hz, 1 H), 6.32 (d, J = 2.8 Hz, 1 H), 3.77 (s, 3 H), 1.03 (s, 9 H), 0.17 (s, 6 H); 13C NMR δ 152.2, 149.8, 135.8, 125.7, 108.2, 99.2, 55.5, 26.1 (3 C), 19.0, −4.0 (2 C); HRMS calcd for C13H22ClO3Si (M+H+) 289.1027, found 289.1000.
2-Bromo-4-(tert-butyldimethylsilyloxy)-3-chloro-5-methoxyphenol (21) & 2-bromo-3-chloro-5-methoxy-1,4-benzoquinone (16)
To a solution of 84 mg (0.29 mmol) of phenol 20 in 2 mL of DMF under argon was added 57 mg (0.32 mmol) of N-bromosuccinimide, and the solution was stirred at 25°C for 12 h. The reaction solution was diluted with diethyl ether and washed with water and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a gradient mixture of hexane and diethyl ether as eluant to give 27 mg (31% yield) of bromophenol 21 and 35 mg (48% yield) of p-benzoquinone 16. Bromophenol 21: 1H NMR δ 6.56 (s, 1 H), 5.38 (bs, 1 H, OH), 3.78 (s, 3 H), 1.03 (s, 9 H), 0.18 (s, 6 H); 13CNMR δ 151.5, 147.4, 136.8, 125.8, 101.5, 98.4, 55.6, 26.1, 19.0, −3.9; HRMS calcd for C13H19BrClO3Si (M-H) 364.9975, found 365.0076; C13H20BrClO3SiNa (M+Na+) 388.9951, found 388.9934. p-Benzoquinone 16: light yellow solids; Mp. 167 – 170°C; IR (neat) ν 2953, 2913, 2839, 1683, 1638, 1613, 1556, 1454, 1229, 1168 cm−1; 1H NMR δ 6.16 (s, 1 H), 3.89 (s, 3 H); 13C NMR δ 177.5, 172.4, 158.9, 137.4, 107.9, 107.2, 57.3; HRMS calcd for C7H5BrClO3 (M+1+) 250.9105, found 251.0593.
Conversion of silyl ether 21 to p-benzoquinone 16
A solution of 25 mg (68 μmol) of silyl ether 21 and 68 μL (68 μmol) of n-Bu4NF (1 M in THF) in 2 mL of THF was stirred under argon from 0°C for 30 min., diluted with 10 mL of aqueous NH4Cl, and extracted with ethyl acetate twice. The combined extract was washed with brine, dried (MgSO4), concentrated and column chromatographed on silica gel using a mixture of hexane:diethyl ether (1:1) as eluent to give 14 mg (81% yield) of p-benzoquinone 16, which spectral data are identical with that described above.
2-Bromo-3-chloro-5-methoxy-1,4-dihydroxybenzene (1)
A mixture of 0.10 g (0.40 mmol) of quinone 16 and 20 mg of 10% palladium over carbon in 10 mL of ethanol under 1 atm. of hydrogen was stirred at 25°C for 15 min., filtered through Celite, and washed with ethanol. The filtrate was concentrated to give 99 mg (98% yield) of dihydroxybenzene 1: IR (neat) ν 3303, (s), 2935, 2851, 1601, 1497, 1441, 1210, 1071, 1046, 994, 864, 842, 825 cm−1; 1H NMR δ 6.60 (s, 1 H), 5.50 (bs, 1 H, OH), 5.30 (bs, 1 H, OH), 3.89 (s, 3 H, OMe); 13C NMR δ 148.2, 147.7, 136.7, 121.1, 100.2, 99.4, 56.0; HRMS calcd for C7H6BrClO3 (M+) 251.9189, found 252.0427.
2-Bromo-3-chloro-4,5-dibenzyloxyphenol (23)
A solution of 0.20 g (0.37 mmol) of silyl ether 22 [20] and 0.37 mL of n-Bu4NF (1 M solution in THF) in 4 mL of THF was stirred under argon at 0°C for 1 h. The solution was diluted with 30 mL of saturated aqueous NaCl solution, extracted twice with ethyl acetate (30 mL each), and the organic extract was dried (MgSO4), filtered, concentrated to dryness, and column chromatographed on silica gel using a gradient mixture of hexane and diethyl ether as eluant to give 0.12 g (77% yield) of compound 23 as white solids, mp. 108 – 110°C; 1H NMR δ 7.40 – 7.20 (m, 10 H), 6.66 (s, 1 H), 5.07 (s, 2 H), 4.96 (s, 2 H); 13C NMR δ 153.1, 149.9, 139.8, 137.0, 136.1, 129.2, 128.8, 128.5, 128.44, 128.40, 127.6, 102, 100.6, 75.4, 71.3; HRMS calcd for C20H17BrClO3 (M+H+) 419.0044, found 419.0051.
5-Bromo-6-chlorobenzene-1,2,4-triol (4)
To a solution of compound 23 (25 mg, 0.06 mmol) in 1 mL of ethanol was added 2.5 mg of 10% palladium over carbon, and the mixture was stirred under 1 atmosphere of hydrogen at 25°C for 3 h. The reaction mixture was filtered through Celite, concentrated to dryness to give 13 mg (93% yield) of compound 4: Mp. >350°C; IR (neat) ν 3382 (bs, OH stretch), 2932, 2843, 1614, 1437, 1285, 1170, 1070 cm−1; UV (in methanol) λ 209.6 (εmax = 30300), 291.8 (1250; likely derived from a partial oxidation of the polyphenol functions), 332.5 (4640), 397.1 (1785) nm; 1H NMR (CD3OD) δ 6.66 (s, 1 H, Ar), 5.58 (bs, 1 H, OH), 5.27 (bs, 1 H, OH), 5.21 (bs, 1 H, OH); 13C NMR (CD3OD) δ 149.4, 147.6, 137.4, 123.1, 103.2, 100.6; HRMS calcd for C6H5BrClO3 (M+H+) 238.9105, found 238.9111.
2-Bromo-4,5-bis-(tert-butyldimethylsilyloxy)-3-chloro-1-methoxybenzene (25)
To a solution of 0.10 g (0.21 mmol) of 24 [20] in 2 mL of dichloromethane at 0°C under argon, were added 55 mg (0.26 mmol) of proton sponge 38 mg (0.26 mmol) of trimethyloxonium tetrafluoroborate, and the mixture was stirred at 0°C for 8 h. It was diluted with water and extracted twice with diethyl ether, the combined extract was washed with water and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a mixture of hexane and diethyl ether (10:1) as eluant to give 67 mg (94% yield based on reacted 24) of 25 along with 31 mg of recovered 24. 1H NMR δ 6.43 (s, 1 H), 3.80 (s, 3 H), 1.03 (s, 9 H), 0.98 (s, 9 H), 0.23 (s, 6 H), 0.17 (s, 6 H); 13C NMR δ 151.0, 147.5, 138.9, 128.9, 104.7, 104.5, 56.9, 26.3 (6 C), 18.9, 18.8, −3.3 (2 C), −3.5 (2 C); HRMS calcd for C19H35BrClO3Si2 (M+H+) 481.0996, found 481.0951.
4-Bromo-3-chloro-5-methoxybenzene-1,2-diol (5)
To a solution of 51 mg (0.10 mmol) of 25 in 2 mL of THF under argon at 0°C, was added 0.20 mL (0.20 mmol) of n-Bu4NF, and the solution was stirred for 30 min. It was diluted with diethyl ether, washed with water and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a gradient mixture of dichloromethane and methanol as eluant to give 19 mg (71% yield) of 5. IR (neat) ν 3436, 3219 (broad & s), 2917, 2851, 1580, 1462, 1417, 1315, 1188, 1070, 984, 820 cm−1; 1H NMR δ 6.59 (s, 1 H), 5.60 (bs, 1 H, OH), 5.24 (bs, 1 H, OH), 3.83 (s, 3 H); 13C NMR δ 151.4, 144.4, 134.2, 121.8, 101.7, 99.7, 57.1; HRMS calcd for C7H7BrClO3 (M+H+) 252.9267, found 252.9254.
3-(tert-Butyldimethylsilyloxy)-5-chloro-4-hydroxybenzaldehyde (26)
To a solution of 3.2 g (19 mmol) of aldehyde 3, 2.9 mL (22 mmol) of triethylamine, and 0.45 g (3.7 mmol) of 4-dimethylaminopyridine in 60 mL of dichloromethane at 0°C under argon, was added 3.4 g (22 mmol) of t-butyldimethylsilyl chloride, and the solution was stirred at 25°C for 8 h. It was diluted with diethyl ether, washed by water and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using hexane and dichloromethane (1:1) as eluant to give 3.7 g (70% yield) of compound 26 and 0.84 g (11% yield) of 5-chloro-3,4-bis-(t-butyldimethylsilyloxy)benzaldehyde. Compound 26 was crystallized from diethyl ether to provide single crystals and its structure was unequivocally identified by a single-crystal X-ray analysis. 1H NMR δ 9.76 (s, 1H, CHO), 7.51 (d, J = 2.8 Hz, 1 H), 7.27 (d, J = 2.8 Hz, 1 H), 6.24 (s, 1 H, OH), 1.03 (s, 9 H), 0.32 (s, 6 H); 13C NMR δ 189.9, 149.9, 144.2, 129.4, 126.8, 120.7, 115.9, 25.8, 18.4, −4.2; HRMS calcd for C13H20ClO3Si (M+H+) 287.0870, found 287.0858. 5-Chloro-3,4-bis-(t-butyldimethylsilyloxy)benzaldehyde: 1H NMR δ 9.78 (s, 1 H), 7.50 (d, J = 2 Hz, 1 H), 7.29 (d, J = 2 Hz, 1 H), 1.05 (s, 9 H), 0.99 (s, 9 H), 0.27 (s, 6 H), 0.24 (s, 6 H); 13C NMR δ 190.1, 149.9, 149.4, 130.3, 128.0, 126.0, 119.0, 26.3 (3 C), 26.2 (3 C), 19.0, −3.2, −3.4; HRMS calcd for C19H34ClO3Si2 (M+H+) 401.1735, found 401.1747.
3-(tert-Butyldimethylsilyloxy)-5-chloro-4-methoxybenzaldehyde (27)
To a solution of 0.17 g (0.59 mmol) of compound 26 in 3 mL of dichloromethane at 0°C under argon, were added 0.26 g (1.2 mmol) of proton sponge and 0.18 g (1.2 mmol) of trimethyloxonium tetrafluoroborate and the mixture was stirred at 0°C for 8 h. The reaction mixture was diluted with water and extracted twice with diethyl ether. The combined extract was washed with water and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a mixture of hexane and diethyl ether (10:1) as eluant to give 0.35 (79% yield) of 27: 1H NMR δ 9.67 (s, 1H, CHO), 7.31 (d, J = 2 Hz, 1 H), 7.06 (d, J = 2 Hz, 1 H), 3.69 (s, 3 H), 0.81 (s, 9 H), 0.02 (s, 6 H); 13C NMR δ 190.1, 153.4, 150.9, 132.8, 129.8, 125.5, 119.9, 60.7, 25.8, 18.4, −4.4; HRMS calcd for C14H22ClO3Si (M+H+) 301.1027, found 301.1421; negative ion detection mode: C14H20ClO3Si (M-H) 299.0870, found 298.9978.
3-(tert-Butyldimethylsilyloxy)-5-chloro-4-methoxyphenyl formate (28)
To a solution of 0.21 g (0.70 mmol) of aldehyde 27 in 2 mL of dichloromethane under argon, was added 0.18 g (1.1 mmol) of m-chloroperbenzoic acid (70%), and the solution was heated to reflux for 8 h. The reaction solution was diluted with 5 mL of aqueous sodium thiosulfate and extracted twice with diethyl ether. The combined extract was washed with saturated aqueous NaHCO3, water, and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a mixture of hexane and diethyl ether (10:1) as eluant to give 0.16 g (71% yield) of 28. 1H NMR δ 8.23 (s, 1 H, OCHO), 6.83 (d, J = 2.6 Hz, 1 H), 6.59 (d, J = 2.6 Hz, 1 H), 3.81 (s, 3 H), 1.01 (s, 9 H), 0.21 (s, 6 H); 13C NMR δ 158.9, 150.7, 146.6, 145.4, 129.1, 115.8, 113.6, 60.7, 25.8 (3 C), 18.4, −4.5; HRMS calcd for C14H22ClO4Si (M+H+) 317.0976, found 317.1355.
3-(tert-Butyldimethylsilyloxy)-5-chloro-4-methoxyphenol (29)
A solution of 0.16 g (0.50 mmol) of compound 28 and 0.34 g (2.50 mmol) of K2CO3 in 5 ml of methanol was stirred at 25°C for 4 h and diluted with diethyl ether. The mixture was washed with water and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a mixture of hexane and diethyl ether (10: 1) as eluant to give 0.14 g (95% yield) of 29: 1H NMR δ 6.48 (d, J = 2.6 Hz, 1 H), 6.29 (d, J = 2.6 Hz, 1 H), 3.75 (s, 3 H), 1.01 (s, 9 H), 0.20 (s, 6 H); 13C NMR δ 152.0, 150.7, 142.6, 128.1, 110.8, 108.6, 60.7, 25.8 (3 C), 18.4, −4.5; HRMS calcd for C13H22ClO3Si (M+H+) 289.1027, found 289.1041.
2-Bromo-5-(tert-butyldimethylsilyloxy)-3-chloro-4-methoxyphenol (30)
To a solution of 90 mg (0.31 mmol) of 29 in 2 mL of DMF under argon was added 61 mg (0.34 mmol) of N-bromosuccinimide, and the solution was stirred at 25°C for 12 h. It was diluted with diethyl ether, washed with water and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a mixture of hexane and diethyl ether (10:1) as eluant to give 85 mg (74% yield) of 30: Mp. 44 – 45°C; 1H NMR δ 6.55 (s, 1 H), 5.42 (s, 1 H, OH), 3.77 (s, 3 H), 1.01 (s, 9 H), 0.20 (s, 6 H); 13C NMR δ 150.2, 149.6, 143.1, 124.2, 107.2, 103.0, 60.8, 25.8 (3 C), 18.5, −4.5; HRMS calcd for C13H21BrClO3Si (M+H+) 367.0132, found 367.0142.
4-Bromo-5-chloro-6-methoxybenzene-1,3-diol (6)
To a solution of 50 mg (0.14 mmol) of 30 in 2 mL of THF under argon at 0°C, was added 0.14 mL (0.14 mmol) of n-Bu4NF (1 M solution in THF), and the solution was stirred at 0°C for 30 min. It was diluted with dichloromethane, washed with water and brine, dried (MgSO4), concentrated, and column chromatographed on silica gel using a gradient mixture of dichloromethane and methanol as eluents to give 23 mg (67% yield) of 6. IR (neat) ν 3321 (bs), 3260, 2917, 1589, 1421, 1241, 984, 800; 1H NMR δ 6.65 (s, 1 H), 3.87 (s, 3 H); 13C NMR δ 150.5, 150.0, 138.7, 127.5, 101.8, 101.7, 61.5; HRMS calcd for C7H7BrClO3 (M+H+) 252.9267, found 252.9279.
2-Amino-4-methylphenol (7) and 2-amino-4-(hydroxymethyl)phenol (8)
A mixture of 0.30 g (1.8 mmol) of 4-hydroxy-3-nitrobenzaldehyde (31) and 0.15 g of 10% Pd/C in 30 mL of ethanol was shaken on a hydrogenator under 30 psi atmosphere of hydrogen for 4 h. The reaction mixture was filtered through Celite and carefully washed with ethyl acetate. The filtrate was concentrated and column chromatographed on silica gel using a mixture of dichloromethane and methane (9:1) as eluant to give 91 mg (41 % yield) of 7 [23] and 0.10 g (41% yield) of 8 [24]. Compound 7: IR (neat) ν 3370 (sharp, m), 3301 (sharp, m), 2921, 1601, 1519, 1458, 1388, 1286, 878, 800 cm−1; 1H NMR δ 6.62 (d, J = 7.6 Hz, 1 H), 6.58 (d, J = 1.6 Hz, 1 H), 6.48 (dd, J = 7.6, 1.6 Hz, 1 H), 2.21 (s, 3 H); 13C NMR δ 141.9, 134.4, 131.2, 120.0, 118.1, 115.4, 20.9. Compound 8: IR (neat) ν 3387 (sharp, m), 3313 (sharp, m), 3047 (broad), 2802, 1605, 1515, 1454, 1364, 1286, 1221, 1155, 1008, 816 cm−1; 1H NMR (D2O) δ 6.88 (d, J = 1.2 Hz, 1 H), 6.85 (d, J = 8 Hz, 1 H), 6.77 (dd, J = 8, 1.2 Hz, 1 H), 4.49 (s, 2 H); 13C NMR (DMSO-d6) δ 142.9, 136.1, 133.5, 114.9, 113.8, 113.4, 63.3.
2-(3,5-Di-tert-butyl-4-hydroxyphenyl)-2-methylpropanal (11)
A solution of 0.17 g (0.61 mmol) of 2,6-di-t-butyl-4-(1,1-dimethyl-2-hydroxyethyl)phenol (10) [25] and 0.21 g (0.73 mmol) of IBX in 3 mL of DMSO was stirred under argon at 25°C for 12 h, diluted with dichloromethane, washed with water and brine, dried (MgSO4), and concentrated to give 0.17 g (99% yield) of aldehyde 11. Spectral data and TLC indicated the compound is pure and was used in subsequent reactions without purification. IR (neat) ν 3603 (OH), 2952, 2900, 2700 (C-H aldehyde), 1714 (C=O aldehyde), 1435, 1360, 1230, 1141, 1120, 903, 826, 744; 1H NMR δ 9.45 (s, 1 H, CHO), 7.06 (s, 2 H, Ar), 5.22 (s, 1 H, OH), 1.44 (s, 24 H, Me); 13C NMR δ 202.8 (C=O), 153.1, 136.2 (2 C), 131.4, 123.6 (2 C), 50.4, 34.8, 30.4 (t-Bu), 22.7; MS negative mode: m/z 275.6 (M-1); positive mode: m/z 299.4 (M+Na+).
2-(3,5-Di-tert-butyl-4-hydroxyphenyl)-2-methylpropanoic acid (12) [19]
A silver oxide solution was prepared from 0.50 g (2.94 mmol) of silver nitrate and 0.24 g (5.88 mmol) of NaOH in 2 mL of water. To it, a solution of 0.16 g (0.59 mmol) of aldehyde 11 in 10 mL of 1,4-dioxane was added, and the solution was stirred at 25°C for 12 h. The reaction solution was acidified with 1 N HCl to pH ~ 2 and extracted with diethyl ether twice. The combined ether extract was washed with water and brine, dried (MgSO4), concentrated, and filtered through a short silica gel column to give 0.14 g (82% yield) of carboxylic acid 12 [19]. IR (neat) ν 3200 (v. broad), 2953, 2864, 1646 (s, C=O), 1593 (C=C aromatic), 1454, 1360, 1307, 1237, 918, 874 cm−1; 1H NMR δ 6.52 (s, 1 H, Ar), 1.29 (s, 18 H, t-Bu), 1.25 (s, 6 H, Me); 13C NMR δ 189.3, 187.9, 158.0 (2 C), 137.3, 130.3 (2 C), 35.7 (2 C, Me), 30.3, 29.5 (6 C, t-Bu), 22.4; MS m/z 293.2 (M+1).
2-(3,5-Di-tert-butyl-4-hydroxyphenyl)-2-methylpropanal oxime (13)
To a solution of 40 mg (0.14 mmol) of aldehyde 11 in 3 mL of acetonitrile and water (2:1), were added 25 mg (0.35 mmol) of NH2OH•HCl and 60 mg (0.43 mmol) of sodium acetate, and the solution was stirred at 25°C for 1.5 h. The reaction solution was diluted with water and extracted twice with diethyl ether. The combined extract was concentrated to give 41 mg (97% yield) of oxime 13: 1H NMR δ 7.49 (s, 1 H, CH=N), 7.12 (s, 2 H, Ar), 1.47 (s, 6 H, Me), 1.44 (s, 18 H, t-Bu); 13C NMR δ 158.7 (C=N), 152.5, 135.9, 135.7, 122.9 (2 C), 41.0, 34.7, 30.5 (6 C), 26.9; MS m/z 314 (M+Na+).
4-[(2-Benzylamino)-1,1-dimethylethyl]-2,6-di-tert-butylphenol (14)
A solution of 47 mg (0.17 mmol) of aldehyde 11 and 18 mg (0.17 mmol) of benzylamine in 2 mL of methanol was stirred under argon at 25°C for 12 h. To it, 16 mg (0.26 mmol) of NaCNBH3 was added, and the resulting solution was stirred for 2 h, diluted with water, and extracted twice with diethyl ether. The combined extract was washed with water and brine, dried (MgSO4), concentrated to give 60 mg of amine 14 (97% yield). 1H NMR δ 7.35 – 7.18 (m, 5 H, Ph), 7.13 (s, 2 H, Ar), 5.08 (broad s, 1 H), 3.72 (s, 2 H, CH2N), 2.68 (s, 2 H, CH2N), 1.43 (s, 18 H, t-Bu), 1.33 (s, 6 H, Me); 13C NMR δ 151.92, 140.4, 137.8, 135.4, 128.5, 128.1, 127.0, 122.7, 61.5, 54.2, 38.6, 34.7, 30.6, 27.9; MS m/z 368.5 (M+1).
2,4-Di-tert-butyl-6-(3-methyl-2-butenyl)phenol (15) and 6,8-di-tert-butyl-2,2-dimethyl-3,4-dihydro-2H-chromene (33)
To a cold (−78°C) solution of 0.12 g (1.4 mmol) of 2-methyl-3-buten-2-ol in 3 mL of dichloromethane under argon was added 0.20 g (1.4 mmol) of BF3 ether followed by 0.20 g (0.96 mmol) of 2,4-di-t-butylphenol. The solution was warmed to 25°C and stirred for 1 h, diluted with aqueous NaHCO3, and extracted twice with diethyl ether. The combined extract was washed with water and brine, dried (anhydrous Na2SO4), concentrated, and column chromatographed on silica gel using 1% diethyl ether in hexane as eluant to give 0.12 g (46% yield) of 15 and 26 mg (10% yield) of 33. Compound 15: 1H NMR δ 7.22 (d, J = 3 Hz, 1 H, Ar), 6.99 (d, J = 3 Hz, 1 H, Ar), 5.34 (t, heptet, J = 7, 1 Hz, 1 H, =CH), 3.37 (d, J = 7 Hz, 2 H, CH2), 1.85 (s, 3 H, Me), 1.80 (s, 3 H, Me), 1.43 (s, 9 H, t-Bu), 1.31 (s, 9 H, t-Bu); 13C NMR δ 151.5, 142.3, 135.9, 135.7, 126.0, 125.0, 122.6, 122.3, 35.1, 34.4, 31.9 (3 C), 31.7, 30.0 (3C), 26.0, 18.2; MS negative mode: m/z 273.8 (M-1).
Compound 33: 1H NMR δ 7.14 (d, J = 3 Hz, 1 H, Ar), 6.93 (d, J = 3 Hz, 1 H, Ar), 2.79 (t, J = 7 Hz, 2 H, CH2), 1.79 (t, J = 7 Hz, 2 H, CH2), 1.39 (s, 9 H, t-Bu), 1.36 (s, 6 H, Me), 1.30 (s, 9 H, t-Bu); 13C NMR δ 150.3, 141.0, 136.9, 124.2, 121.8, 119.9, 73.9, 35.2, 34.3, 33.0, 31.9, 30.0, 27.3, 23.5; MS positive mode: m/z 275.2 (M+1).
Cyclic voltammetry
Cyclic voltammetric experiments [8,27] were carried out with a three-electrode setup as described in General Methods. The glassy carbon electrode was polished with an alumina paste on a clean microcloth prior to the experiment. The electrochemical studies were performed in a 20% ethanol aqueous PBS buffer solution, and a concentration of 10 mM of the substrate was used. Ethanol is needed to dissolve some of the substrates. Experiments at pH 7.0 and 5.5 (acetic acid was added to adjust the pH to 5.5) were conducted. The solution was deoxygenated with nitrogen prior to the addition of the substrate. Cyclic voltammetric experiments for each substrate were carried out in triplicate and consistent results were obtained.
Laccase assays
Laccase purified from the fungus Trametes versicolor was purchased from Sigma (product #53739). Laccase was dissolved in 50 mM ammonium acetate (pH 5), 50% glycerol, and was stored at −20°C. We used peptide mass fingerprinting to determine that the most abundant protein in the laccase preparation was T. versicolor laccase IIIb, a 53 kDa enzyme (accession number AAL93622, data not shown). Product literature from Sigma stated that laccase was 10% of the total protein content. This was used to calculate the laccase concentration for determination of kcat values. The most abundant protein was laccase, which was confirmed by peptide mapping.
Extinction coefficients for the oxidized products of compounds 1, 3 – 8, 34, and 35 were estimated by fully oxidizing 100 μM compound with 25 ng laccase, using a spectrophotometer to measure absorbance of the product at a particular wavelength (see Table 1), and converting the units to M−1•cm−1.
Enzymatic reactions used to determine kinetic constants contained 0.58 nM laccase plus various concentrations of substrate in 100 mM sodium citrate, pH 5, in a volume of 200 μL. The substrate concentrations used were typically 0.01, 0.02, 0.1, 0.2, 0.4, 0.6, 1, 2, and 4 mM. A spectrophotometer was used to monitor product formation (using wavelengths listed in Table 1). GraphPad Prism 4 was used to fit data to the Michaelis-Menten equation or a substrate inhibition equation. The Michaelis-Menten equation is:
The substrate inhibition equation is:
Akaike’s Information Criterion was used to select the equation with a better fit to the data. The appropriate equation was then used to estimate two kinetic constants, Km and kcat.
Anti-Larval Assay
Mosquito
A laboratory colony of Anopheles gambiae was initially obtained from the Malaria Research and Reference Reagent Resource Center (MR4) (Manassas, VA) and was cultured in the Department of Entomology at Kansas State University (Manhattan, KS) based on the method of Benedict [41] with modifications [42,43].
Bioassay of Chemicals
The toxicities of the chemicals were screened using third-instar mosquito larvae at 25°C as described [43]. After a group of 12–15 larvae was transferred into a glass beaker containing 99.5 mL of distilled water and 0.5 mL of larval food [41], the stock solution of each chemical was added to the beaker to obtain final concentrations of 50 or 1000 μg/L. Each bioassay also included controls under the same conditions except that the same volume of acetone or water depending on the solvent used to dissolve the chemical was added to each beaker. All the treatments and controls were repeated four times and larval mortalities were assessed at 72 h.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (AI070864). We thank Ms. Sharon R. Starkey for her technical assistance in carrying out the anti-larval studies, Mr. Stewart Gardner for his technical assistance in carrying out some of the laccase activity assays, Dr. Yasuaki Hiromasa for peptide mass fingerprinting, and Dr. Sundeep Rana for technical assistance in the synthesis.
Footnotes
Supplementary Data. Table S1, Figures S1 – S4, and HPLC graphs of compounds 1 – 15 are available. Supplementary data associated with this article can be found, in the online version, at …
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kunamneni A, Camarero S, Garcia-Burgos C, Plou FJ, Ballesteros A, Alcalde M. Microbial Cell Factories. 2008;7:32. doi: 10.1186/1475-2859-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quaratino D, Federici F, Petruccioli M, Fenice M, D’Annibale A. Antnoie van Leeuwenhoek. 2007;91:57. doi: 10.1007/s10482-006-9096-4. [DOI] [PubMed] [Google Scholar]
- 3.Baldrian P. FEMS Microbiol Rev. 2006;30:215. doi: 10.1111/j.1574-4976.2005.00010.x. [DOI] [PubMed] [Google Scholar]
- 4.Rebrikov DN, Stepanova EV, Koroleva OV, Budarina AhI, Zakharova MV, Yurkova TV, Solonin AS, Belova OV, Pozhidaeva ZA, Leont’evsky AA. Appl Biochem Microbiol. 2006;42:564. [Google Scholar]
- 5.Smirnov SA, Koroleva OV, Gavrilova VP, Belova AB, Klyachko NL. Biochem. 2001;66:774. doi: 10.1023/a:1010216829856. [DOI] [PubMed] [Google Scholar]
- 6.Xu F, Deussen H-JW, Lopez B, Lam L, Li K. Eur J Biochem. 2001;268:4169. doi: 10.1046/j.1432-1327.2001.02328.x. [DOI] [PubMed] [Google Scholar]
- 7.Garzillo AMV, Colao MC, Caruso C, Caporale C, Celletti D, Buonocore V. Appl Microbiol Biotechnol. 1998;49:545. doi: 10.1007/s002530051211. [DOI] [PubMed] [Google Scholar]
- 8.Kramer KJ, Kanost MR, Hopkins TL, Jiang H, Zhu YC, Xu R, Kerwin JL, Turecek F. Tetrahedron. 2001;57:385. [Google Scholar]
- 9.Dittmer NT, Kanost MR. Insect Biochem Mol Biol. 2010;40:179. doi: 10.1016/j.ibmb.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Couto SR, Toca JL. Curr Enzyme Inhibition. 2006;2:343. [Google Scholar]
- 11.Johannes C, Majcherczyk A. J Biotech. 2000;78:193. doi: 10.1016/s0168-1656(00)00208-x. [DOI] [PubMed] [Google Scholar]
- 12.Paterson RRM, Meon S, Zainal Abidin MA, Lima N. Curr Enzyme Inhibition. 2008;4:172. [Google Scholar]
- 13.Lyashenko AV, Bento I, Zaitsev VN, Zhukhlistova NE, Zhukova Y, Gabdoulkhakov AG, Morgunova EY, Voelter W, Kachalova GS, Stepanova EV, Koroleva OV, Lamzin VS, Tishkov VI, Betzel C, Lindley PF, Mikhailov AM. J Biol Inorg Chem. 2006;11:963. doi: 10.1007/s00775-006-0158-x. [DOI] [PubMed] [Google Scholar]
- 14.Giardina P, Faraco V, Pezzella C, Piscitelli A, Vanhulle S, Sannia G. Cell Mol Life Sci. 2010;67:369. doi: 10.1007/s00018-009-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walton BT, Sanborn JR, Metcalf RL. Pesticide Biochem Physiol. 1979;12:23. [Google Scholar]
- 16.Semensi V, Sugumaran M. Pesticide Biochem Physiol. 1986;26:220. [Google Scholar]
- 17.Biggers WJ, Laufer H. Biol Bull. 2004;206:13. doi: 10.2307/1543194. [DOI] [PubMed] [Google Scholar]
- 18.Nishinaga A, Shimizu T, Matsuura T. Tetrahedron Lett. 1981;22:5293. [Google Scholar]
- 19.Lai JT. Tetrahedron Lett. 2001;42:557. [Google Scholar]
- 20.Hua DH, Huang X, Chen Y, Battina SK, Tamura M, Noh SK, Koo SI, Namatame I, Tomoda H, Perchellet EM, Perchellet J-P. J Org Chem. 2004;69:6065. doi: 10.1021/jo0491399. [DOI] [PubMed] [Google Scholar]
- 21.Godfrey IM, Sargent MV, Elis JA. J Chem Soc, Perkin Trans I. 1974:1353. [Google Scholar]
- 22.Barakat MZ, El-Wahab MFA, El-Sadr MM. J Am Chem Soc. 1955;77:1670. [Google Scholar]
- 23.Kikugawa Y, Tsuji C, Miyazawa E, Sakamoto T. Tetrahedron Lett. 2001;42:2337. [Google Scholar]
- 24.Touzeau F, Arrault A, Guillaumet G, Scalbert E, Pfeiffer B, Rettori P, Merour J-Y. J Med Chem. 2003;46:1962. doi: 10.1021/jm021050c. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz LH, Flor RV. J Org Chem. 1969;34:1499. [Google Scholar]
- 26.Masagutov RM, Tolstikov GA, Kirichenko GN, Grigor’eva NG, Tsypysheva LG. Neftekhimiya. 1985;25:481. [Google Scholar]
- 27.Kramer KJ, Nuntnarumit C, Aso Y, Hawley MD, Hopkins TL. Insect Biochem. 1983;13:475. [Google Scholar]
- 28.Brunet PC. J Insect Biochem. 1980;10:467. [Google Scholar]
- 29.Arakane Y, Muthukrishnan S, Beeman RW, Kanost MR, Kramer K. J PNAS. 2005;102:11337. doi: 10.1073/pnas.0504982102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hearing VJ, Jr, Ekel T, Montaque PM, Nicholson JM. Biochim et Biophys Acta. 1980;611:251. doi: 10.1016/0005-2744(80)90061-3. [DOI] [PubMed] [Google Scholar]
- 31.Hearing VJ, Jimenez M. Int J Biochem. 1987;19:1141. doi: 10.1016/0020-711x(87)90095-4. [DOI] [PubMed] [Google Scholar]
- 32.Bard AJ, Faulkner LR. Electrochemical Methods: Fundamental and Applications. 2. Wiley; New York: 2001. [Google Scholar]
- 33.Osman AM, Wong KKY, Fernyhough A. Biochem Biophys Res Commun. 2006;346:321. doi: 10.1016/j.bbrc.2006.05.118. [DOI] [PubMed] [Google Scholar]
- 34.Eggert C, Temp U, Eriksson K-EL. Appl Envir Microbiol. 1996;62:1151. doi: 10.1128/aem.62.4.1151-1158.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu PK-L, Abuzakhm S, Turro C. Photochem Photobiol. 2005;81:89. doi: 10.1562/2004-07-20-RA-237. [DOI] [PubMed] [Google Scholar]
- 36.Giurg M, Piekielska K, Gebala M, Ditkowski B, Wolanski M, Peczynska-Czoch W, Mlochowski J. Syn Commun. 2007;37:1779. [Google Scholar]
- 37.Xu F. Biochem. 1996;35:7608. doi: 10.1021/bi952971a. [DOI] [PubMed] [Google Scholar]
- 38.Xu F, Shin W, Brown SH, Wahleithner JA, Sundaram UM, Solomon EIA. Biochim Biophys Acta. 1996;1292:303. doi: 10.1016/0167-4838(95)00210-3. [DOI] [PubMed] [Google Scholar]
- 39.Tadesse MA, D’Annibale A, Galli C, Gentili P, Sergi F. Org Biomol Chem. 2008;6:868. doi: 10.1039/b716002j. [DOI] [PubMed] [Google Scholar]
- 40.Spafford H, Jardine A, Carver S, Tarala K, van Wees M, Weinstein PJ. Am Mosquito Control Ass. 2007;23:304. doi: 10.2987/8756-971X(2007)23[304:LDOEOA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 41.Benedict MQ. In: In The Molecular Biology of Insect Disease Vectors; Crampton JM, Beard CB, Louis C, editors. Chapman and Hall; London: 1997. pp. 3–13. [Google Scholar]
- 42.Zhu KY, Heise S, Zhang J, Anderson TD, Starkey SR. J Med Entomol. 2007;44:1047. doi: 10.1603/0022-2585(2007)44[1047:csoeot]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Zhu KY. Insect Biochem Mol Biol. 2006;36:712. doi: 10.1016/j.ibmb.2006.06.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.