

Abstract
Cryo-elecron microscopy (Cryo-EM) can provide important structural information of large macromolecular assemblies in different conformational states. Recent years have seen an increase in structures deposited in the Protein Data Bank (PDB) by fitting a high-resolution structure into its low-resolution cryo-EM map. A commonly used protocol for accommodating the conformational changes between the X-ray structure and the cryo-EM map is rigid body fitting of individual domains. With the emergence of different flexible fitting approaches, there is a need to compare and revise these different protocols for the fitting. We have applied three diverse automated flexible fitting approaches on a protein dataset for which rigid domain fitting (RDF) models have been deposited in the PDB. In general, a consensus is observed in the conformations, which indicates a convergence from these theoretically different approaches to the most probable solution corresponding to the cryo-EM map. However, the result shows that the convergence might not be observed for proteins with complex conformational changes or with missing densities in cryo-EM map. In contrast, RDF structures deposited in the PDB can represent conformations that not only differ from the consensus obtained by flexible fitting but also from X-ray crystallography. Thus, this study emphasizes that a “consensus” achieved by the use of several automated flexible fitting approaches can provide a higher level of confidence in the modeled configurations. Following this protocol not only increases the confidence level of fitting, but also highlights protein regions with uncertain fitting. Hence, this protocol can lead to better interpretation of cryo-EM data.
Keywords: Flexible fitting, Rigid fitting, X-ray structure, Electron microscopy, Protein data bank
Introduction
Cryo-electron microscopy (cryo-EM) has become a common tool for structure determination of large macromolecular assemblies (Campbell, 2002; Saibil, 2000; Sali et al., 2003) and more specifically to access different conformational states from which important insight into function can be gained (Frank, 2002; Henderson, 2004; Subramaniam and Milne, 2004). For example, cryo-EM has been successfully applied to the structural and functional elucidation of the ribosome (Frank and Agrawal, 2000; Valle et al., 2003a; Valle et al., 2003b), GRoEL (Falke et al., 2005; Ranson et al., 2001), RNA polymerase (Darst et al., 2002), myosin (Wendt et al., 2001) and viruses (Conway et al., 2001; Lee and Johnson, 2003). Since electron microscopy allows for the computational identification of structural heterogeneities within a sample population, its application to dynamic assemblies is gaining popularity.
Electron density maps obtained by cryo-EM techniques are of intermediate to low resolutions (usually 6–30 Å). However, a high-resolution structure can be modeled from the map by using atomic level information of the individual components from X-ray crystallography or NMR structures (Fabiola and Chapman, 2005; Rossmann et al., 2005) or by comparative modeling (Topf and Sali, 2005). This can be achieved by rigid body fitting, which performs a global search in six translation/rotation degrees of freedom to get the best fitting orientation of the structure in the map (Chacon and Wriggers, 2002; Volkmann and Hanein, 1999; Wriggers et al., 1999). Since Cryo-EM is often used to study different functional and conformational states, fitting becomes challenging when the map corresponds to a state in a considerably different conformation than the template structure used for the fitting. To deal with this issue, further refinement approaches that can account for the conformational changes between the structure and the map are required.
Traditional attempts to consider such conformational differences between the cryo-EM map and the template structure, divide a macromolecule into rigid parts (usually domains) and fit them independently as individual rigid bodies. These approaches have been successfully applied to different biological systems (Gao and Frank, 2005; Gao et al., 2003; Rawat et al., 2003; Volkmann et al., 2000; Wendt et al., 2001). However, such approaches might lead to a conformation inconsistent with the map since the collective and restricted nature of domain motions in macromolecules is ignored in these approaches. Independent fitting of domains only depends on the shape features, and not on the underlying energetics. Hence, at low resolution (~20 Å), the electron density may not have sufficiently distinctive features for an unambiguous placement of a component (Fabiola and Chapman, 2005). Furthermore, ad hoc partitioning of macromolecules into rigid parts is also subjective.
To move beyond rigid fitting, several automated flexible fitting approaches have been developed in recent years to account for molecular flexibility. Such flexible fitting approaches based on coarse-grained normal modes (NMA) have been very successful in quantitative flexible fitting (Delarue and Dumas, 2004; Hinsen et al., 2005; Suhre et al., 2006; Tama et al., 2004a; Tama et al., 2004b) and have been applied to different biological systems (Falke et al., 2005; Hinsen et al., 2005; Mitra et al., 2005; Suhre et al., 2006; Tama et al., 2006). Several “biased” molecular dynamics (MD) techniques have also been used for flexible fitting a high-resolution structure into its low-resolution cryo-EM map (Grubisic et al., 2009; Orzechowski and Tama, 2008; Trabuco et al., 2008). These MD based methods rely on a standard molecular mechanics force field that uses additional biasing forces to conform the structure to the cryo-EM map. Successful applications include the ribosome (Trabuco et al., 2009; Villa et al., 2009) as well as protein-induced membrane curvature (Hsin et al., 2009; Sener et al., 2009). For computational efficiency, a coarse-grained MD approach (Grubisic et al., 2009) has also been developed that uses Cα atom Go-model potentials (Clementi et al., 2000; Taketomi et al., 1975; Ueda et al., 1978), or all-atom Go-model potentials (Ratje et al., 2010; Whitford et al., 2009; Whitford et al., 2011). Another approach, YUP.SCX, uses a simplified potential energy function and simulated annealing to efficiently reach to the global minimum of the energy function (Tan et al., 2008).
Several other methods based on different computational methods also exist. Random-walk displacements with a simplified force field, elastic network models, and distance restraints have been implemented in DireX (Schroder et al., 2007). Monte Carlo and simulated annealing have also been incorporated into Flex-EM, which uses heuristic optimization for multilevel subdivisions of the structure from domain to secondary structure elements (Topf et al., 2008). Structural variability of protein domains within the same family can be considered for flexible fitting (Velazquez-Muriel and Carazo, 2007; Velazquez-Muriel et al., 2006). Finally, dynamics can be obtained by geometric simulation, which uses graph theory for identifying rigid and flexible regions in a structure (Jacobs et al., 2001; Jolley et al., 2008).
Diverse, quantitative and objective approaches for flexible fitting are now available. Until recently, rigid body fitting of individual domains was the commonly used approach (Fabiola and Chapman, 2005) to build models which are available in the Protein Data Bank (PDB) (Berman et al., 2000) while the corresponding density maps are available from the Electron Microscopy Data Bank (EMDB) (Henrick et al., 2003; Tagari et al., 2002). As structural information obtained from cryo-EM is increasing every year (Lawson et al., 2010), the trend of fitting of molecular structures into cryo-EM map needs validation and revision. Therefore important questions arise: Do models predicted by different approaches show consensus in their conformations? How reliable is rigid domain fitting (RDF)? Are automated flexible fitting approaches a more accurate alternative? The answers to these questions would certainly help to evolve a more robust and reliable protocol for flexible fitting and, thus, improve the quality of the fitted models.
In this study, we focus on single protein structures that require considerable conformational changes to fit into their cryo-EM maps. We have applied three diverse automated flexible fitting approaches on a small dataset of proteins for which models obtained from RDF have been deposited in the PDB. The flexible-fitted structures are subsequently compared with each other as well as with the corresponding RDF structure in the PDB. Recently, a X-ray crystal structure (Gao et al., 2009) of Elongation Factor G (EFG) bound to the ribosome was solved. For validation purpose, this structure is compared with EFG models derived from an electron density map (Valle et al., 2003a) using either automated computational or RDF approach. In general, a consensus in conformations is observed among the flexibly fitted structures, which is an indication of convergence to the most probable solution corresponding to the cryo-EM map by these approaches. In addition, RDF structures deposited in the PDB using rigid body fitting of individual domains may represent conformations that not only differ from the consensus obtained by flexible fitting but also from X-ray crystallography. Consensus found among several fitted conformations using automated flexible fitting approaches provides a robust alternative to RDF. Therefore we emphasize the need for multiple flexible fittings with different approaches as a standard protocol, in order to gain better understanding and confidence about the fitted structure.
Materials and Methods
General strategy and protein data set
To investigate consensus between computational approaches and the reliability of the models obtained from rigid body fitting of individual domains of their high-resolution structures into low-resolution cryo-EM maps, we refitted these structures using three diverse flexible fitting approaches. The flexible fitted structures were compared with each other and with the rigid domain fitted structures (RDF structure) deposited in Protein Data Bank (PDB) (Berman et al., 2000).
To obtain a dataset of single protein structures for the analysis, the PDB was searched for structures deposited with experimental method as “Electron Microscopy” and with molecular type as “Proteins” and not as “DNA/RNA”. This search query resulted in a list of 184 PDB entries as of March 2010. Since this study focuses on single protein structures, structures with viruses and proteins in oligomeric states (142) were rejected. After discarding these structures, the remaining 42 PDB entries were then cross-referenced with all of the available entries in the Electron Microscopy Data Bank (EMDB; 719 entries on the day) in order to search for corresponding EM maps. This resulted in a list of 19 unique PDB and EMDB entries matches (the list is provided in the Supporting Information). Initial PDB files used for the RDF fitting were also identified. Since this study focuses on the conformational differences between fitted structures, 10 entries were discarded based on small or no conformational differences (< 4 Å rmsd) and one entry (PDB ID: 2BK1) was discarded due to complex conformational changes involving secondary structure rearrangements between the initial and RDF structures. For the case where isolated protein densities were not available, they were extracted from the complex densities if the RDF structure was already fitted in the cryo-EM map (i.e., for EMDB ID: 1302 and 1343). Otherwise, (i.e., in three cases) the pair was discarded. This resulted in five single proteins with a large conformational change for which both EM maps, initial X-ray structure and a RDF atomic model were available through the EMDB and PDB. The proteins in the dataset and related information are given in Table 1.
Table 1.
A list of the studied proteins.
| Protein | Initial PDBa) | RDF PDBb) | RMSD (Å)c) | No. of residues | EMDBd) | Resolution (Å)d) | Residues in domainse) |
|---|---|---|---|---|---|---|---|
| Release factor 2 (RF2) | 1GQE | 1MI6 | 16.7 | 362 | 1010 | 12.8 | I: 4–124; II: 125–225; III: 226–320; IV: 321–365; |
| Elongation factor 2 (EF2) | 1N0U | 3DNY | 10.2 | 654 | 5017 | 12.6 | G′, I, II: 3–48, 67–480; IV: 560–595, 640–726; V: 727–759, 763–800. |
| Elongation factor G (EFG) | 1FNM | 1PN6 | 9.5 | 655 | 1364 | 10.9 | I: 6–39, 68–284; II: 285–403; III: 404–482; IV: 483–603, 676–688; V: 604–675. |
| Release factor 3 (RF3) | 2H5E | 2O0F | 7.2 | 488 | 1302 | 15.5 | I: 3–38,70–278; II: 279–350, 356–387; III: 388–406, 410–529. |
| Elongation factor 2 (EF2′) | 1U2R | 2P8X | 9.9 | 814 | 1343 | 9.7 | G′, I, II: 3–48, 67–481; III: 482–558; IV: 559–724, 801–842; V: 730–759, 763–800. |
Starting structures used for flexible fitting.
Structures reported in the PDB by rigid body fitting of individual domains of the initial structures into the corresponding cryo-EM maps.
RMSD between initial and RDF structures.
Target cryo-EM maps obtained from the Electron Microscopy Data Bank (EMDB).
Domains that do not show relative movements are grouped together for simplicity. A small patch of underlined residues was not structurally close enough to be considered as part the domain for RMSD and rotation/translation calculations.
Different flexible fitting approaches are used to build models and compare them with RDF structures deposited in the PDB. In this manuscript, we focus on fully automated flexible fitting methods therefore only methods that do not require partitioning of the molecule (i.e. domains or secondary structure segmentation) were chosen. In addition, techniques differing in their underlying framework, computational algorithms, and protein models to describe protein dynamics were used. The following three approaches were chosen: normal mode flexible fitting (NMFF) (Tama et al., 2004a; Tama et al., 2004b), All-atom Structure-based molecular dynamics flexible fitting (MDfit) (Grubisic et al., 2009; Whitford et al., 2009) and flexible fitting based on simulated annealing optimization (YUP.SCX) (Tan et al., 2008). It is important to mention here that these methods do not represent all of the available flexible fitting techniques (Grubisic et al., 2009; Jolley et al., 2008; Orzechowski and Tama, 2008; Schroder et al., 2007; Topf et al., 2008; Trabuco et al., 2008); nevertheless they are diverse in their theory.
The starting structures (i.e., initial structures) and the cryo-EM maps used for flexible fittings are given in Table 1. Since flexible fitting using these approaches requires a suitable guess of the fitted orientation into the map, an initial rigid body fitting of the structures was performed. The flexible-fitted structures from these approaches and RDF structures from PDB were compared in terms of overall RMSD between the structures. Differences in domain orientations between fitted structures were calculated by first RMS fitting two structures from the three different approaches and then calculating the rotation/translation needed to fit individual domains. For validation, a recently deposited X-ray structure (Gao et al., 2009) of elongation factor G (EFG) at 3.6 Å resolution is also compared with flexible-fitted and RDF structures.
Initial rigid body fitting
The Situs package (Wriggers et al., 1999) was used for the initial rigid body fitting of the initial structures into the respective cryo-EM maps. In this fitting, 11 codebook vectors were used to represent a PDB structure and a cryo-EM map. The highest-ranked model from Situs was then used to start the flexible fitting (the initial structure). Subsequently, refinement procedures were needed to improve the fitting by taking into account the conformational differences between the structure and the map. The three different refinement methods used in this study are briefly described below.
Normal mode flexible fitting (NMFF)
To flexibly fit a structure into its map, NMFF recursively deforms the structure along a linear combination of low-frequency normal modes. An elastic network model (ENM) (Bahar et al., 1997; Hinsen, 1998; Tama and Sanejouand, 2001) is used for efficient normal mode calculations, which uses a simplified representation of the system and its potential energy function. For building ENM, every pair of Cα atoms within a cutoff distance (i.e., 8 Å) is connected by a Hookean spring of certain strength (i.e., 1 kcal mol−1 Å−2). In this study, the first 20 low-frequency modes were used for the fitting, including the six zero-frequency translational/rotational modes for refining the overall orientation of the structure.
The quality of fitting is measured by the correlation between the calculated electron density map of the structure being fitted and the experimental density map from EM (Tama et al., 2004a; Tama et al., 2004b). One can use the correlation coefficient (CC) to measure the agreement between the simulated map (from the structure being fitted) and the experimental density map. During fitting, the objective is to maximize the CC following the normal mode directions. In order to determine the optimal amplitude and phase of a mode l in a linear combination that increases the CC, the gradient of CC is calculated with respect to the amplitude ql, Fl = ∂CC/∂ql. Since displacement of atoms in normal mode directions causes local distortions in the structure, a suitable displacement is needed that expedites convergence while minimizing local distortions. The size of the displacements is set to be proportional to the gradient, ql = λFl. Here, λ is the parameter that controls the distortions in the structure during refinement iterations and it is chosen such that the maximum displacement of any atom during any iteration of refinement is less than a cutoff value, i.e., 1 Å in this study. In order to maximize the CC, the optimization procedure starts with the steepest decent and switches to the Newton-Raphson method as the solution approaches a local maximum.
All-atom structure-based molecular dynamics flexible fitting (MDfit)
Several “biased” molecular dynamics (MD) simulation techniques have been used for flexibly fitting a high-resolution structure into low-resolution EM map (Grubisic et al., 2009; Orzechowski and Tama, 2008; Trabuco et al., 2008). These approaches mainly differ in the form of the biasing potential that is applied. In this study, we used a MD approach based on structure-based (i.e. Go-like) potentials (Grubisic et al., 2009). Initially structure-based models employed a lattice representation of the protein (Taketomi et al., 1975; Ueda et al., 1978); however, subsequent models employed an off-lattice representation of the Cα atoms (Clementi et al., 2000; Karanicolas and Brooks, 2003), which has recently been extended to accommodate all-atom based interactions (Whitford et al., 2009).
Using a structure-based model for the template configuration, the MD is then biased to the EM density by modifying the force field potential, V, to include the sum of a classical (structure-based) force field, Vff, and an effective potential VFit. The additional effective potential is a function of the correlation coefficient (CC), VFit = k(1 − CC). Here, k is the constant that regulates the magnitude of the effective potential and determines the degree of biasing for the MD simulation. Since, the structure-based force field includes 1 unit of stabilizing energy per heavy atom in the system (Whitford et al., 2009), the constant k is set equal to the number of heavy atoms in the protein being fit. The input topology files for the simulations were obtained using the SMOG@ctbp web-server (Noel et al., 2010; Whitford et al., 2009). Default parameters, as on the server, are used. Reduced units were used for all calculations, as described previously (Whitford et al., 2010). Each simulation was performed for 100,000 timesteps of size 0.0005. We employed a reduced temperature of 0.083 (10 in gromacs units) during fitting. Since, in this model, the folding temperature is typically around 1 (Noel et al., 2010; Whitford et al., 2009), the secondary and tertiary structure encoded in the initial configuration remains stable during the fit. The best-fit structure in the trajectory, as given by the highest correlation coefficient with the density map, was used in this study. It is important to note that the best-fit structure is a good representative of the best-fit ensemble of the MDFit and the conformational variability with in the best-fit ensemble is not more than 0.5 Å Cα-atoms RMSD.
YUP.SCX flexible fitting
YUP.SCX (Tan et al., 2008) is another refinement procedure for fitting molecular structure into cryo-EM maps and has been implemented as part of YUP (Tan et al., 2006), a general-purpose molecular mechanics program. YUP.SCX, in contrast to the above two approaches, does not use the gradient of correlation coefficient (CC) in order to steer simulation. Instead, YUP.SCX translates the refinement problem into a simplified potential energy function and uses Simulated Annealing (SA) to efficiently reach to the global minimum of the energy function. The potential energy function contains three terms; a SCX (SwissCheeseX) term, a Hooke term, and a SSX (SoftSphereX) term. The purpose of the SCX term is to steer the atoms of the structure into the density map. The Hooke’s term is used, in place of conventional molecular mechanics, to approximately restrain steriochemistry for the structure. A modified all-atom based elastic network model (ENM) is built where cut-off lengths and strengths of the network connections are varied according to atom types. The SSX term is used to maintain non-bonded separation (the sum of the van der Waals radii) between pairs of atoms that are not linked by the ENM. The total potential energy is the sum of the three terms. The global minimum of the function will be at the optimal refolding of a stereochemically correct structure into the target map.
The overall procedure in YUP.SCX is divided into 10 intervals. The system is heated to a maximum temperature (10 K by default) over four intervals, kept at the maximum temperature over one interval and then cooled down to a low temperature (1 K by default). The overall duration of the procedure is the number of atoms rounded to the nearest 1000, times the stepfactor (stepfactor = 2 is used in this study in order to run the method large enough to reach solution, default is 0.5). All other parameters are kept at their default values. The step size is set at default 5 fs. The radius ratio, which affects the range of each well and smoothness of the objective function, is also kept at its default value of 4. YUP.SCX fitting was performed using the latest available version 1.080613.
The three methods used in this study rely on different computational and optimization techniques, i.e. normal mode analysis and molecular dynamics. The potential energy functions that model the intramolecular interactions are also different, elastic network Cα based model and two all atom models types; however, it should be noted that these biased molecular dynamics simulations are not sensitive to the choice of the force fields, since the conformational transition is guided by the cryo-EM density map and not by the force fields (Grubisic et al., 2009). In addition, for each of these approaches, no partitioning or additional constraint on the protein structure is needed. Comparison of models obtained from this diverse set of techniques should provide useful information on the validity and accuracy of these models.
Results and discussion
Consensus in the models from different flexible fitting approaches
Despite the fact that the three flexible fitting approaches are diverse in their underlying techniques, in general a consensus is found among the flexible-fitted structures. Figure 1 shows Cα-atoms RMSD’s among the fitted structures. In three cases, fitted structures from all three approaches are only, on average, ~2.4 Å RMSD away from each other. MDfit and YUP.SCX fitted structures of EF2′ are as near as 1.4 A of RMSD. These values are encouraging considering that, not only are the required conformational changes large (on average ~9 Å, Table 1), but also the initial structures, in some cases, needed to undergo a closed-to-open transition in order to fit into the EM maps. It has been shown previously that NMFF is less accurate in describing the closed-to-open transitions due to inadequate description of normal modes (Grubisic et al., 2009; Tan et al., 2008). This is seen in two cases (EFG and EF2′, Figure 1) out of the three cases described above. The NMFF structures of EFG and EF2′ show relatively higher RMSD with the other two approaches. Despite this NMFF limitation, a consensus is found in the three cases with all these approaches. In addition, better consensus appears to be achieved with higher resolution maps (EF2′ at 9.7 A resolution). This would not be surprising as each of these methods is more accurate at higher resolution (Tama et al., 2004a; Tama et al., 2004b; Tan et al., 2008). However, due to the limited dataset presented in this study, this observation would need a more throughout validation on a larger dataset using simulated maps from known X-ray structure.
Figure 1.
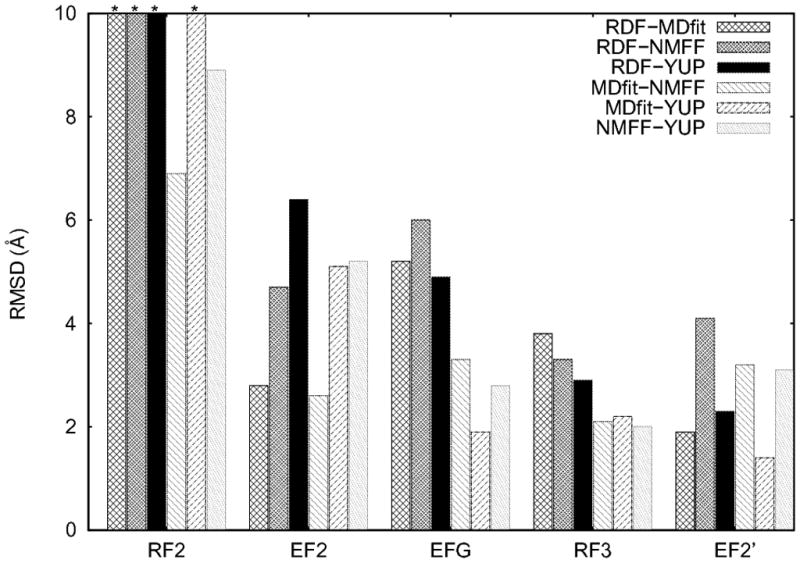
RMSD between different RDF/flexible-fitted structures of the four studied proteins obtained from different flexible-fitting approaches (MDfit, YUP.SCX, and NMFF) and from the PDB database fitted using rigid -body fitting (RDF) into Cryo-EM maps. For RF2, bars with * on the top have RMSD values > 10 Å (i.e., 14.6, 15.6, 17.8 and 11.6 Å between RDF and MDfit, RDF and NMFF, RDF and YUP, and MDfit and YUP, respectively).
To get a better understanding of the conformational variation in the fitted structures, differences in domain orientations between the structures were calculated in terms of rotation/translation. In the case of RF3, all three structures are very similar (Figure 2) and show consensus among all the three fitted domain orientations (Table 2; complete data for all domains is provided in Table S5 in the Supporting Information). Smaller rotation/translation values (average over 9 values is ~6/1 degrees/Å) are observed in all the three RF3 domains of the three pairs of structures from different approaches. Similar consensus is also found in the EFG and EF2′ cases, however, domain III and V of the NMFF fitted structures have different relative orientations than the fitted structures from the other two approaches. For example, in EF2′ the rotation/translation needed for fitting domain III between NMFF structure and MDfit or YUP.SCX structure is ~32/4 degrees/Å, whereas these values are only ~2/1 between MDfit and YUP.SCX structures (Table 2). As discussed above, this small variation in domain orientations is due to the limitations of NMFF in describing conformational changes that require closed-to-open transitions. Interestingly, rotation/translation values show that only domain III and V are affected. This can be explained by the fact that these domains are in close contact with domain I/II of EFG and EF2′, which restricts the relative movements of these domains in NMFF. Furthermore, the largest moving domain (domain IV) in the NMFF fitted structure, where the tip of the domain (residue Val 575 in EFG) moves by approximately 34 Å, is oriented in consensus with the other two structures (Table S5).
Figure 2.
A comparison of rigidly fitted (RDF, red, PDB: 2O0F) and flexibly fitted (MDfit, green; NMFF, blue; YUP.CSX, cyan) structures of release factor 3 (RF3) after aligning domain I with the RDF structure.
Table 2.
Rotation (translation) differences in the selected domains between the fitted structuresa)
| Protein domains | RDF b) |
MDfit
|
NMFF
|
|||
|---|---|---|---|---|---|---|
| MDfit | NMFF | YUP.SCX | NMFF | YUP.SCX | YUP.SCX | |
| EF2 | ||||||
| IV | 17.7 (3.2) | 37.1 (5.0) | 30.2 (7.3) | 23.4 (1.9) | 17.1 (4.5) | 25.5 (3.5) |
| V | 18.6 (3.1) | 38.5 (5.0) | 28.5 (7.3) | 27.1 (2.5) | 16.0 (5.7) | 15.2 (3.6) |
| EFG | ||||||
| II | 18.7 (2.0) | 23.8 (2.9) | 20.9 (2.5) | 5.6 (1.1) | 2.5 (0.6) | 4.7 (0.8) |
| V | 97.3 (2.2) | 87.8 (6.2) | 79.1 (2.3) | 21.5 (5.2) | 19.2 (1.7) | 15.8 (4.1) |
| RF3 | ||||||
| II | 20.9 (2.1) | 18.2 (2.2) | 16.0 (1.6) | 2.6 (1.4) | 6.7 (1.7) | 4.9 (1.0) |
| III | 16.2 (2.2) | 11.3 (1.3) | 4.3 (1.9) | 9.6 (1.2) | 12.5 (0.7) | 9.4 (0.8) |
| EF2′ | ||||||
| III | 17.3 (1.9) | 46.5 (5.3) | 16.3 (2.2) | 31.9 (4.2) | 1.8 (0.9) | 31.7 (3.4) |
| V | 7.3 (1.6) | 30.9 (6.8) | 17.8 (2.4) | 29.4 (5.4) | 13.7 (1.1) | 17.7 (4.7) |
Rotations (translations) differences in domains orientations between two fitted structures in degrees (Å) calculated after removing the overall transitional/rotational degrees of freedom by root mean square fitting the two structures. These are the rotations and translations needed for the domains to fit them individually to accommodate the conformational changes of domain orientations between the two structures. Rotation (translation) values > 20° (> 4 Å) are in bold and values > 15° (> 3 Å) are underlined. A complete rotation/translation table containing all domains in the proteins is provided in the Supporting Information (Table S5).
Structures reported in the PDB by rigid body fitting of individual domains of the initial structures into the corresponding cryo-EM maps.
These results show that understanding the strengths and limitations (Table S7) of different approaches can help to resolve the disagreements among fitted structures. For example, NMFF is known to work relatively well with low-resolution (>15 Å) EM maps; however, it is less accurate in fitting structures that require a closed-to-open transition (Lindert et al., 2009; Tama et al., 2004a; Tama et al., 2004b). YUP.SCX is tailored towards medium-resolution (6–15 Å) EM maps; however, since it uses a global optimization approach, it has a better chance of attaining the global minimum of the solution (Tan et al., 2008). Additionally, both MDFit and YUP.SCX generate good quality structures when compared to the initial X-ray structure (NMFF generates only Cα atoms models; Table S6).
Limitations in the models from different flexible fitting approaches
While a consensus between models was obtained, we also found cases for which not all the three approaches agree. For EF2, because of missing densities corresponding to domain III and IV insert region (residues 481–559 and 801–842) in the map (12.6 Å resolution), these domains were excluded from the RDF structure, reported by Sengupta et al. (Sengupta et al., 2008). As ignoring domains might affect the dynamics and hence the fitting of the structure, we performed flexible fitting with and without the domains with missing densities (domains III and IV insert region). The RMSD between flexibly fitted models are shown in Table 3 for both cases. The NMFF fitted structure is considerably different (~4.5 Å) which is due to a change in the normal modes when the additional domains are included. In contrast, MDfit and YUP.SCX fitted structures do not show considerable differences (0.6 and 1.7 Å, respectively), which indicates that the allowable modes of motion with these methods are less sensitive to the initial structure of EF2.
Table 3.
RMSD (Å) between RDF/flexible-fitted structures for EF2 with and without the domains having missing densities in the cryo-EM map.
| Proteins | RDFa) |
MDfit
|
NMFF
|
|||
|---|---|---|---|---|---|---|
| MDfit | NMFF | YUP.SCX | NMFF | YUP.SCX | YUP.SCX | |
| EF2 (domain III and IV insert region excluded) | 2.5 | 6.0 | 6.4 | 4.9 | 5.4 | 2.9 |
| EF2 (domain III and IV insert region included ) | 2.8 | 4.7 | 6.4 | 2.6 | 5.1 | 5.2 |
The RDF structure reported in the PDB by rigid body fitting of individual domains of the initial structure into the corresponding cryo-EM map.
Since removing domains can considerably affect the flexible fitting results, for further analysis in this study we used results obtained from flexible fittings of EF2 including the domain III and IV insert region (Figure 1 and Table 2). Interestingly, the NMFF fitted structure when including these domains agrees well with the MDfit fitted structure (RMSD ~2.6 Å, Table 3) while the YUP.SCX fitted structure does not agree with the other two flexible fitted structures. Assuming consensus in these two approaches is an indication of the most probable solution, it shows that YUP.SCX is more sensitive to the provided density maps and does not work as well when some densities are missing. In contrast, MDfit is most robust among the three approaches. Despite missing densities for some domains, consensus is found between MDfit and NMFF fitted structures. This example illustrates a limitation that might be inherent in the flexible fitting techniques. Cryo-EM maps are more difficult to interpret with missing densities, as dynamics of biomolecules will be affected by removing domains during the fitting. For a more accurate modeling of the conformational change all domains should be included in the initial structure.
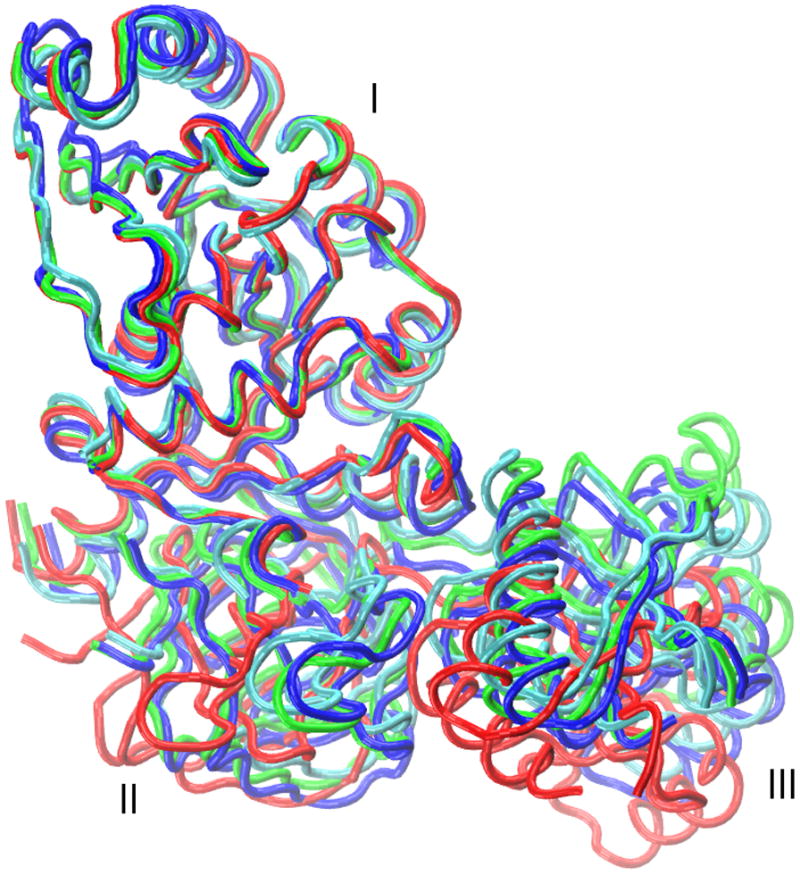
Finally, in the case of RF2, no consensus is found between the fitted models. A closed-to-open transition of ~17 Å is required to fit the initial structure into the cryo-EM map (12.8 Å resolution) such that domain III is peeled away from domains II & IV and domain I is also considerably shifted away, according to the RDF model (Figure 3). The initial Situs fitting into the map oriented the initial structure quite differently than the fitted RDF model. Considering the large conformational differences between the two states with completely different shape features, it is not surprising that rigid protein fitting failed to optimally orient the initial structure into the map. Therefore fitting was also performed using the initial structure in the RDF aligned orientation. Either way, this complex transition could not be achieved using any of the three flexible fitting approaches. Despite protein deformation of flexibly fitted structures (Table S2 and S3) good fits with the cryo-EM map were not observed (Figure 3). Although in each case the correlation coefficients (CC) increase (Table S4), values are lower than when a consensus is achieved. Large RMSD values between all flexibly fitted structures are also observed (Figure 1). While domain I seems to fit reasonably well, (Figure 3), domain III could not be peeled away from the two interacting domains II & IV, even by removing all contacts between domains that might obstruct the opening using MDFit (data not shown). There have also been discussions on whether the closed structure of RF1 and RF2 reflect functional states of the factors or if they occur in crystals only (Vestergaard et al., 2005; Weixlbaumer et al., 2008; Zoldak et al., 2007). In the later case, the inability of the computational approaches to describe the conformational change might be due to a faulty initial X-ray structure.
Figure 3.

A comparison of initial (yellow, PDB: 1GQE), rigidly fitted (RDF, red, PDB: 1MI6) and flexibly fitted (MDfit, green) structures of release factor 2 (RF2). A closed-to-open transition of ~17 Å is required to fit the initial structure into the cryo-EM map (12.8 Å resolution) such that the domain III is peeled away from domains II & IV and the domain I is also considerably shifted away, according to the RDF model. This complex transition could not be achieved using any of the three flexible fitting approaches.
Consensus in the flexible fitting approaches, despite being theoretically different, point towards the most probable solution corresponding to the Cryo-EM map. As described above, disagreement between the approaches reflects their limitations. Furthermore, the data presented here emphasize the need for multiple flexible fittings with different approaches, in order to gain better understanding and confidence about the fitted structure. The strengths and limitations of the three approaches used in this study are summarized in Table S7 based on our experiences and literature review. The results discussed above show that a general consensus among the fitted structures can be expected by the different approaches and disagreements can be explained. The extent to which these consensus fittings agree with RDF is discussed below.
Comparison between flexibly fitted structures and rigidly fitted structures deposited in the PDB
Flexibly fitted structures obtained from the three different approaches are also compared with structures deposited in the PDB for which individual domains were separately and rigidly fitted in the cryo-EM map (the RDF structures). For four proteins (excluding RF2), the average RMSD between a RDF structure and a flexibly fitted structure is ~4.0 Å (Figure 1). In contrast, average RMSD between a pair of flexibly fitted structures is only ~2.9 Å. This shows in general that the consensus, in conformation, found within the flexibly fitted structures is not seen when these structures are compared with the RDF structures. In the two cases where a consensus among all the three flexible fitting approaches is observed (EFG and RF3), disagreement between the flexible fitted structures and the RDF structures is prominent. On average, the three flexibly fitted structures of EFG and RF3 are ~5.4 and ~3.3 Å away from corresponding RDF structures, respectively (Figure 1). Table 2 shows that the disagreements between the RDF and the flexibly fitted structures mainly come from domain II and V in EFG and domain II in RF3. An extremely high value of rotation/translation (~88/3.5 degrees/Å, on average) for domain V in EFG between the flexibly fitted structures and the RDF structure shows that this domain is incorrectly oriented in the RDF structure. This has been pointed out earlier by Grubisic et al. (Grubisic et al., 2009) and Velazquez-Muriel et al. (Velazquez-Muriel et al., 2006). The differences in domain orientations of the RDF and the flexibly fitted structures in EFG and RF3 also comes from domain II, which shows a rotation/translation of ~21/2.5 and ~18/2.0 degrees/Å (on average), respectively. It should be noted that, in the EFG RDF structure (PDB ID: 1PN6) domains I and II were considered as a single rigid body, with the assumption that relative movement is not required (Valle et al., 2003a). This assumption seems to be in contradiction with the flexibly fitted structures. Furthermore, disagreements between RDF and flexibly fitted structures are also found in relative movements of domains III–V with respect to domains I–II for EFG (Figure 4a) and domains II–III with respect to domain I for RF3 (Figure 2). This can be seen when the RDF and flexibly fitted structures of EFG and RF3 are aligned with respect to domains I–II and domain I, respectively (Figure 4a and Figure 2).
Figure 4.

A comparison of the rigidly fitted structure (RDF, red, PDB: 1PN6), the flexibly fitted structures (MDfit, green; NMFF, blue; YUP.CSX, cyan), and the X-ray structure (PDB: 2WRI, magenta) of elongation factor G (EFG) after aligning domains I–II with the X-ray structure. (a) Flexible-fitted structures show a consensus in the fitting, which is significantly different from the RDF structure. (b) The flexibly fitted structures are also near to the crystal structure in comparison to the RDF structure. Domain V of EFG, which shows the largest orientation differences between the flexibly fitted and the RDF structures, is zoomed in on the right.
In the case of EF2 – in which MDfit and NMFF fitted structures agree (Figure 1) – the RDF structure agrees with MDfit fitted structure with an RMSD ~2.9 Å, but not with NMFF fitted structure (RMSD ~4.7 Å). Table 2 shows that these differences in orientations come from domains IV and V. As discussed above, densities for the domain III and IV insert region are missing in the EF2 map (12.6 Å resolution), which might affect the flexible fitting results. Moreover, as shown for the EFG and RF3 cases above, rigidly fitting individual domains might lead to an unrealistic conformation. As backbone connectivity and excluded volume are not accounted for in RDF, correlation between domain motions is not accounted for. Therefore, rigid fitting results in the EF2 case should be interpreted with caution. In contrast, EF2′ can be regarded as the only instance in which the RDF structure agrees with the consensus found in flexible fitting (except for NMFF). The RDF structure is only ~1.9 and ~2.3 Å away from MDfit and YUP.SCX fitted structures. As pointed out earlier, NMFF is less accurate in describing the conformational changes in EF2′, as it requires a closed-to-open transition to fit into the cryo-EM map.
The results presented above indicate that the RDF structures deposited in the PDB can differ from the consensus obtained by flexible fitting approaches, as observed for EFG and RF3. This disagreement is probably due to the uncorrelated fittings of the domains in RDF. This is also supported by the observation that the RDF structures, compared to the flexibly fitted structures, are relatively far in term of RMSD from the initial structures in all the cases (Table S2). The collective and restrictive nature of domain motions in macromolecules is completely ignored in RDF and, hence, might lead to a conformation that is invalid or inaccessible with respect to the underlying conformational transition.
Comparison of EFG models with a known X-ray structure
The consensus found among the diverse flexible fitting approaches is a good indication that the fitted structures represent the most probable solution corresponding to the cryo-EM map. Yet, comparison to high-resolution structures, in the same conformational state as observed in the EM map, would be valuable to validate these methods. Recently, a high-resolution crystal structure (3.6 Å resolution) of ribosome bound EFG was reported in its posttranslocation state using the antibiotic fusidic acid (70S•EFG•GDP•fusidic acid) (Gao et al., 2009). Fusidic acid allows GTP hydrolysis and translocation by EFG but prevents its release from the ribosome. Here we compare the X-ray structure (PDB ID: 2WRI) with the RDF structure (PDB ID: 1PN6) as well as the three different flexibly fitted structures of EFG in order to investigate the differences between RDF and flexibly fitted structures. It should be noted that the cryo-EM map (10.9 Å resolution) used for the EFG fitting is GTP-bound (70S•EFG•GDPNP, where GDPNP is a non-hydrolysable GTP analog) (Valle et al., 2003a), in contrast the crystal structure used for the comparison is GDP-bound. However, apart from minor relative domain movements, no drastic conformational changes have been reported between the EFG structures before and after GTP hydrolysis (Frank et al., 2007; Li et al., 2011; Taylor et al., 2007).
Overall, the crystal structure is nearer to all the flexibly fitted structures than the RDF structure (Figure 4). Cα-atom RMSDs between the crystal structure and the MDfit, YUP.SCX and NMFF fitted structures are 2.8, 3.1 and 4.0 Å, respectively. In contrast the RMSD between the crystal structure and the RDF structure is 5.4 Å. Interestingly, the domain V orientation predicted by the flexibly fitted structures, which results in the largest disagreement with the RDF structure, agrees very well with the crystal structure. Table 4 shows domain orientation differences between the crystal structure and RDF/flexible-fitted structures. A difference in rotation of ~100° is observed in domain V between the RDF and crystal structures, whereas this value is ~16° on average for the three flexible fitting structures and as low as ~6° in the MDfit fitted structure. The orientation of domain V in MDfit fitted structure and the RDF structure along with the crystal structure is emphasized in Figure 4b. The large differences in domain V of the RDF structure are clearly seen. This is in agreement with a recent study that elucidated conformational changes between the two states using a similar flexible fitting by a MD-based approach (Li et al., 2011). This further supports the improbable orientation of domain V in the RDF structure.
Table 4.
Rotation (translation) differences in elongation factor G (EFG) domains between the X-ray structure and the RDF/flexible-fitted structures.
| Domains | X-ray structurea) |
|||
|---|---|---|---|---|
| RDFb) | MDfit | NMFF | YUP.SCX | |
| I | 12.2 (1.4) | 3.8 (1.3) | 3.6 (0.9) | 3.4 (1.1) |
| II | 12.8 (1.9) | 7.0 (1.2) | 11.9 (1.4) | 8.5 (1.5) |
| III | 13.9 (1.1) | 22.6 (1.7) | 34.6 (3.3) | 17.5 (1.2) |
| IV | 11.7 (2.4) | 8.8 (2.1) | 14.6 (1.7) | 10.8 (1.7) |
| V | 100.5 (3.9) | 6.3 (2.7) | 19.8 (3.0) | 21.1 (2.3) |
Rotations in degrees (translations in Å) needed to align domains of different models, individually, with the X-ray crystal structure (PDB ID: 2WRI).
EFG structures reported in PDB (ID: 1PN6) by rigid body fitting of individual domains of the initial structure into the corresponding cryo-EM map.
Table 4 also reports other differences between the crystal structure and the flexibly fitted structures, mainly in domains III–V. After aligning the domains I–II, all the flexible-fitted structures show consensus in orientations of domains III–IV, which is different from the crystal structure (Figure 4). For example, between MDfit fitted structure and crystal structure, rotation/translation of ~26/2 and ~12/6 degrees/Å are needed to fit domains III and IV, respectively, with fixed domains I–II (Figure 4b). These differences – particularly those where there are consensuses among the flexible fitting approaches – could be explained by the fact that the cryo-EM map (70S•EFG•GDPNP) and the crystal structure (70S•EFG•GDP•fusidic acid) correspond to two different states, before and after GTP hydrolysis, respectively and/or ribosome-EFG interactions. It has been previously suggested (Frank et al., 2007) that the conformational changes in EFG upon GTP hydrolysis are likely similar to those observed in elongation factor 2 (EF2) bound to the 80S ribosome (Taylor et al., 2007), which includes a ~6 Å shift in domain IV and some reorientations in domains III–V with respect to domains I–II. This is in general agreement with the flexibly fitted structures, as discussed above; however, the directions of orientations of these domains are different (data not shown). In contrast, the RDF structure, when compared to the crystal structure, shows relatively large conformational differences in domains III–IV with respect to domains I–II (rotation/translation of ~18/8 and 16/14 degrees/Å in domain III and IV, respectively, Figure 4b). These large domain shifts, compared to the flexibly fitted structures of EFG and previous studies for EF2, show that not only domain V but also domains III–IV may be incorrectly oriented in the RDF structure. In contrast, the flexibly fitted structures show consensus in the fitting and agree well with the crystal structure. The small differences can be attributed to suggested conformational changes upon GTP hydrolysis.
Conclusion
This study focused on comparing rigid domain fitting (RDF) and automated flexible fitting approaches using a dataset of models already submitted in PDB using RDF. The following three main conclusions can be drawn from this work: 1) By comparing flexibly fitted structures from three different methods, it was shown that there is, in general, a consensus in conformations among the flexible-fitted structures, which is an indication of convergence to the most probable solution by these approaches. 2) In contrast, structures deposited in the PDB using rigid body fitting of individual domains (RDF) not only differ from the consensus obtained by flexible fitting, but also differ from X-ray crystallography. This deviation can be as high as ~100 degrees from the most probable orientation of domain V in elongation factor G. Thus, RDF fitting is comparatively less reliable, as it may lead to a conformation that is inaccessible with respect to the underlying conformational transition and chain connectivity. This result also emphasizes that the visual inspection of the fitted structures as well as the correlation coefficient between the structure and the cryo-EM map, which are commonly used to measure the quality of fit, should be interpreted with caution in RDF fitting. 3) Automated flexible fitting approaches provide a robust alternative to RDF, especially in cases where consensus among the fitted conformation is obtained from different approaches. However, these flexible-fitting approaches might not be successful in extreme cases where fitting of the initial structure requires a complex transition like RF2. Interestingly, these cases can be identified based on the large disagreements among the flexibly fitted models.
Based on the above three conclusions, we suggest using multiple flexible fitting with different approaches for improved and robust fitted atomic models. Overall, when a “consensus” is achieved between different fits, the confidence level of the fitting is higher. Additionally, regions of the protein where consensus is not achieved can be attributed to regions of uncertainty in the model, providing a more comprehensive interpretation of cryo-EM data. Furthermore, the three approaches used in this study are computationally fast and do not require any domain segmentation, hence, these multiple flexible fitting techniques could easily be used in an automated fashion.
Supplementary Material
Acknowledgments
We thank Dr. Steve Harvey for making the YUP.SCX program available to us. Financial support from National Science Foundation grant 0744732 (Molecular Cellular and Biosciences) to FT is greatly appreciated. This work was also supported by the LANL LDRD program and NIH Grant R01GM072686. PCW is funded by a LANL Director’s Postdoctoral Fellowship.
Abbreviations
- Cryo-EM
cryo-electron microscopy
- RDF
rigid domain fitted
- EMDB
electron microscopy data bank
- NMFF
normal mode flexible fitting
- MDfit
molecular dynamics flexible fitting
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Aqeel Ahmed, Email: aqeel@email.arizona.edu.
Paul C. Whitford, Email: whitford@lanl.gov.
Karissa Y. Sanbonmatsu, Email: kys@lanl.gov.
References
- Bahar I, Atilgan AR, Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell ID. Timeline: the march of structural biology. Nat Rev Mol Cell Biol. 2002;3:377–381. doi: 10.1038/nrm800. [DOI] [PubMed] [Google Scholar]
- Chacon P, Wriggers W. Multi-resolution contour-based fitting of macromolecular structures. J Mol Biol. 2002;317:375–384. doi: 10.1006/jmbi.2002.5438. [DOI] [PubMed] [Google Scholar]
- Clementi C, Nymeyer H, Onuchic JN. Topological and energetic factors: what determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? An investigation for small globular proteins. J Mol Biol. 2000;298:937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- Conway JF, Wikoff WR, Cheng N, Duda RL, Hendrix RW, et al. Virus maturation involving large subunit rotations and local refolding. Science. 2001;292:744–748. doi: 10.1126/science.1058069. [DOI] [PubMed] [Google Scholar]
- Darst SA, Opalka N, Chacon P, Polyakov A, Richter C, et al. Conformational flexibility of bacterial RNA polymerase. Proc Natl Acad Sci U S A. 2002;99:4296–4301. doi: 10.1073/pnas.052054099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delarue M, Dumas P. On the use of low-frequency normal modes to enforce collective movements in refining macromolecular structural models. Proc Natl Acad Sci U S A. 2004;101:6957–6962. doi: 10.1073/pnas.0400301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiola F, Chapman MS. Fitting of high-resolution structures into electron microscopy reconstruction images. Structure. 2005;13:389–400. doi: 10.1016/j.str.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Falke S, Tama F, Brooks CL, 3rd, Gogol EP, Fisher MT. The 13 angstroms structure of a chaperonin GroEL-protein substrate complex by cryo-electron microscopy. J Mol Biol. 2005;348:219–230. doi: 10.1016/j.jmb.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Frank J. Single-particle imaging of macromolecules by cryo-electron microscopy. Annu Rev Biophys Biomol Struct. 2002;31:303–319. doi: 10.1146/annurev.biophys.31.082901.134202. [DOI] [PubMed] [Google Scholar]
- Frank J, Agrawal RK. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature. 2000;406:318–322. doi: 10.1038/35018597. [DOI] [PubMed] [Google Scholar]
- Frank J, Gao H, Sengupta J, Gao N, Taylor DJ. The process of mRNA-tRNA translocation. Proc Natl Acad Sci U S A. 2007;104:19671–19678. doi: 10.1073/pnas.0708517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Frank J. Molding atomic structures into intermediate-resolution cryo-EM density maps of ribosomal complexes using real-space refinement. Structure. 2005;13:401–406. doi: 10.1016/j.str.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Gao H, Sengupta J, Valle M, Korostelev A, Eswar N, et al. Study of the structural dynamics of the E coli 70S ribosome using real-space refinement. Cell. 2003;113:789–801. doi: 10.1016/s0092-8674(03)00427-6. [DOI] [PubMed] [Google Scholar]
- Gao YG, Selmer M, Dunham CM, Weixlbaumer A, Kelley AC, et al. The structure of the ribosome with elongation factor G trapped in the posttranslocational state. Science. 2009;326:694–699. doi: 10.1126/science.1179709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubisic I, Shokhirev MN, Orzechowski M, Miyashita O, Tama F. Biased coarse-grained molecular dynamics simulation approach for flexible fitting of X-ray structure into cryo electron microscopy maps. J Struct Biol. 2009;169:95–105. doi: 10.1016/j.jsb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Henderson R. Realizing the potential of electron cryo-microscopy. Q Rev Biophys. 2004;37:3–13. doi: 10.1017/s0033583504003920. [DOI] [PubMed] [Google Scholar]
- Henrick K, Newman R, Tagari M, Chagoyen M. EMDep: a web-based system for the deposition and validation of high-resolution electron microscopy macromolecular structural information. J Struct Biol. 2003;144:228–237. doi: 10.1016/j.jsb.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Hinsen K. Analysis of domain motions by approximate normal mode calculations. Proteins. 1998;33:417–429. doi: 10.1002/(sici)1097-0134(19981115)33:3<417::aid-prot10>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Hinsen K, Reuter N, Navaza J, Stokes DL, Lacapere JJ. Normal mode-based fitting of atomic structure into electron density maps: application to sarcoplasmic reticulum Ca-ATPase. Biophys J. 2005;88:818–827. doi: 10.1529/biophysj.104.050716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin J, Gumbart J, Trabuco LG, Villa E, Qian P, et al. Protein-induced membrane curvature investigated through molecular dynamics flexible fitting. Biophys J. 2009;97:321–329. doi: 10.1016/j.bpj.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs DJ, Rader AJ, Kuhn LA, Thorpe MF. Protein flexibility predictions using graph theory. Proteins. 2001;44:150–165. doi: 10.1002/prot.1081. [DOI] [PubMed] [Google Scholar]
- Jolley CC, Wells SA, Fromme P, Thorpe MF. Fitting low-resolution cryo-EM maps of proteins using constrained geometric simulations. Biophys J. 2008;94:1613–1621. doi: 10.1529/biophysj.107.115949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanicolas J, Brooks CL., 3rd Improved Go-like models demonstrate the robustness of protein folding mechanisms towards non-native interactions. J Mol Biol. 2003;334:309–325. doi: 10.1016/j.jmb.2003.09.047. [DOI] [PubMed] [Google Scholar]
- Lawson CL, Baker ML, Best C, Bi C, Dougherty M, et al. EMDataBank.org: unified data resource for CryoEM. Nucleic Acids Res. 2010;39:D456–464. doi: 10.1093/nar/gkq880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KK, Johnson JE. Complementary approaches to structure determination of icosahedral viruses. Curr Opin Struct Biol. 2003;13:558–569. doi: 10.1016/j.sbi.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Li W, Trabuco LG, Schulten K, Frank J. Molecular dynamics of EF-G during translocation. Proteins. 2011;79:1478–1486. doi: 10.1002/prot.22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindert S, Stewart PL, Meiler J. Hybrid approaches: applying computational methods in cryo-electron microscopy. Curr Opin Struct Biol. 2009;19:218–225. doi: 10.1016/j.sbi.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K, Schaffitzel C, Shaikh T, Tama F, Jenni S, et al. Structure of the E. coli protein-conducting channel bound to a translating ribosome. Nature. 2005;438:318–324. doi: 10.1038/nature04133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel JK, Whitford PC, Sanbonmatsu KY, Onuchic JN. SMOG@ctbp: simplified deployment of structure-based models in GROMACS. Nucleic Acids Res. 2010;38:W657–661. doi: 10.1093/nar/gkq498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzechowski M, Tama F. Flexible fitting of high-resolution x-ray structures into cryoelectron microscopy maps using biased molecular dynamics simulations. Biophys J. 2008;95:5692–5705. doi: 10.1529/biophysj.108.139451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranson NA, Farr GW, Roseman AM, Gowen B, Fenton WA, et al. ATP-bound states of GroEL captured by cryo-electron microscopy. Cell. 2001;107:869–879. doi: 10.1016/s0092-8674(01)00617-1. [DOI] [PubMed] [Google Scholar]
- Ratje AH, Loerke J, Mikolajka A, Brunner M, Hildebrand PW, et al. Head swivel on the ribosome facilitates translocation by means of intra-subunit tRNA hybrid sites. Nature. 2010;468:713–716. doi: 10.1038/nature09547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat UB, Zavialov AV, Sengupta J, Valle M, Grassucci RA, et al. A cryo-electron microscopic study of ribosome-bound termination factor RF2. Nature. 2003;421:87–90. doi: 10.1038/nature01224. [DOI] [PubMed] [Google Scholar]
- Rossmann MG, Morais MC, Leiman PG, Zhang W. Combining X-ray crystallography and electron microscopy. Structure. 2005;13:355–362. doi: 10.1016/j.str.2005.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saibil HR. Conformational changes studied by cryo-electron microscopy. Nat Struct Biol. 2000;7:711–714. doi: 10.1038/78923. [DOI] [PubMed] [Google Scholar]
- Sali A, Glaeser R, Earnest T, Baumeister W. From words to literature in structural proteomics. Nature. 2003;422:216–225. doi: 10.1038/nature01513. [DOI] [PubMed] [Google Scholar]
- Schroder GF, Brunger AT, Levitt M. Combining efficient conformational sampling with a deformable elastic network model facilitates structure refinement at low resolution. Structure. 2007;15:1630–1641. doi: 10.1016/j.str.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sener M, Hsin J, Trabuco LG, Villa E, Qian P, et al. Structural model and excitonic properties of the dimeric RC-LH1-PufX complex from Rhodobacter sphaeroides. Chem Phys. 2009;357:188–197. doi: 10.1016/j.chemphys.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta J, Nilsson J, Gursky R, Kjeldgaard M, Nissen P, et al. Visualization of the eEF2-80S ribosome transition-state complex by cryo-electron microscopy. J Mol Biol. 2008;382:179–187. doi: 10.1016/j.jmb.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Milne JL. Three-dimensional electron microscopy at molecular resolution. Annu Rev Biophys Biomol Struct. 2004;33:141–155. doi: 10.1146/annurev.biophys.33.110502.140339. [DOI] [PubMed] [Google Scholar]
- Suhre K, Navaza J, Sanejouand YH. NORMA: a tool for flexible fitting of high-resolution protein structures into low-resolution electron-microscopy-derived density maps. Acta Crystallogr D Biol Crystallogr. 2006;62:1098–1100. doi: 10.1107/S090744490602244X. [DOI] [PubMed] [Google Scholar]
- Tagari M, Newman R, Chagoyen M, Carazo JM, Henrick K. New electron microscopy database and deposition system. Trends Biochem Sci. 2002;27:589. doi: 10.1016/s0968-0004(02)02176-x. [DOI] [PubMed] [Google Scholar]
- Taketomi H, Ueda Y, Go N. Studies on protein folding, unfolding and fluctuations by computer simulation. I The effect of specific amino acid sequence represented by specific inter-unit interactions. Int J Pept Protein Res. 1975;7:445–459. [PubMed] [Google Scholar]
- Tama F, Sanejouand YH. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- Tama F, Miyashita O, Brooks CL., 3rd Flexible multi-scale fitting of atomic structures into low-resolution electron density maps with elastic network normal mode analysis. J Mol Biol. 2004a;337:985–999. doi: 10.1016/j.jmb.2004.01.048. [DOI] [PubMed] [Google Scholar]
- Tama F, Miyashita O, Brooks CL., 3rd Normal mode based flexible fitting of high-resolution structure into low-resolution experimental data from cryo-EM. J Struct Biol. 2004b;147:315–326. doi: 10.1016/j.jsb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Tama F, Ren G, Brooks CL, 3rd, Mitra AK. Model of the toxic complex of anthrax: responsive conformational changes in both the lethal factor and the protective antigen heptamer. Protein Sci. 2006;15:2190–2200. doi: 10.1110/ps.062293906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan RK, Devkota B, Harvey SC. YUP.SCX: coaxing atomic models into medium resolution electron density maps. J Struct Biol. 2008;163:163–174. doi: 10.1016/j.jsb.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan RKZ, Petrov AS, Harvey SC. YUP: A Molecular Simulation Program for Coarse-Grained and Multiscaled Models. J Chem Theory Comput. 2006;2:529–540. doi: 10.1021/ct050323r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DJ, Nilsson J, Merrill AR, Andersen GR, Nissen P, et al. Structures of modified eEF2 80S ribosome complexes reveal the role of GTP hydrolysis in translocation. EMBO J. 2007;26:2421–2431. doi: 10.1038/sj.emboj.7601677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topf M, Sali A. Combining electron microscopy and comparative protein structure modeling. Curr Opin Struct Biol. 2005;15:578–585. doi: 10.1016/j.sbi.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Topf M, Lasker K, Webb B, Wolfson H, Chiu W, et al. Protein structure fitting and refinement guided by cryo-EM density. Structure. 2008;16:295–307. doi: 10.1016/j.str.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabuco LG, Villa E, Schreiner E, Harrison CB, Schulten K. Molecular dynamics flexible fitting: a practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods. 2009;49:174–180. doi: 10.1016/j.ymeth.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Taketomi H, NG Studies on protein folding, unfolding, and fluctuations by computer simulation. II. A. Three-dimensional lattice model of lysozyme. Biopolymers. 1978;17:1531–1548. [Google Scholar]
- Valle M, Zavialov A, Sengupta J, Rawat U, Ehrenberg M, et al. Locking and unlocking of ribosomal motions. Cell. 2003a;114:123–134. doi: 10.1016/s0092-8674(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Valle M, Zavialov A, Li W, Stagg SM, Sengupta J, et al. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat Struct Biol. 2003b;10:899–906. doi: 10.1038/nsb1003. [DOI] [PubMed] [Google Scholar]
- Velazquez-Muriel JA, Carazo JM. Flexible fitting in 3D-EM with incomplete data on superfamily variability. J Struct Biol. 2007;158:165–181. doi: 10.1016/j.jsb.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Velazquez-Muriel JA, Valle M, Santamaria-Pang A, Kakadiaris IA, Carazo JM. Flexible fitting in 3D-EM guided by the structural variability of protein superfamilies. Structure. 2006;14:1115–1126. doi: 10.1016/j.str.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Vestergaard B, Sanyal S, Roessle M, Mora L, Buckingham RH, et al. The SAXS solution structure of RF1 differs from its crystal structure and is similar to its ribosome bound cryo-EM structure. Mol Cell. 2005;20:929–938. doi: 10.1016/j.molcel.2005.11.022. [DOI] [PubMed] [Google Scholar]
- Villa E, Sengupta J, Trabuco LG, LeBarron J, Baxter WT, et al. Ribosome-induced changes in elongation factor Tu conformation control GTP hydrolysis. Proc Natl Acad Sci U S A. 2009;106:1063–1068. doi: 10.1073/pnas.0811370106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann N, Hanein D. Quantitative fitting of atomic models into observed densities derived by electron microscopy. J Struct Biol. 1999;125:176–184. doi: 10.1006/jsbi.1998.4074. [DOI] [PubMed] [Google Scholar]
- Volkmann N, Hanein D, Ouyang G, Trybus KM, DeRosier DJ, et al. Evidence for cleft closure in actomyosin upon ADP release. Nat Struct Biol. 2000;7:1147–1155. doi: 10.1038/82008. [DOI] [PubMed] [Google Scholar]
- Weixlbaumer A, Jin H, Neubauer C, Voorhees RM, Petry S, et al. Insights into translational termination from the structure of RF2 bound to the ribosome. Science. 2008;322:953–956. doi: 10.1126/science.1164840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt T, Taylor D, Trybus KM, Taylor K. Three-dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc Natl Acad Sci U S A. 2001;98:4361–4366. doi: 10.1073/pnas.071051098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitford PC, Noel JK, Gosavi S, Schug A, Sanbonmatsu KY, et al. An all-atom structure-based potential for proteins: bridging minimal models with all-atom empirical forcefields. Proteins. 2009;75:430–441. doi: 10.1002/prot.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitford PC, Geggier P, Altman RB, Blanchard SC, Onuchic JN, et al. Accommodation of aminoacyl-tRNA into the ribosome involves reversible excursions along multiple pathways. RNA. 2010;16:1196–1204. doi: 10.1261/rna.2035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitford PC, Yu Y, Hennelly SP, Ahmed A, Tama F, et al. Excited States of Ribosome Translocation Revealed Through Integrative Molecular Modeling. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1108363108. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wriggers W, Milligan RA, McCammon JA. Situs: A package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol. 1999;125:185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- Zoldak G, Redecke L, Svergun DI, Konarev PV, Voertler CS, et al. Release factors 2 from Escherichia coli and Thermus thermophilus: structural, spectroscopic and microcalorimetric studies. Nucleic Acids Res. 2007;35:1343–1353. doi: 10.1093/nar/gkl696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.