

Abstract
The Pdss2 gene product is needed for the isoprenylation of benzoquinone to generate coenzyme Q (CoQ). A fatal kidney disease occurs in mice that are homozygous for a missense mutation in Pdss2, which can be recapitulated in conditional Pdss2 knockouts targeted to glomerular podocytes. We now report that homozygous missense mutants also demonstrate significant neuromuscular deficits, as validated by behavioral and coordination assays, and these deficits are recapitulated in conditional Pdss2 knockouts targeted to dopaminergic neurons. Both conditional knockout and missense mutant mice demonstrate deficiencies in tyrosine hydroxylase-positive neurons in the substantia nigra, implicating a pathology similar to sporadic Parkinson’s disease (PD).
Keywords: Parkinson’s disease, coenzyme Q, dopaminergic neurons
1. Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease, affecting 1% of the population above the age of 65. The characteristic pathology involves degeneration of dopaminergic neurons in the substantia nigra pars compacta and the appearance of intracytoplasmic inclusions known as Lewy bodies (Beal, 2001). The specific etiology of PD is unknown, although both genetic and environmental factors that influence this disease have been identified. Genetic forms of PD can be caused by mutations in SCNA (PARK1), which encodes alpha-synuclein, a key component of Lewy bodies; PARK2, which encodes parkin, a ubiquitin E3 ligase; PINK1 (PARK6), which encodes a serine-threonine kinase; DJ-1 (PARK7), an oxidative stress sensor; or leucine-rich repeat kinase 2 (LRRK2, PARK8), which encodes a serine-threonine kinase (Henchcliffe and Beal, 2008).
A number of animal models of PD have been developed that either involve mutations at candidate gene loci or injection of neurotoxins such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), 6-hydroxydopamine (6-OHDA), or rotenone (Bloem et al., 1990; Sauer and Oertel, 1994; Abeliovich et al., 2000; Betarbet et al., 2000; Goldberg et al., 2003). While each of these models has made a significant contribution to our understanding of PD, the complexity of the disease suggests multiple levels of pathogenesis. Among the many etiologies of PD that have been proposed, one common pathogenic factor is mitochondrial dysfunction (Henchcliffe and Beal, 2008). Reduced coenzyme Q (CoQ) levels have been observed in platelets of PD patients (Shults et al., 1997; Beal, 2001), and some studies have shown that patients with PD may benefit from CoQ10 supplementation (Beal, 2003). CoQ10 supplementation has also been shown to protect against MPTP-induced damage in aged mice (Beal, 2001). CoQ is an essential co-factor that shuttles electrons between complexes I or II and complex III in the electron transport chain, and also functions as a potent antioxidant in mitochondria. An NIH-sponsored Phase 3 clinical research trial is in progress to test the effectiveness of CoQ10 in slowing the progression of Parkinson’s disease (http://www.pdtrials.org/en/browse/all/1/view/238).
To further investigate how CoQ might impact a PD phenotype, we have studied two mouse models of primary CoQ deficiency. These animals have either a homozygous missense mutation in a gene encoding a CoQ biosynthetic pathway enzyme with resultant CoQ deficiency in all tissues, or a conditional knockout of the same gene that is specifically targeted to the dopaminergic neurons. This gene was initially identified as a spontaneous mutation designated kd for kidney disease (Lyon and Hulse, 1971), and was subsequently shown to encode a mitochondrial enzyme with prenyltransferase-like activity (Peng et al., 2004). It is now called prenyl diphosphate synthase subunit 2 (Pdss2), and its gene product is one of two enzymes needed for the isoprenylation of CoQ (Saiki et al., 2005). Upon targeted knockout of Pdss2 specifically in glomerular podocytes, the kidney disease phenotype of kd/kd (B6.Pdss2kd/kd) mice was recapitulated (Peng et al., 2008). This renal phenotype could be significantly mitigated when mutant B6.Pdss2kd/kd mice were supplemented from weaning throughout adulthood with CoQ10 in their drinking water (Saiki et al., 2008). In contrast, when Pdss2 was deleted specifically in hepatocytes, myeloid cells, or renal proximal tubular epithelial cells, no disease phenotype was evident (Peng et al., 2008), suggesting that differentiated cells differ significantly with regard to their susceptibility to CoQ deficiency. We therefore examined whether behavioral deficiencies that resemble PD might appear in either the Pdss2 conditional knockouts or missense mutants.
2. Materials and methods
2.1. Mice
The B6.Pdss2kd/kd mice were derived by backcrossing the original kd mutation onto the B6 background in the course of positional cloning (Dell et al., 2000). B6;SJL-Slc6a3tm1.1(cre)Bkmn/J mice that express Cre recombinase under the control of the dopamine transporter (DAT) promoter (Backman et al., 2006) were obtained from The Jackson Laboratory (Bar Harbor, ME) and mated with B6.Pdss2loxP/loxP mice (Peng et al., 2008). The F1 hybrids were backcrossed to the B6.Pdss2loxP/loxP strain, and Cre-positive Pdss2loxP/loxP homozygotes (B6.DAT/cre,Pdss2loxP/loxP) were selected for behavioral testing.
Animals were housed 1–5 per cage. Food and water were provided ad libitum. Lights were maintained on a 12:12 h light/dark cycle. All behavioral testing was carried out between the hours of 8:00 and 14:00. All animal care and experiments were carried out in accordance with NIH guidelines and were fully approved by the IACUCs of the University of Pennsylvania and the Children’s Hospital of Philadelphia.
2.2. Motor Function Testing
Motor function was tested using the rotarod, a rotating cylinder on which animals must balance for as long as they are able (Colotla et al., 1990; Rozas et al., 1998). Testing was performed between 8:00 and 11:00 on 3–5 consecutive days. Mice were placed in the testing room and allowed to habituate for 30 minutes. After the habituation period on the first day of every testing session, the mice were “pre-trained” and acclimated to the rotating cylinder. Each mouse was placed on the beam facing away from the experimenter so that forward ambulation was necessary to maintain balance. With constant rotation of 4 rpm for 30 seconds, the mouse was replaced if falls occurred and the number of falls during pre-training was recorded.
Mice were tested 30 minutes after pre-training. The animals were placed on the beam facing away from the experimenter, and the rotarod was set to “accelerating mode” from 4–40 rpm over 5 minutes. The falling time and falling speed of each mouse was recorded. The testing period was repeated 3 times on each testing day with 30-minute inter-trial intervals. Statistical analysis was performed using two-way ANOVA, Bonferroni-Dunn post hoc tests, and Student’s t-test.
2.3. Locomotor Activity Analysis
Locomotor activity was analyzed in a “home cage” activity monitoring system (MedAssociates, St. Albans, VT). The testing cage was placed in a photobeam frame (28.9 cm × 17.8 cm × 12 cm) with two levels of sensors arranged in an 8 beamarray strip with 1.25 inch spacing. All locomotor activity assessment was carried out between 8:00 and 11:00. Mice were acclimated to the testing room 30 minutes prior to testing and were then individually placed in cages for 30-minute monitoring periods. Beam break data were analyzed using MedAssociates software and quantified to represent crosses, rears, and total activity in 5-minute intervals. Statistical analysis was performed using 2-way ANOVA, Bonferroni-Dunn post hoc test, and Student’s t-test. This assessment was repeated every 10–14 days for studies involving akinesia progression and the effects of CoQ10 supplementation.
2.4. Akinesia analysis
Akinesia was assessed by determining the subjects’ latency to move four limbs (Sedelis et al., 2001). After habituation to the testing room for 30 minutes, each mouse was removed from their home cage and placed in a neutral position, facing away from the experimenter on a 69.85 cm × 85.09 cm white pad. The latency to move all four limbs was recorded. The akinesia test was repeated over 3 trials with 30-minute inter-trial periods, and assessed every 14 days. Statistical analysis was performed by 2-way ANOVA and Bonferroni-Dunn post hoc tests. The experimenter was blind to the subject’s genotype.
2.5. Limb movement and balance assessment
The pole test was performed to detect impairment of limb movement and balance. The animal was placed on a metal pole (height 45.7 cm, diameter 8 mm) covered in gauze, with a small Styrofoam ball (diameter ~ 1.5 cm) on the top. Each mouse was placed on the Styrofoam ball at t=0. Latencies to turn downward, descend halfway down the pole (both hind limbs crossing the half-way point), and fully descend to the bottom of the pole (both hind limbs reaching the ground) were recorded. The trial was excluded from analysis if the mouse fell and failed to successfully traverse the pole. This test was administered between the hours of 8:00 and 12:00 and each animal was acclimated to the testing room for 30 minutes prior to testing. Three trials were carried out per time point, with a 30-minute inter-trial period. To allow for pure assessment of the animal’s balance, limb movement, and overall bradykinesia while eliminating bias due to positioning on the pole and novelty-induced delay in descending from the pole, analyses used time intervals for descending from the halfway mark to the ground. Statistical analysis was performed by two-way ANOVA and Bonferroni-Dunn post hoc tests (Matsuura et al., 1997; Sedelis et al, 2001).
2.6. Coenzyme Q10 supplementation and probucol treatment
Mice were given CoQ10 (Tishcon LiQsorb) in the drinking water from the time of weaning at a concentration of 1 mg/ml, which approximates a final dosage of 400 mg CoQ10 per kg body weight per day (Saiki et al., 2008). Other mice received probucol in the chow at a concentration of 1% wt/wt, obtained from Animal Specialties and Provisions (Quakertown, PA).
2.7 Mitochondrial respiratory capacity analysis
Immediately following euthanasia, skeletal muscle was dissected on each of three separate days from the proximal hind limbs of three pairs of missense mutant male mice aged 125–143 days and wild-type B6 male controls aged 150–155 days. Skeletal muscle was collected in an ice cold relaxing and biopsy preservation solution, BIOPS, which contained 10 mM Ca-EGTA buffer, 0.1 uM free calcium, 20 mM imidazole, 20 mM taurine, 50 mM K-MES (2-[N-morpholino]ethane sulfonic acid), 0.5 mM DTT (dithiothreitol), 6.56 mM MgCl, 5.77 mM ATP, 15 mM phosphocreatine, pH 7.1.
Permeabilized muscle fibers were freshly prepared for respiratory capacity analysis by high resolution polarography using an Oxygraph-2K, as previously described (Lemieux and Gneiger, 2008). Briefly, individual muscle bundles were separated and transferred to fresh ice cold BIOPS solution containing saponin in 50 ug/ml final concentration. Muscle bundles were shaken by gentle agitation for 30 minutes at 4°C. Fiber bundles were then transferred into mitochondrial respiration medium (MiR05) and shaken by gentle agitation for 10 minutes in the cold room at 4°C. After permeabilization, wet weight was measured. 1–4 mg of permeabilized muscle tissue from wild-type and B6.Pdss2kd/kd mutant mice were analyzed in parallel in adjacent chambers. Assessment of respiratory capacity was performed at 37°C in MiR05, as previously described (Lemieux et al., 2009). Substrates and inhibitors were added to the permeabilized muscle tissue in the following order: glutamate 20 mM, malate 10 mM, adenosine diphosphate (ADP) 0.2 mM, cytochrome C 10 uM, succinate 20 mM, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) step-wise titration in 0.125 uM increments, rotenone 0.5 uM, and antimycin A 2.5 uM. Data were analyzed using DatLab4 (Oroboros, Austria). Statistical analysis between state-specific respiratory capacity rates of each group was performed by Student’s t-test. All experiments were performed by a single individual (E.P.).
2.8 Brain histology analysis
Antibodies against tyrosine hydroxylase (TH) and the dopamine transporter (MAB369 1:500; Chemicon, Bedford MA) were used to evaluate dopaminergic neurons. Nissl staining was employed to evaluate cell death. For histological analysis, mice were deeply anesthetized and perfused transcardially with saline followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (pH 7.3). The brains were removed and kept immersed in fixative overnight, transferred to 30% sucrose solution in phosphate buffer for cryoprotection, subsequently frozen with cold 2-methylbutane, and stored at −80°C until dissection. The substantia nigra was dissected through its entire rostro-caudal axis in 30-μm coronal sections using a cryostat. Sections were collected free-floating and every fourth section was stained with tyrosine hydroxylase antibody (rabbit polyclonal; Calbiochem, 657012) diluted 1:1000, followed by incubation with biotin-conjugated anti-rabbit antibody, ABC reagents, and DAB Peroxidase substrate (Vector Laboratories, Burlingame, CA). The slides were counterstained with cresyl violet, dehydrated, and coverslips were applied. Digital scans of the slides were created using the Scanscope scanner (Aperio, Vista, CA). Imagescope software (Aperio) was used to annotate the substantia nigra region in both hemispheres and to assess the total number of tyrosine hydroxylase-positive and Nissl-positive neurons within the annotated areas of 4 animals per group. The results are expressed as the percent of tyrosine hydroxylase positive neurons relative to the total neuron number in the region. Images of tissue sections on glass slides were obtained using confocal microscopy (Leica Microsystems, Inc., Deerfield, IL).
2.9 Kidney disease evaluation
B6.Pdss2kd/kd mice were placed in metabolic cages without food for 24 hours with 0.45% NaCl and 2.5% sucrose in their drinking water. Total urine volumes were measured, and aliquots of urine were tested for albumin concentration by ELISA. Mice were then euthanized and their kidneys were fixed and stained with hematoxylin and eosin. Histologic sections were scored blindly according to the following scale: 0 = no tubular dilatation and no mononuclear cell infiltrates; 1 = small focal areas of cellular infiltration and tubular dilatation involving less than 10% of the cortex; 2 = involvement of up to 25% of the cortex; 3 = involvement of up to 50% of the cortex; 4 = extensive damage involving more than 75% of the cortex.
3. Results
Alterations in motor coordination and locomotor activity were examined in B6.Pdss2kd/kd mice that expressed the missense mutation globally as well as in Pdss2loxP/loxP;Slc6a3tm1.1(cre)Bkmn knock-out (K.O.) mice that lacked this gene specifically in dopaminergic neurons. Animals were reanalyzed over time to monitor for potential neuromuscular effects, since it is known that their lethal kidney disease phenotype does not present in missense mutants before approximately 4 months of age (Peng et al., 2008).
3.1 Motor coordination
In rotarod testing of age-matched B6.Pdss2kd/kd and wild-type B6 mice, the B6.Pdss2kd/kd animals consistently performed worse (Fig. 1a), which indicated that missense mutant mice have impaired motor coordination. Testing was repeated every 10 days between 11 and 13 weeks of life beginning at age 75 +/− 5.9 days. B6.Pdss2kd/kd mice initially exhibited the same rotarod performance as wild-type mice. However, their performance worsened after subsequent trials within a single testing session. By day 4 of time point 1 (mean age: 11 weeks) the mutant animals displayed significant inability to maintain balance on the rotating rod at levels comparable to those achieved by wild-type animals (p<0.001). As additional rotarod testing periods lapsed, B6.Pdss2kd/kd mice maintained a significantly lower latency to fall (p<0.001). This deviation between the two groups remained constant over time.
Fig. 1.



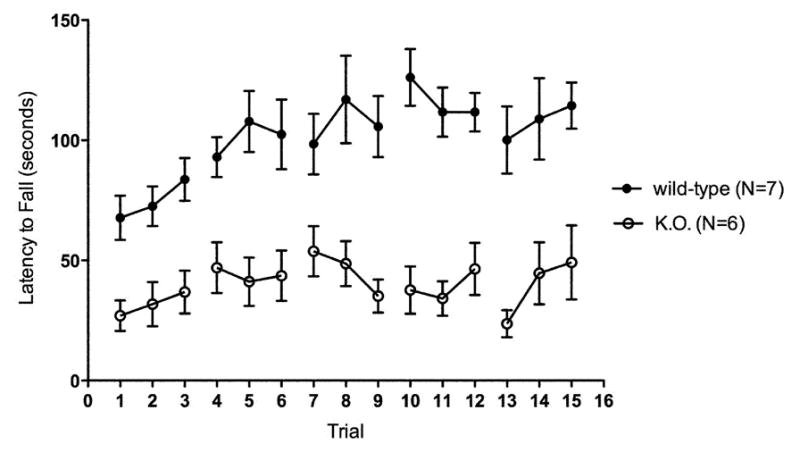
Rotarod Performance of Untreated kd/kd and K.O. Mice.
(a) Behavioral testing assessing latency to fall was significantly different across multiple timepoints between B6 (wild-type) and B6.Pdss2kd/kd (kd/kd) animals and also (b) between B6.Pdss2loxP/loxP (wild-type) and B6.DAT/cre,Pdss2loxP/loxP (K.O.) animals. Three trials occurred per day over 5 consecutive days. Mean ages at each timepoint (TP) for both cohorts are as follows: TP1: 11 weeks, TP2: 12 weeks, TP3: 13 weeks. (c) Significant deviations are also reported within a single trial between age-matched kd/kd and wild-type animals (p<0.001) and (d) between age-matched wild-type and K.O. animals (p<0.001). Statistical analyses were performed using two-way ANOVA followed by Bonferroni-Dunn post-hoc tests. Data points represent mean +/− S.E.M., N values denote animal group size. *p<0.05, **p<0.01, ***p<0.001.
In order to determine whether this phenotype could be attributed to a defect in dopaminergic neurons, mice that had the Pdss2 gene conditionally targeted by the DAT/cre construct were also evaluated by rotarod testing. As shown in Fig. 1b and 1d, the Pdss2loxP/loxP;Slc6a3tm1.1(cre)Bkmn (K.O.) mice demonstrated a phenotype that was very similar to that of B6.Pdss2kd/kd missense mutants of the same age (Fig 1c), although it tended to be somewhat more severe.
3.2 Locomotor Activity
Baseline locomotor activity of the B6.Pdss2kd/kd mice was significantly lower than that of wild-type mice in each testing period (Fig 2a and 2c), which was repeated approximately every 10 days for 4 weeks starting at age 69 +/− 5.9 days. Furthermore, B6.Pdss2kd/kd mice had a significantly lower number of crosses and rears in the 30-minute testing period (p<0.01). Not only do these observations indicate missense mutant mice had decreased motor function relative to age-matched wild-type animals, but also signify their decreased initiation of spontaneous movement and decreased response to a novel environment. B6.Pdss2kd/kd mice did not seem to show a similar temporal pattern of habituation to the novel environment of the open-field as did the wild-type animals, which was most evident in the third and fourth testing periods (Fig. 2a). Similar to the decline in motor function and balance coordination observed in rotarod performance after subsequent trials, significant impairment in locomotor activity was maintained relative to wild-type animals and did not continue to worsen with animal age. Locomotor activity sharply declined at approximately 8 months of age (data not shown), which likely reflects overall ill health due to end-stage renal disease rather than motor control degradation.
Fig. 2.
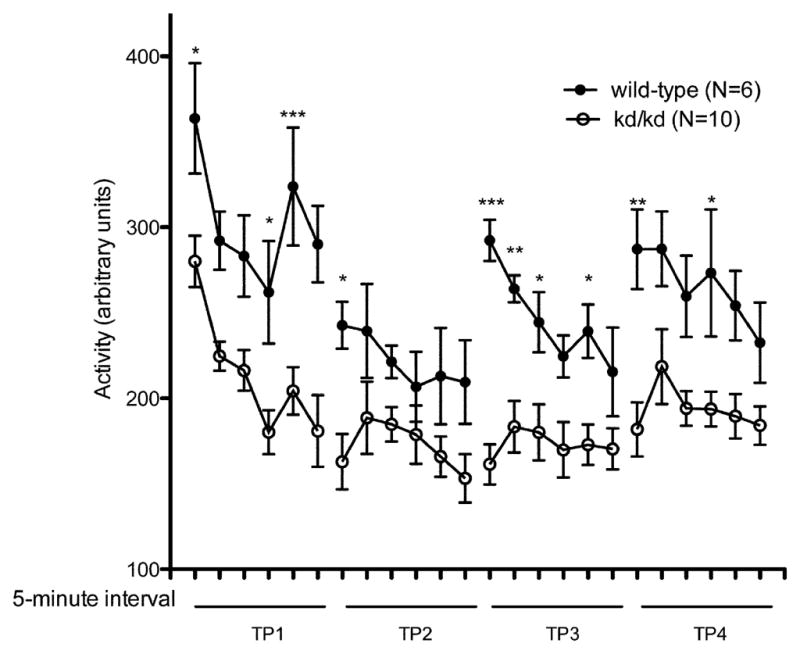
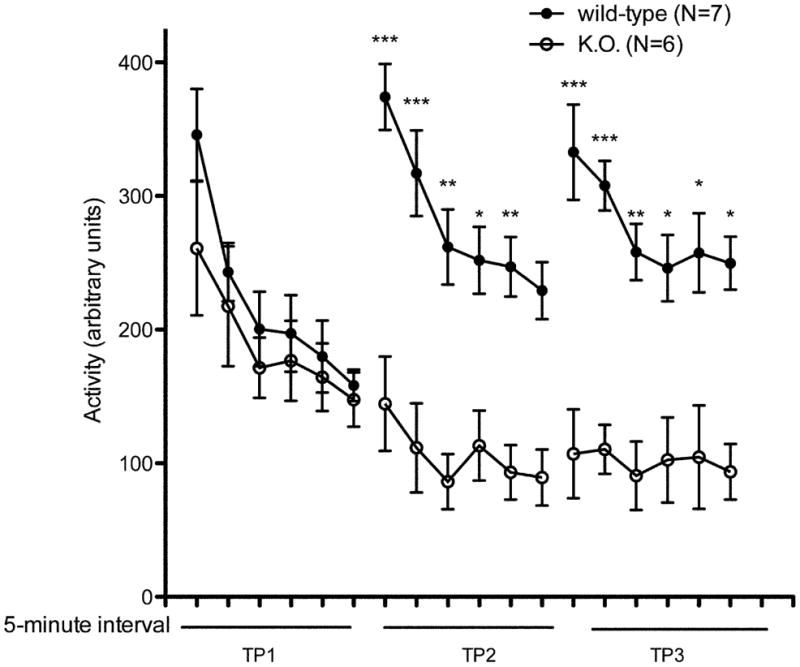
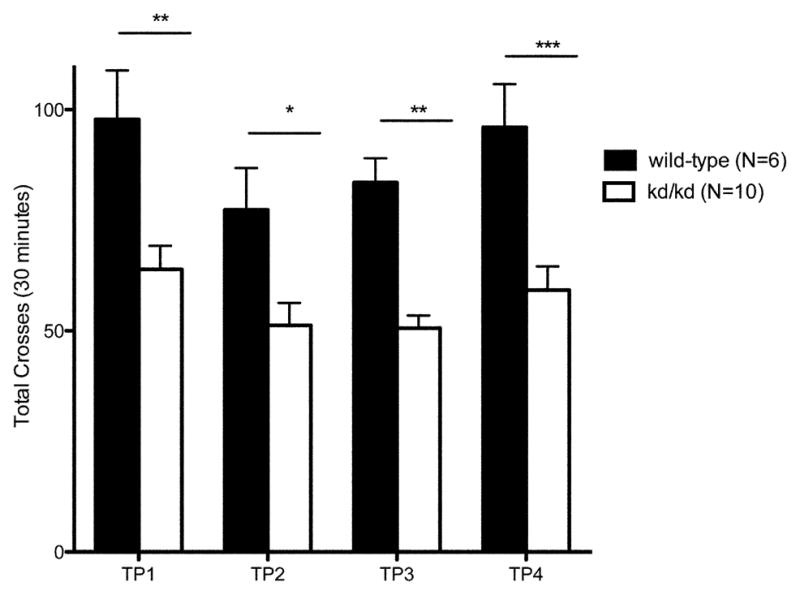

Locomotor Performance in Untreated kd/kd Mice and K.O. mice.
(a) B6.Pdss2kd/kd (kd/kd) and (b) B6.DAT/cre,Pdss2loxP/loxP (K.O.) mice exhibited significantly lower locomotor activity when tested using the “home cage” activity monitoring system (MedAssociates). Ambulations are reported as mean +/− S.E.M. (arbitrary units) in 5-minute intervals for a total testing period of 30 minutes. The discrete groupings indicate different testing dates separated by 10-day intertrial intervals. (c) kd/kd mice and (d) K.O. mice exhibited significantly lower total cage-crosses when assessed using the “home cage” paradigm (MedAssociates). Statistical analyses were performed using two-way ANOVA followed by Bonferroni-Dunn post-hoc tests. N values denote animal group size. Ages at each timepoint (TP) are as follows: TP1: 10 weeks, TP2: 12 weeks, TP3: 13 weeks, TP4: 15 weeks. *p<0.05, **p<0.01, ***p<0.001.
In accordance with our observations using the rotarod paradigm, the Pdss2loxP/loxP;Slc6a3tm1.1(cre)Bkmn (K.O.) mice lacking this gene in dopaminergic neurons exhibited a similar phenotype, in that their locomotor activity as quantified by beam-breaks (Fig 2b) and total crosses in the 30 minute testing period (Fig 2d) were significantly lower than age-matched wild-type controls (p<0.001).
3.3 Akinesia
Through subsequent trials and retesting, B6.Pdss2kd/kd animals showed an increased latency to move (p<0.05) (Fig. 3a). This decreased spontaneous movement in an open field and new environment complements findings from the locomotor activity assessments (Fig 2a). Testing carried out on Pdss2loxP/loxP;Slc6a3tm1.1(cre)Bkmn (K.O.) animals reflects similar akinetic behavior when compared to age-matched wildtype controls (Fig 3b).
Fig. 3.


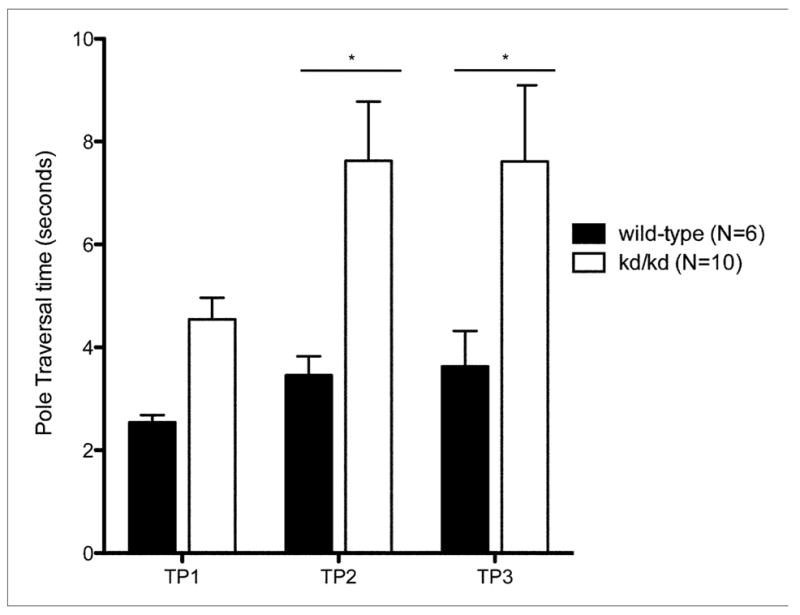

Balance and Coordination in kd/kd mice and K.O. mice.
Akinesia tests show significant deficiency (p<0.05) in (a) B6.Pdss2kd/kd (kd/kd) animals and (b) B6.DAT/cre,Pdss2loxP/loxP (K.O.) animals. Pole tests confirm significant deviations (p<0.01) in coordination and locomotor activity in (c) kd/kd and (d) K.O. animals. Statistical analyses were performed using two-way ANOVA followed by Bonferroni-Dunn post-hoc tests. Data points represent mean +/− S.E.M., N values denote animal group size. Ages at each timepoint (TP) are as follows: TP1: 10 weeks, TP2: 12 weeks, TP3: 13 weeks. *p<0.05, **p<0.01, ***p<0.001
3.4 Pole test
B6.Pdss2kd/kd mice exhibited increased difficulty in traversing the vertical pole compared to wild-type animals, as evidenced by a significantly longer time required to move from a predetermined location on the pole to the ground (p<0.01) (Fig. 3c). Furthermore, they showed marked difficulty in coordinating the transition from the pole to the ground relative to wild-type animals. These results further indicate the impaired balance and coordination of Pdss2 missense mutant mice. Pdss2loxP/loxP;Slc6a3tm1.1(cre)Bkmn (K.O.) impairment on the pole tests parallels the behavioral phenotype observed in the missense mutants, with significantly longer pole traversal time by the second time point (p<0.01) (Fig. 3d).
3.5 Effects of CoQ supplementation and probucol treatment
As CoQ10 supplementation has been shown to improve kidney pathology caused by the missense mutation (Saiki et al, 2008), we examined whether this treatment also rescued locomotor deficits. However, B6.Pdss2kd/kd mice that received CoQ10 supplementation beginning at 4 weeks of age failed to show recovery of motor control impairment when tested using the rotarod paradigm (Fig. 4a). The latency to fall recorded for CoQ10-treated B6.Pdss2kd/kd mice showed no significant change from the rotarod performance of untreated, age-matched B6.Pdss2kd/kd animals (p>0.05). Similarly, no significant effect of treatment was found when comparing the CoQ10 treated to untreated wild-type controls (p>0.05). Additionally, the latency to fall for both CoQ10-treated and untreated B6.Pdss2kd/kd animals was significantly lower than that of both the treated and untreated wild-type mice (p<0.01), similar to the rotarod performance noted in previous cohorts of B6.Pdss2kd/kd mice. These trends were consistent over three testing periods of mice aged 14 to 19 weeks old.
Fig. 4.

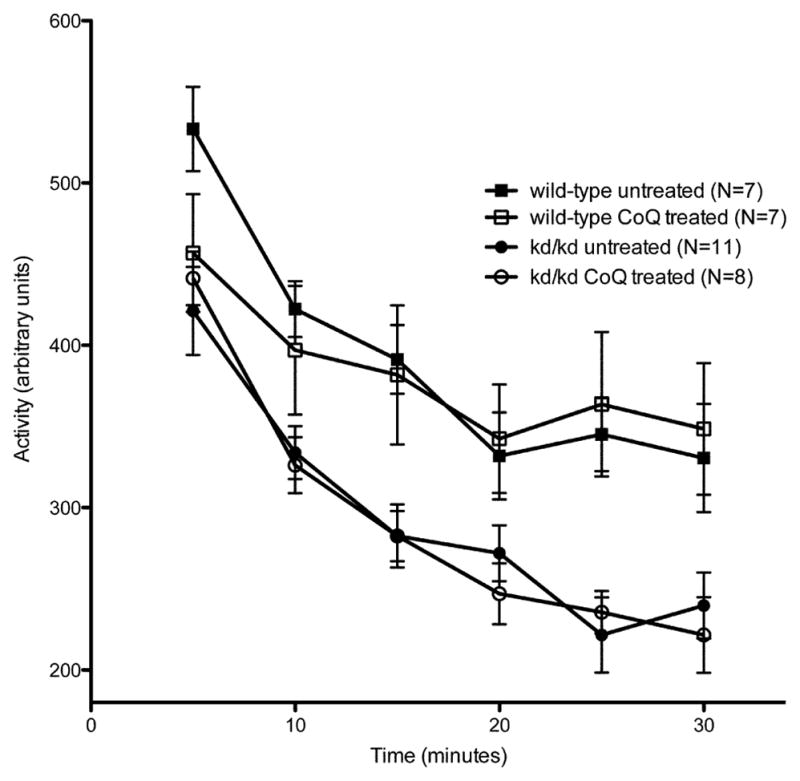

Effect of Coenzyme Q supplementation on kd/kd Motor Activity.
(a) Coenzyme Q (CoQ) treated B6.Pdss2kd/kd (kd/kd) mice showed no significant changes in performance on the rotarod when compared to untreated kd/kd controls (p>0.05). Data points represent mean +/− S.E.M. for each day of testing, assessment at each timepoint involved three trials per day over three consecutive days. Ages at each time point (TP) are as follows: TP1: 14 weeks, TP2: 17 weeks, TP3: 19 weeks. (b) Coenzyme Q (CoQ) treated kd/kd mice showed no significant changes in locomotor activity when tested using the “home cage” activity monitoring system (MedAssociates) in comparison with untreated kd/kd animals (p>0.05). Ambulations are reported as mean +/− S.E.M. (arbitrary units) in 5-minute intervals for a total testing period of 30 minutes. (c) kd/kd mice with dietary probucol supplementation showed no significant changes in performance on the rotarod when compared to untreated kd/kd controls (p>0.05). Data points represent mean +/− S.E.M. for each day of testing, assessment at each timepoint involved three trials per day over three consecutive days. Ages at each timepoint (TP) are as follows: TP1: 17 weeks, TP2: 19 weeks. (d) Probucol treated kd/kd mice showed no significant changes in locomotor activity when tested using the “home cage” activity monitoring system in comparison with untreated kd/kd animals. Ambulations are reported as mean +/− S.E.M. (arbitrary units) in 5-minute intervals for a total testing period of 30 minutes. Statistical analyses were performed using two-way ANOVA followed by Bonferroni-Dunn post-hoc tests. N values denote animal group size.
CoQ10 supplementation also had no effect on locomotor performance, as CoQ10 treated B6.Pdss2kd/kd mice showed no significant rescue of behavioral deficits (p>0.05) when tested using the “home cage” activity monitoring paradigm (Fig 4b). Furthermore, no effect was seen based on locomotor performance between treated and untreated B6 animals (p>0.05). Similar to results from rotarod performance assays, a significant difference in locomotor activity was again seen in this cohort of wild-type and B6.Pdss2kd/kd mice, with both CoQ10 treated and untreated wild-type animals maintaining significantly increased overall ambulations during the testing period compared to CoQ10 treated and untreated B6.Pdss2kd/kd mice (p<0.01).
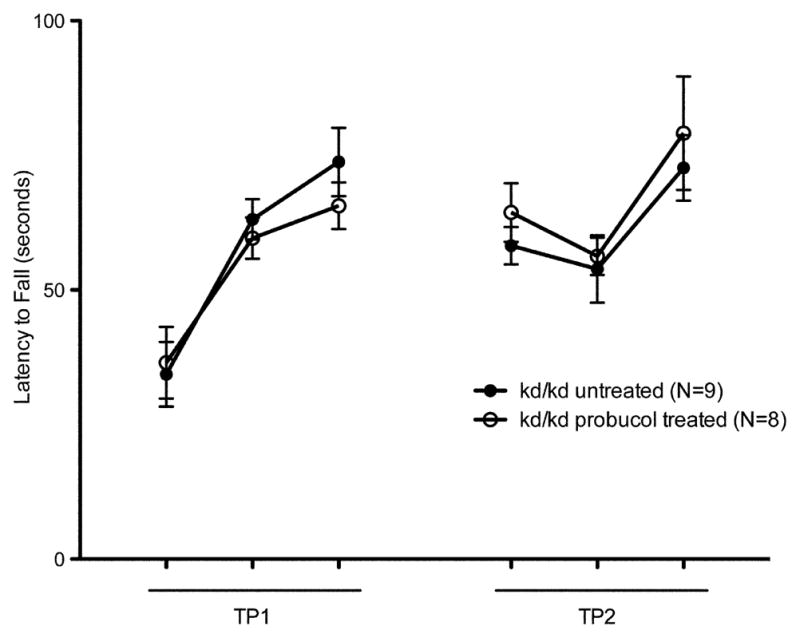
As probucol treatment was known to ameliorate the renal disease in a similar mouse model (Binder et al., 1999), B6.Pdss2kd/kd mice were given the same treatment (Zhang et al., 2010). There was no improvement in either the rotorod test (Fig. 4c) or locomotor performance (Fig. 4d), despite the fact that this treatment was extremely effective in preventing the kidney disease, especially in males. Five males that received probucol from birth to sacrifice at 206 days excreted 0.76 +/− 0.14 mg albumin per 24 hours and had histological scores of 0.6 +/− 0.49, compared to 60.21 mg of albumin per 24 hours and histological scores of 2.33 +/− 0.47 for 3 untreated B6.Pdss2kd/kd males of the same age. These 5 males were included in the data shown in Table 1 of a recently-published manuscript in which the probucol effect is discussed more fully (Falk et al., 2011). Representative H&E sections are shown in Fig. 5.
Fig. 5.
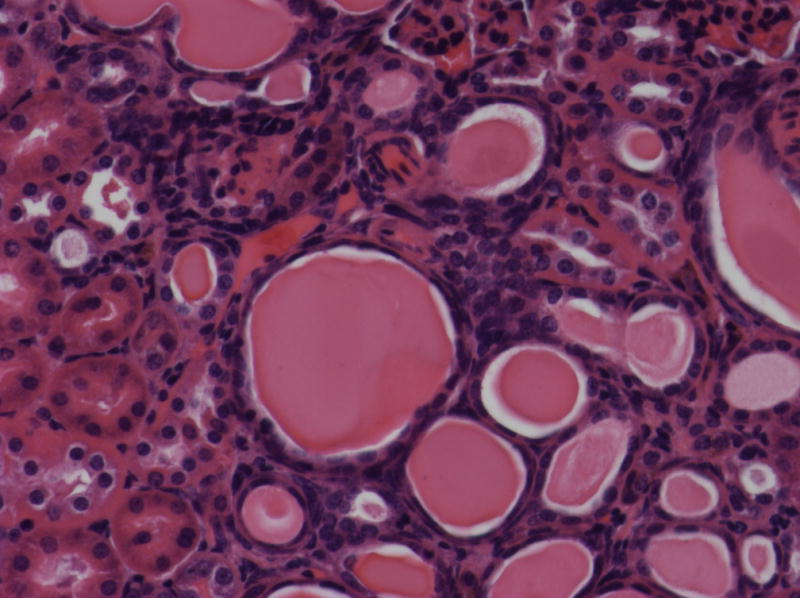
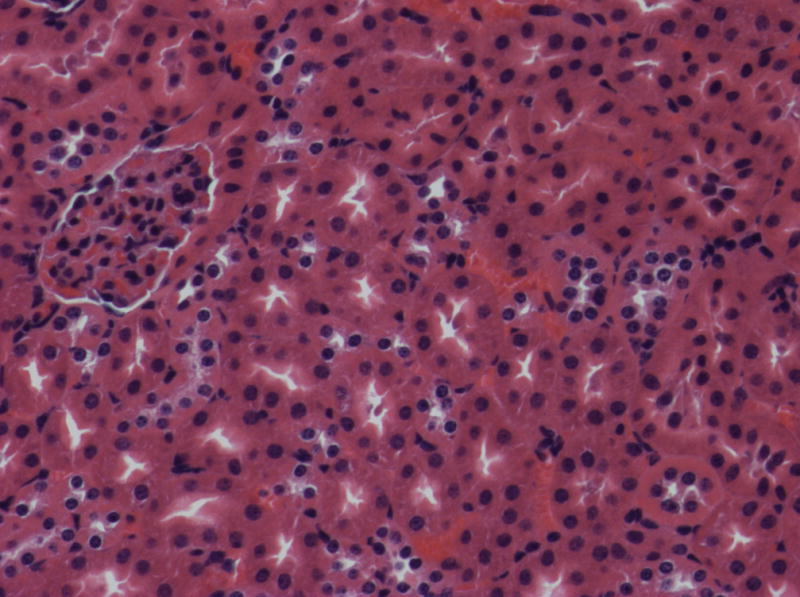
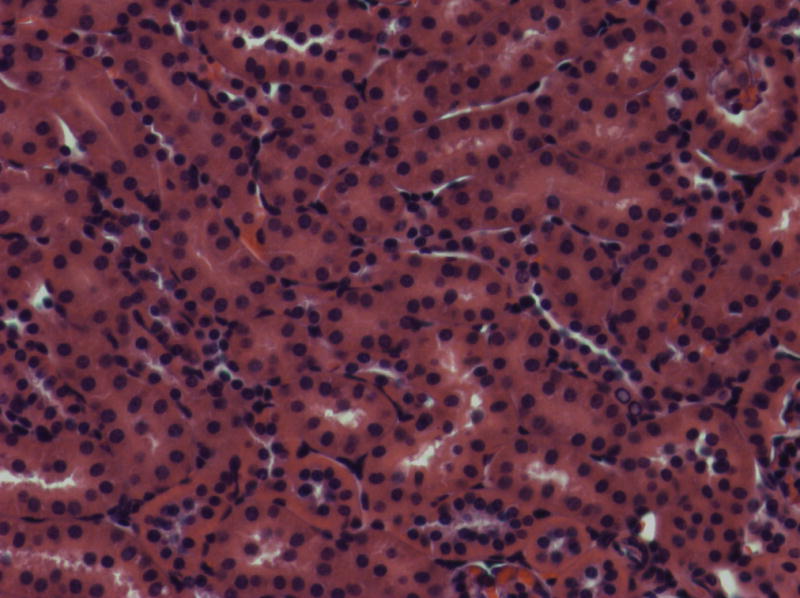
Histologic features of renal disease in kd/kd mice.
(a) Untreated B6.Pdss2kd/kd (kd/kd) mouse (213 days old; 81.9 mg albumin). Note the prominent dilated tubules, in which urinary protein is stained, and interstitial infiltrates. (b) kd/kd mouse treated from birth with probucol (206 days old; 0.85 mg albumin). (c) Wild-type, untreated B6 mouse (151 days old; 0.39 mg albumin).
3.6 Skeletal muscle respiratory capacity
Complex I-dependent (malate+glutamate+ADP) oxygen flux was reduced by 35% to 58% between each age-matched pair of male B6.Pdss2kd/kd mice and B6 animals studied on a given day, with a significant reduction by a mean of 49% in kd/kd mutants across all three pairs (p = 0.041) (Fig 6). Addition of cytochrome C (Cyt C) showed no increased rate to suggest altered permeability of the outer mitochondrial membrane in the permeabilized muscle fiber preparations, with significantly lower oxygen flux maintained in permeabilized muscle of B6.Pdss2kd/kd mutants relative to that of B6 control mice (p = 0.0288). Trends toward a 46% mean decrease in combined complex I plus complex II-dependent respiratory capacity were also seen (p = 0.0858), which did not reach a level of statistical significance perhaps due to small sample size. Similar trends of decreased B6.Pdss2kd/kd mice skeletal muscle fiber respiratory capacity were seen for all other respiratory states, including background respiratory capacity (malate + glutamate without supplemental ADP, p = 0.0631), maximal respiratory capacity in the presence of an uncoupler (FCCP, p = 0.1013), and complex I-inhibited respiratory capacity (rotenone, p = 0.2973). Thus, B6.Pdss2kd/kd missense mutant mice have decreased mitochondrial integrated respiratory capacity evident in skeletal muscle. These data in permeabilized skeletal muscle fibers are consistent with similar magnitude decreases in integrated respiratory capacity that we previously reported in isolated liver mitochondria from B6.Pdss2kd/kd missense mice relative to B6 wild-type controls (Peng et al., 2008). Therefore, decreased motor performance, particularly with provocative exercise testing, in B6.Pdss2kd/kd missense mutant animals may also be reflective of their inherently reduced skeletal muscle mitochondrial respiratory capacity.
Fig. 6.

Integrated mitochondrial respiratory capacity analysis in permeabilized skeletal muscle fibers from B6.Pdss2kd/kd mice relative to B6 (wild-type), age-matched controls. Background (malate + glutamate) oxygen flux was modestly reduced in permeabilized muscle fibers from three pairs of male missense mutant mice aged 125–143 days relative to wild-type controls aged 150–155 days (p = 0.0631). Complex I dependent (+ADP) oxygen flux was significantly reduced between 35% and 58% between each age-matched animal pair studied on a given day, and by a mean of 49% across all three pairs (p = 0.041). Assay of muscle fiber membrane permeability (cyt C) showed no increased rate to suggest poor tissue quality in any animal, with significantly lower rate seen overall in kd/kd mutants relative to B6 control (p = 0.0288). Non-significant trends toward decreased rates in kd/kd mutants were seen for complex I + complex II dependent respiratory capacity (succinate, mean 46% decrease, p = 0.0858), maximal respiratory capacity in the presence of an uncoupler (FCCP, p = 0.1013), and complex I- inhibited respiratory capacity (rotenone, p = 0.2973).
CI, complex I; Cyt C, cytochrome C; CI + CII, complex I and complex II; FCCP, carbonyl cyanide-ptrifluoromethoxyphenylhydrazone; B6, wild-type control mice; kd/kd, B6.Pdss2kd/kd missense mutant mice. Bars represent mean and standard error of three independent experiments per strain. *p < 0.05.
3.7. Brain Histology
To assess the histological effects of depletion of Pdss2 from the dopaminergic neurons, midbrain slices from conditional-knockout (B6.DAT/cre,Pdss2loxP/loxP), B6.Pdss2kd/kd missense mutants and B6.Pdss2loxP/loxP control mice were evaluated. This analysis revealed a striking loss of tyrosine hydroxylase positive neurons from the substantia nigra of the conditional knockout animals as compared to the B6.Pdss2loxP/loxP control (Figs 7a, 7b, and 7c). Further stereologic analysis of the substantia nigra confirmed a 62.4% decrease (p<0.001) in the number of tyrosine hydroxylase positive neurons (Fig 7c). Interestingly, staining for tyrosine hydroxylase was not completely lost from the ventral tegmental area (VTA) neurons (Fig 7b). VTA neurons that are also dopaminergic express DAT (the promoter used to generate the B6.Pdss2 knock-out), so the greater effect observed in the substantia nigra does not appear to be promoter specific. Analysis of the striatum region that is innervated from the nigral neurons corroborated the results, showing almost complete lack of staining for tyrosine hydroxylase and the dopamine transporter (data not shown) in the knockout mice. Conversely, the region of nucleus accumbens receiving connections from VTA was positive for tyrosine hydroxylase. Histological analysis thus revealed that knocking out Pdss2 from dopaminegic neurons resulted in relatively selective damage of the nigrostriatal innervations, whereas the mesolimbic pathway was less affected. This implies that the nigral neurons are most susceptible to CoQ10 deficiency. Overall, quantitative histological analysis confirms that the B6.DAT/cre,Pdss2loxP/loxP knockout mouse model recapitulates the selective nigral degeneration observed in human Parkinson’s disease. Similarly, analysis of the B6.Pdss2kd/kd missense mutants reveals modest, yet statistically significant decrease in TH immunoreactive neurons in midbrain sections (p<0.004), corresponding to an 18.3% reduction in TH positive neurons (Fig 7d, 7e and 7f).
Fig. 7.
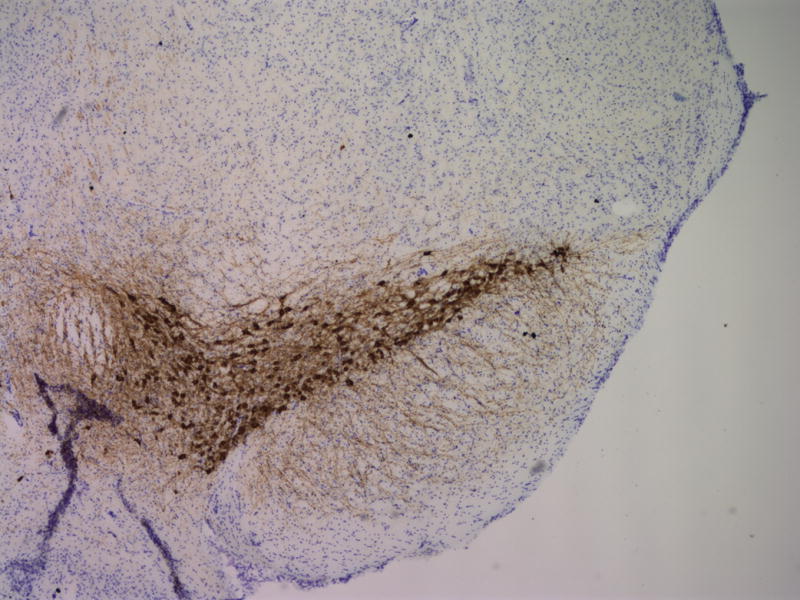
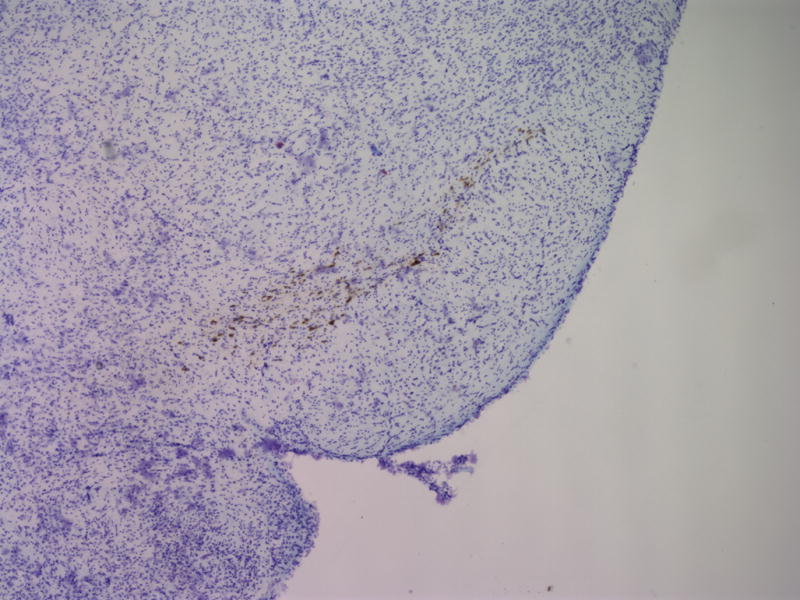
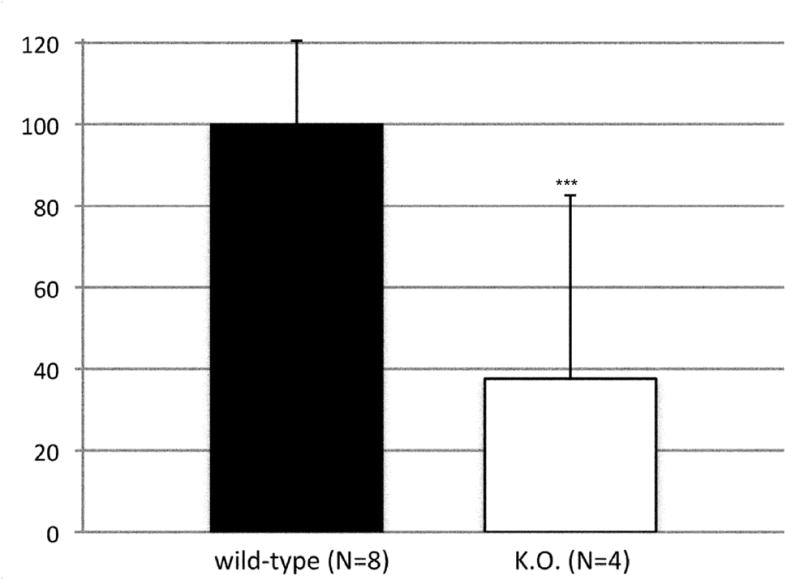
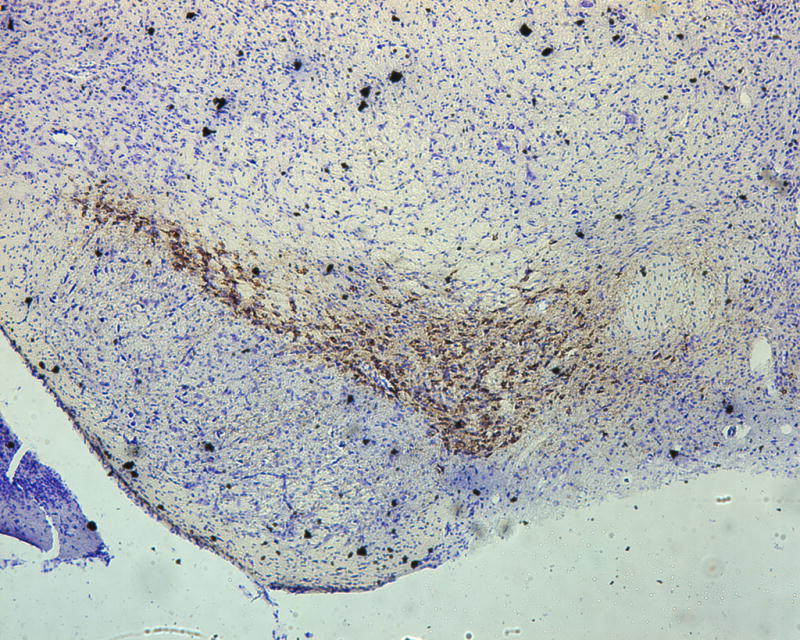
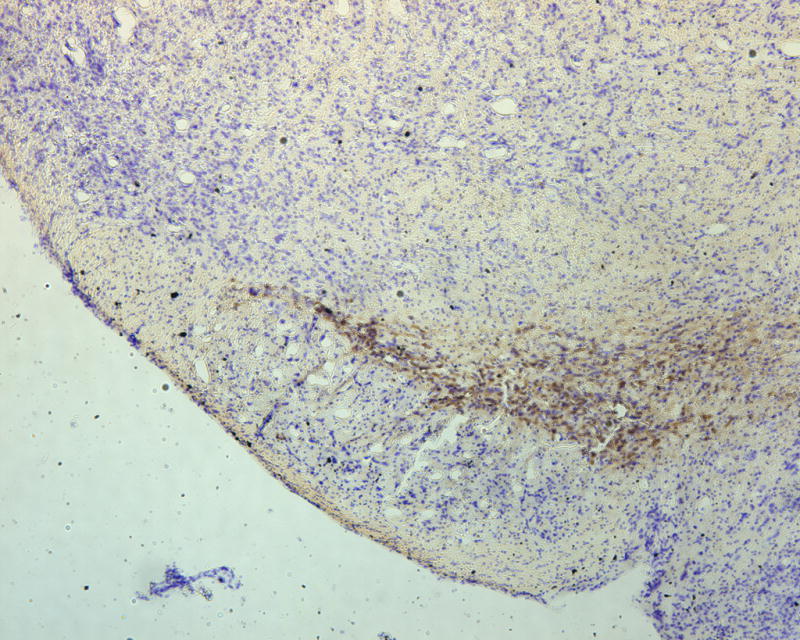
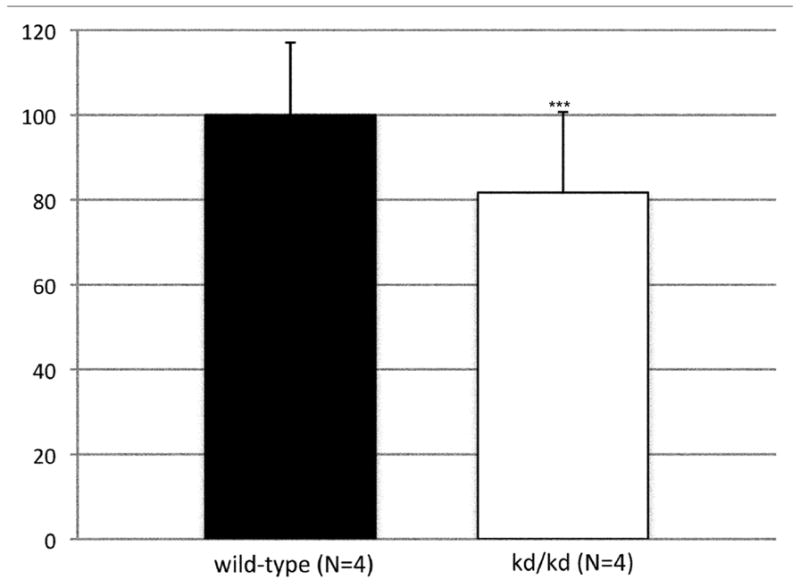
Brain Histology in K.O. and kd/kd mice. TH immunoreactive neurons and Nissl stained cells in SNpc from (a) B6.Pdss2loxP/+.Slc6a3+/− (control) and (b) B6.DAT/cre,Pdss2loxP/loxP (K.O.) mice. (c) Stereological analysis of reduced number of TH-positive neurons in knock out mice, represented as percent reduction compared to control (p<0.0004). (d) B6 (wild-type) control (e) B6.Pdss2kd/kd (kd/kd) mice TH immunoreactive neurons and Nissl stained cells in Snpc. (f) Stereological analysis of reduced number of TH-positive neurons in kd/kd mice, represented as percent reduction compared to wild-type control (p<0.004). ***p<0.001
4. Discussion
The only overt clinical disease phenotype that has been previously observed in studies of Pdss2kd/kd missense mutant mice was restricted to severe renal glomerular disease. Only one human patient with a Pdss2 defect has been reported to date, and this was a severely affected infant who had Leigh syndrome with nephropathy (Lopez et al., 2006). The patient had refractory seizures, became progressively hypotonic, had difficulties feeding because of exhaustion, and died at 8 months of age from severe, refractory, focal status epilepticus. Because of the neurologic symptoms that were evident in this patient, we sought to evaluate B6.Pdss2kd/kd mice for functional evidence of a central neurologic defect.
The results in this report demonstrate that B6.Pdss2kd/kd mice do indeed manifest significant deficits in motor coordination and locomotor activity by 2 months of age. They have significant impairment in several behavioral tests, and these defects are recapitulated with increased severity in the B6.DAT/cre,Pdss2loxP/loxP knockout model. As the rotarod challenge paradigm measures balance and coordination and has been commonly used as an indicator of brain insult in various disease states, the data suggest that the mutation in both the B6.Pdss2kd/kd mice missense and B6.DAT/cre,Pdss2loxP/loxP knockout mice causes neuromuscular impairment. Further locomotor and balance testing confirmed these findings of altered motor coordination.
Although the results with the conditional knockouts can be attributed to a mutation that was targeted only to dopaminergic neurons, the findings with the B6.Pdss2kd/kd mice could involve either neurological or muscular deficits. Integrated mitochondrial respiratory capacity was assayed in freshly permeabilized skeletal muscle fibers from these mice, and significantly reduced complex I-dependent respiratory capacity was observed. There was also a trend toward decreased complex I plus complex II-dependent respiratory capacity relative to age- and gender-matched B6 wild-type controls. Similar magnitude reductions in complex I and complex II-dependent respiratory capacity, as well as tissue CoQ content, were previously observed in freshly isolated liver mitochondria from B6.Pdss2kd/kd missense mutant mice (Peng et al., 2008). We conclude that mitochondrial respiratory capacity is indeed reduced in skeletal muscle from missense mutant mice, as is expected given the global tissue distribution of Pdss2 and the central role of CoQ in electron transport. Thus, skeletal muscle respiratory dysfunction likely contributes to the impaired motor coordination and locomotor activity observed in B6.Pdss2kd/kd mice.
Once mitochondrial dysfunction and severely reduced motor coordination were both clearly established, midbrain histologic sections were analyzed for dopaminergic nigrostriatal degeneration. Both the B6.Pdss2kd/kd missense mutants and B6.DAT/cre,Pdss2loxP/loxP conditional knockout animals exhibited significantly reduced TH-positive neurons by immunohistochemical TH-staining. This corresponded to a 62.4% reduction in animals for which the Pdss2 gene was selectively deleted in dopaminergic neurons (knockout model) and an 18.3% reduction in B6.Pdss2kd/kd missense mice.
Despite the fact that CoQ10 supplementation and probucol treatment significantly ameliorate the kidney disease phenotype, this did not occur with regard to the neuromuscular tests. The probucol effect was especially informative. Despite the fact that the defect in B6.Pdss2kd/kd is a deficiency of coenzyme Q, probucol is a more effective treatment of the renal disease than CoQ10 supplementation (Falk et al., 2011). The 5 probucol-treated males discussed in section 3.5 excreted only 0.76 +/− 0.14 mg albumin per 24 hours, which is the most objective indication that these mice were not experiencing any sign of kidney disease. Nevertheless, their performance in both the rotarod and “home cage” activity measurements were not significantly different from those of untreated B6.Pdss2kd/kd mice (Figure 4). This demonstrates that the neuromuscular difficulties were independent of any renal disease.
The fact that two treatments that can ameliorate the kidney disease had no effect on the neuromuscular difficulties is indeed disheartening. This could indicate that neither CoQ10 nor probucol was able to reach the target tissue well enough to be effective, or that additional factors that have not yet been identified are involved. Nevertheless, we conclude that the B6.Pdss2kd/kd missense mutant mouse offers a novel model for the study of pathogenesis in Parkinson’s disease. This is supported by the combined evidence of selective degeneration of dopaminergic neurons in the substantia nigra, robust impairment of motor coordination, and recapitulation of the phenotype in conditional knockouts.
Highlights.
Pdss2 deficient mice exhibit severe neuromuscular defects as demonstrated by behavioral testing
Marked reduction in TH-positive cells in the substantia nigra reflects neuromuscular impairment
Probucol prevented kidney disease, but not neuromuscular phenotypes, in Pdss2 missense mutants
Acknowledgments
We thank Dr. M. Flint Beal for helpful discussions, and the members of the Morphology Core for Molecular Studies in Digestive and Liver Diseases for histologic preparations. This work was supported by grants from the National Institutes of Health (R01-DK55852 to D.L.G and K-08-DK073545 to M.J.F) as well as the Gisela and Dennis Alter Chair in Pediatric Neonatology, and the Joseph Strokes Jr. Investigator program (H.I.). The content is solely the responsibility of the authors, and does not necessarily represent the official views of the NIH.
Role of the funding source
The funding sources played no role in determining the study design, analysis or interpretation of the data, or the content of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Backman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, Tomac AC. Characterization of a mouse strain expressing Cre recombinase from the 3′ untranslated region of the dopamine transporter locus. Genesis. 2006;44:383–390. doi: 10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- Beal MF. Experimental models of Parkinson's disease. Nat Rev Neurosci. 2001;2:325–334. doi: 10.1038/35072550. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson's disease. Ann N Y Acad Sci. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Binder CJ, Weiher H, Exner M, Kerjaschki D. Glomerular overproduction of oxygen radicals in Mpv17 gene-inactivated mice causes podocyte foot process flattening and proteinuria: A model of steroid-resistant nephrosis sensitive to radical scavenger therapy. Am J Pathol. 1999;154:1067–1075. doi: 10.1016/S0002-9440(10)65359-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloem BR, Irwin I, Buruma OJ, Haan J, Roos RA, Tetrud JW, Langston JW. The MPTP model: versatile contributions to the treatment of idiopathic Parkinson's disease. J Neurol Sci. 1990;97:273–293. doi: 10.1016/0022-510x(90)90225-c. [DOI] [PubMed] [Google Scholar]
- Colotla VA, Flores E, Oscos A, Meneses A, Tapia R. Effects of MPTP on locomotor activity in mice. Neurotoxicol Teratol. 1990;12:405–407. doi: 10.1016/0892-0362(90)90061-g. [DOI] [PubMed] [Google Scholar]
- Dell KM, Li YX, Peng M, Neilson EG, Gasser DL. Localization of the mouse kidney disease (kd) gene to a YAC/BAC contig on Chromosome 10. Mamm Genome. 2000;11:967–971. doi: 10.1007/s003350010188. [DOI] [PubMed] [Google Scholar]
- Falk MJ, Polyak E, Zhang Z, Peng M, King R, Maltzman JS, Okwuego E, Horyn O, Nakamaru-Ogiso E, Ostrovsky J, Xie LX, Chen JY, Marbois B, Nissim I, Clarke CF, Gasser DL. Probucol ameliorates renal and metabolic sequelae of primary CoQ defieiency in Pdss2 mutant mice. EMBO Mol Med. 2011;3:410–427. doi: 10.1002/emmm.201100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- Lemieux H, Gneiger E. Preparation of permeabilized muscle fibers for diagnosis of mitochondrial respiratory function. Mitochondrial Physiology Network. 2008;12.22:1–4. [Google Scholar]
- Lemieux H, Votion M-D, Gneiger E. Mitochondrial respiration in permeabilized fibers: Needle Biopsies from horse skeletal muscle. Mitochondrial Physiology Network. 2009;12.23:1–4. [Google Scholar]
- Lopez LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, Dimauro S, Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79:1125–1129. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF, Hulse EV. An inherited kidney disease of mice resembling human nephronophthisis. J Med Genet. 1971;8:41–48. doi: 10.1136/jmg.8.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura K, Kabuto H, Makino H, Ogawa N. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J Neurosci Methods. 1997;73(1):45–48. doi: 10.1016/s0165-0270(96)02211-x. [DOI] [PubMed] [Google Scholar]
- Peng M, Falk MJ, Haase VH, King R, Polyak E, Selak M, Yudkoff M, Hancock WW, Meade R, Saiki R, Lunceford AL, Clarke CF, Gasser DL. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. 2008;4:e1000061. doi: 10.1371/journal.pgen.1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Jarett L, Meade R, Madaio MP, Hancock WW, George AL, Jr, Neilson EG, Gasser DL. Mutant prenyltransferase-like mitochondrial protein (PLMP) and mitochondrial abnormalities in kd/kd mice. Kidney Int. 2004;66:20–28. doi: 10.1111/j.1523-1755.2004.00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas G, Lopez-Martin E, Guerra MJ, Labandeira-Garcia JL. The overall rod performance test in the MPTP-treated-mouse model of Parkinsonism. J Neurosci Methods. 1998;83:165–175. doi: 10.1016/s0165-0270(98)00078-8. [DOI] [PubMed] [Google Scholar]
- Saiki R, Lunceford AL, Shi Y, Marbois B, King R, Pachuski J, Kawamukai M, Gasser DL, Clarke CF. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am J Physiol Renal Physiol. 2008;295:F1535–1544. doi: 10.1152/ajprenal.90445.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki R, Nagata A, Kainou T, Matsuda H, Kawamukai M. Characterization of solanesyl and decaprenyl diphosphate synthases in mice and humans. FEBS J. 2005;272:5606–5622. doi: 10.1111/j.1742-4658.2005.04956.x. [DOI] [PubMed] [Google Scholar]
- Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience. 1994;59:401–415. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- Sedelis M, Schwarting RK, Huston JP. Behavioral phenotyping of the MPTP mouse model of Parkinson's disease. Behav Brain Res. 2001;125:109–125. doi: 10.1016/s0166-4328(01)00309-6. [DOI] [PubMed] [Google Scholar]
- Shults CW, Haas RH, Passov D, Beal MF. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann Neurol. 1997;42:261–264. doi: 10.1002/ana.410420221. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Gasser DL, Rappaport EF, Falk MJ. Cross-platform expression microarray performance in a mouse model of mitochondrial disease therapy. Mol Genet Metabol. 2010;99:309–318. doi: 10.1016/j.ymgme.2009.10.179. [DOI] [PMC free article] [PubMed] [Google Scholar]