

Abstract
T cell-dependent development of anti-factor VIII (FVIII) antibodies that neutralize FVIII activity is a major obstacle to replacement therapy in hemophilia A. To create a less immunogenic therapeutic protein, recombinant FVIII can be modified to reduce HLA binding of epitopes based on predicted anchoring residues. Here, we used immunoinformatics tools to identify C2 domain HLA DR epitopes and predict site-specific mutations that reduce immunogenicity. Epitope peptides corresponding to original and modified sequences were validated in HLA binding assays and in immunizations of hemophilic E16 mice, DR3 and DR4 mice and DR3xE16 mice. Consistent with immunoinformatics predictions, original epitopes are immunogenic. Immunization with selected modified sequences lowered immunogenicity for particular peptides and revealed residual immunogenicity of incompletely de-immunized modified peptides. The stepwise approach to reduce protein immunogenicity by epitope modification illustrated here is being used to design and produce a functional full-length modified FVIII for clinical use.
Keywords: Epitope, T cell, Factor VIII, Inhibitors, De-immunization
1. Introduction
Hemophilia A, an X-linked recessive genetic bleeding disorder caused by mutations of coagulation factor VIII (FVIII), is the most common type of bleeding disorder with a prevalence of 1 in 5000–10000 live male births. FVIII is a protein that has a six-domain structure, A1-A2-B-A3-C1-C2. The full-length gene product is processed intracellularly into a heavy chain and light chain in connection with a divalent metal ion. The heavy chain of FVIII contains the A1, A2 and a residual part of the B domain; whereas the light chain contains A3, C1 and C2 domains [1]. Previous studies have shown that sites in the A1, A2, A3 and C2 domains of FVIII are involved in its pro-coagulant interactions [2–4] which lead to the activation of Factor IX in the “intrinsic” coagulation pathway, thrombin activation and fibrin formation. Hence, mutations in FVIII lead to excessive bleeding and can be life-threatening.
Current treatment for hemophilia A consists of repeated intravenous administration of plasma-derived FVIII or recombinant FVIII [5]. However the development of anti-FVIII antibodies that neutralize FVIII activity (called “inhibitors”) is a major obstacle to continued FVIII replacement therapy. Up to 30% of all hemophiliacs develop inhibitors during the course of treatment. Nearly 50% of severe hemophiliacs, who have major deletions in the FVIII gene, develop inhibitors because they have no detectable FVIII activity or circulating FVIII protein [6, 7].
Inhibitor development has been shown to be a T cell dependent process in both animal studies and clinical evidence from hemophiliac AIDS patients [8–13]. This process is dependent on antigen presenting cell (APC) signaling to CD4+ T helper cells [9, 11–13]. Disruption of CD40/CD40L interactions by MR1, an anti-CD40L antibody, completely prevented inhibitor formation and both Th1 and Th2 responses in hemophilic mice after FVIII treatment [10]. T cell epitopes in FVIII have been identified that appear to play a pivotal role in this T cell-dependent immune response to FVIII [14]. In the present study, we focused on T cell epitopes in the C2 domain because it has been reported that this domain contains many B cell epitopes [15]. In addition, T cell epitopes have been mapped to C2 domain residues 2181–2312, [16, 17] 2191–2210, 2241–2290, and 2291–2330 [15, 18]. Some of these T cell epitopes overlap with inhibitory antibody binding sites [15, 18–20].
Because the immunogenicity of FVIII [21] and lack of tolerance to the non-native protein in hemophilia patients both contribute to inhibitor formation, the therapeutic efficacy of FVIII can be limited [5, 22–25]. Methods that successfully reduce the immunogenicity of FVIII would improve quality of life and reduce treatment-associated costs for individuals affected by inhibitors. A number of approaches to induce FVIII tolerance are used in the clinic but have significant limitations [26–28]. Recent studies demonstrate a role for regulatory T cells in controlling antibody responses, including antibodies against FVIII, and offer new immunomodulatory approaches to preventing inhibitor formation [29–31]. Here, we report a novel method to develop a modified version of FVIII by T cell epitope modifications designed to reduce binding of FVIII epitopes to MHCII, leading to a reduction or failure of this antigen being presented to T cells, while preserving the function of FVIII. This process is called de-immunization for functional therapeutics or “DeFT” [32, 33].
An early application of de-immunization by T cell epitope modification came from alanine substitutions to the MHC II anchoring residues Y73, K74, R77, E80, and D82 of Staphylokinase, individually or in combination. Warmerdam et al. showed that mutations to alanine reduced or eliminated T cell response and clinical immunogenicity of Staphylokinase [34], presumably due to reduction in HLA binding affinity.
We employed a stepwise process to identify and de-immunize FVIII C2 epitopes: 1) in silico epitope mapping, 2) validation of computational predictions in vitro and in vivo, 3) selection of epitopes to modify, 4) iterative in silico modification and analysis accounting for potential immunogenicity and conservation and 5) validation of carefully selected modified sequences in vitro and in vivo.
In previous studies, we found that predicted epitope peptides in the C2 domain of FVIII could be modified to reduce MHC II binding, leading to reduced immune recall (antigenicity) in vitro in FVIII KO mice (E16, H-2b) immunized with FVIII [35]. In the current study, we have confirmed and extended these studies to humanized HLA-DR transgenic mice, using state-of-the art immunoinformatics tools to select and de-immunize the immunodominant epitopes. We provide evidence that the approach can be used to design de-immunized peptides that are less likely to cause de novo immunogenic responses. The algorithms EpiMatrix and ClustiMer were used to select promiscuous T cell epitopes that would bind to multiple human MHC II alleles [36]. OptiMatrix was used to iteratively analyze anchor residue substitutions so as to identify modifications that would interfere with MHC binding while accounting for naturally conserved substitutions [33]. Peptides representing the original predicted epitopes (ORG) and their OptiMatrix-defined modifications (MOD) were then evaluated in an HLA binding assay. Finally, the peptides (or FVIII) were used to immunize mice so as to measure their potential for immune recall (antigenicity) and de novo (immunogenicity) responses. We found that 6 ORG peptides (2191-O, 2231-O, 2254-O, 2271-O, 2299-O and 2310-O) were immunogenic in DR3 transgenic mice, in DR4 mice and in DR3 mice crossed to FVIII knockout (KO) hemophilic mice (DR3×E16). After identification of ORG peptides, the MODs were tested in de novo immunogenicity studies. We successfully identified several MODs that were non-immunogenic in our mouse models, although selected MOD peptides retained immunogenicity following immunization of mice with the peptides. This stepwise approach using immunoinformatics tools followed by in vitro and in vivo validation may be of use to develop novel FVIII therapies and for the development of less immunogenic bio-therapeutics.
2. Materials and Methods
2.1 Tools for de-immunization: the EpiMatrix system
The EpiMatrix computational epitope mapping method has been published [36, 37]. Briefly, the sequence of human FVIII C2 domain was parsed into overlapping 9-mer frames and the immunogenic potential of each frame was assessed against a panel of eight archetypal HLA class II alleles (DRB1*0101, DRB1*0301, DRB1*0401, DRB1*0701, DRB1*0801, DRB1*1101, DRB1*1301 and DRB1*1501) that represent >90% of MHC diversity in the human population [38]. A ‘Z’ score for each analyzed 9-mer is assigned by EpiMatrix. Any peptide scoring above 1.64 on the EpiMatrix ‘Z’ scale (approximately the top 5% of the random peptide set) has a significant chance of binding to the MHCII molecule and is known as a ‘hit.’ Peptides scoring above 2.32 on the scale (the top 1%) are extremely likely to bind; most published T cell epitopes fall within this range of scores. These EpiMatrix results were then screened for the presence of T cell epitope clusters with ClustiMer. Regions of high immunogenic potential, defined as having a score above 10 on our immunogenicity scale, were designated “original” (ORG) peptides and verified by in vitro methods [39].
Next, to identify amino acids within the identified immunodominant epitope regions (ORG) that were suitable for modification, we evaluated the contribution of each amino acid in these regions to HLA binding using OptiMatrix (part of the EpiVax tool kit for de-immunization). OptiMatrix begins with looking at “critical” residues, which contribute most to MHC binding affinity across multiple 9-mer frames and multiple HLA alleles, and averages the contribution of each amino acid to binding across 9-mer binding frames and HLA alleles. The program then iteratively substitutes all 19 alternative amino acids in any given position of a protein sequence (with operator-defined input that may limit the list to naturally conserved variants) and then reanalyzes the predicted immunogenicity of the sequence, following that change. The predicted disruptive impact on the overall immunogenic potential of the MOD peptides was verified by in vitro methods as well.
2.2 Synthetic peptides
Original (ORG) and modified (MOD) peptides corresponding to the epitope selections were synthesized (New England Peptide, Gardner, MA, USA) by 9-fluoronylmethoxy-carbonyl (Fmoc) method on an automated Rainen Symphony/Protein Technologies synthesizer. We only used peptides that were purified to 80% or higher by HPLC, as these give more accurate results.
2.3 HLA binding assay
Class II HLA binding assays were used to estimate the affinity of predicted epitope sequences for multiple HLA alleles. We employed a competition-based HLA binding assay initially described by Steere et al [40]. and adapted for higher throughput by EpiVax. In 96-well plates, non-biotinylated test peptides at 100 μM competed for binding to purified Class II molecules (50 nM) against a biotinylated standard peptide at a fixed concentration (0.1 μM) for 24 hours at 37°C to reach equilibrium. Class II complexes were then captured on ELISA plates using pan anti-Class II antibodies (L243, anti-HLA-DR). Plates were washed and incubated with Europium-labeled Streptavidin for 1 hour at room temperature. The Europium activation buffer was added to develop the plates for 15–20 minutes at room temperature before they were read on a Time Resolved Fluorescence (TRF) plate reader. All assays were performed in triplicate. Binding assays were performed for 5 alleles: DRB1*0101, DRB1*0301, DRB1*0401, DRB1*0701 and DRB1*1501, which provided a broad representation of class II HLA allele binding pockets [38].
2.4 Mice
The hemophilic FVIII KO mouse strain (E16) has been characterized previously [41, 42]. Hemizygous adult male and homozygous female E16 mice, aged 6–10 weeks, were used in this study. These mice, on a C57Bl/6 background, have murine MHCII molecules (H-2b). HLA class II (HLA-DR) transgenic mice expressing HLA-DR4 (DRB1*0401) were obtained from Taconic Farms. HLA-DR3 (DRB1*0301) mice were provided by EpiVax; these mice were originally obtained from Dr. Chella David (Mayo Clinic, Rochester, MN) under commercial license. Previous studies have shown that expression of HLA-DR molecules in these mice is comparable with regular murine MHCII molecules. However, it should be noted that these mice completely lack all endogenous murine MHCII genes, so that the T cell responses are restricted by the transgenic HLA-DR [43, 44]. Hemophilic DR3 (DR3×E16) mice were generated by the crossing of E16 mice to DR3 mice and typed for expression of MHCII and FVIII mutation. Female F1, DR3+/H-2b+/FVIII−/−, were used in this study after genotyping. All animals were housed and bred in pathogen-free micro isolator cages at the animal facilities operated by the University of Maryland School of Medicine, and animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Maryland, School of Medicine. The genotypes of hemophilic mice were confirmed by polymerase chain reaction (PCR) analysis of genomic DNA extracted from tail samples.
2.5 Mouse immunizations
Groups of mice were immunized with either rFVIII or 50 μl indicated peptides, 3 μg each, emulsi ed in CFA (Sigma, St. Louis, MO) per injection site via the foot pad and the base of tail. Draining LNs (superficial inguinal and popliteal) were removed 14 days post immunization, and the dissociated cells were washed and cultured in 96-well microculture plates (0.5×106 cells per well) in complete RPMI 1640 (supplemented with 1mM sodium pyruvate, 100 mg/ml penicillin and streptomycin, 2mM glutamine, 50 μM 2-mercaptoethanol, 20 mM HEPES, 1× nonessential amino acids, 2.0% FBS and 0.5% hemophilic mouse serum). 2–3 mice were used per group and experiments repeated 3–4 times with reproducible results.
2.6 T cell Proliferation Assay
Thymidine incorporation and T cell proliferation assays were performed at University of Maryland, as previously described [45]. Briefly, pooled LN cells from each group were cultured in 96-well plates (0.5×106 cells per well) with dilutions of antigen (3-fold dilutions ranging 0.1–10 μg/ml) for 48 hours in RPMI 1640 with 2% FBS and 0.5% hemophilic mouse serum. After 48 hours, cultures were pulsed with 1 μCi/well of [3H]-methyl-thymidine (Amersham Life Sciences, Arlington Heights, IL) and incubated for another 16–20 hours. Cells were then harvested on glass fiber filters. Incorporation of thymidine was measured by a beta counter (MicroBeta TriLux; PerkinElmer Life and Analytical Sciences, Waltham, MA). Values were expressed in counts per minute (CPM) with antigens (peptides). Each sample was run in triplicate or quadruplicate. The mean CPM from each of the wells were calculated and the background subtracted for statistical calculations, and expressed as delta CPM (Δ CPM). Background levels (i.e., CPM without antigen) were typically less than 6000 CPM. Each Δ CPM point is the mean ± SEM of triplicate or quadruplicate cultures.
2.7 Statistical analysis
Statistical differences were determined using a two-tailed unpaired Student’s t-test. A p value < 0.05 was considered to be statistically significant.
3. Results
3.1 Six high HLA binding epitope peptides identified in C2 domain of FVIII
To identify immunogenic “clusters” in T cell epitope regions of FVIII’s C2 domain, we utilized the EpiMatrix and ClustiMer algorithms. The 9-mer epitopes and epitope clusters for eight prevalent HLA class II alleles are shown in Figure. 1. We identified six epitope clusters at amino acid positions 2191–2210, 2231–2251, 2254–2269, 2271–2289, 2299–2317 and 2310–2330. Their corresponding cluster scores are 4.15, 10.69, 5.62, 10.17, 12.92 and 20.86, respectively. All six clusters overlap with published FVIII epitopes as shown in Figure 1 [46, 47] although the in silico tools provided a detailed analysis that more finely maps the epitopes in this FVIII domain.
Fig. 1. HLA Class II epitope map of the FVIII C2 domain as predicted by EpiMatrix.
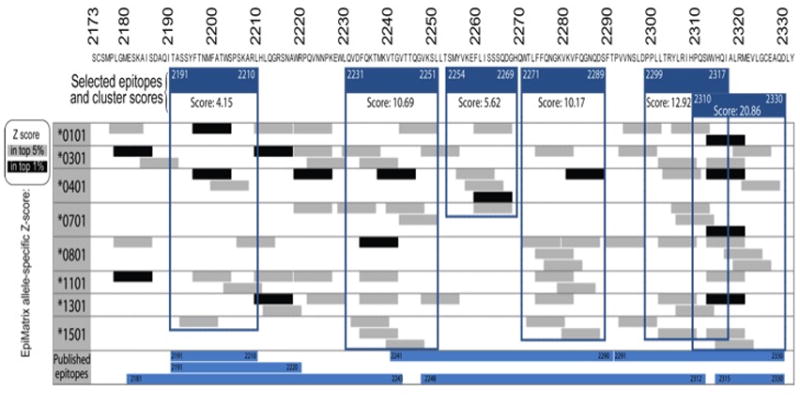
EpiMatrix-predicted 9-mer hits for 8 prevalent HLA class II alleles are aligned along the human FVIII C2 sequence. Any peptide scoring above 1.64 on the EpiMatrix ‘Z’ scale (top 5%) is considered to be a potential epitope (gray bars). Peptides scoring above 2.32 on the scale (top 1%) are extremely likely to bind MHC (black bars). Published epitopes determined by experimental methods are similarly aligned (blue bars). EpiMatrix identified clusters of epitopes spanning residues 2191–2210, 2231–51, 2254–69, 2271–89, 2299–2317 and 2310–30 that are framed in blue. The EpiMatrix algorithm finely maps the positions of published epitopes.
3.2 Epitope modification lowers HLA binding
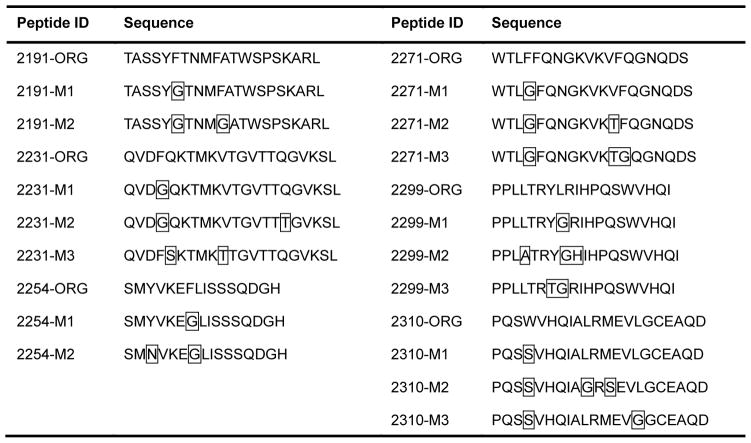
We used OptiMatrix to identify and evaluate the contribution of critical HLA binding amino acid residues in the most immunogenic regions of the FVIII C2 domain. These amino acids were substituted with residues predicted to have a disruptive impact on HLA binding. To minimize perturbations to protein structure and function, substitutions were crafted with the aim of reducing the propensity of the peptide to bind to MHC while using the minimal number of substitutions. Several alternative modifications (MODs) were prepared for each epitope ranging from one to three mutations; their nomenclature reflects the increasing severity of the mutations (M1–4) (Table 1).
Table 1. FVIII C2 domain epitope sequences and modifications.
An EpiMatrix analysis of the FVIII C2 domain was performed to computationally map T-cell epitopes that may contribute to FVIII immunogenicity. OptiMatrix, an algorithm that identifies amino acids that contribute to HLA binding and creates mutants that reduce predicted HLA affinity, was used to identify modified FVIII C2 sequences that may lower FVIII immunogenicity. Original sequences are indicated by ORG; modified by MOD. Boxed amino acids contribute significantly to HLA binding and are considered optimal sites for amino acid substitution. De-immunization was performed by replacing these residues with those predicted to disrupt binding to HLA.
|
Next, we assessed the effects of the modifications by comparing the HLA binding capacity of original (ORG) and MOD peptides in a binding assay screen. Peptide binding to DRB1*0101, DRB1*0301, DRB1*0401, DRB1*0701, and DRB1*1501 was evaluated. As shown in Fig. 2, at a concentration of 100 μM, 2191-O bound strongest, as an average across HLA alleles, and 2231-O weakest among the ORG peptides. 2231-O inhibited competitor binding by an average of only 20%. Lower than predicted HLA interactions may be explained by peptide aggregation or folding that interferes in the binding assay. As predicted, most of the peptide modifications lowered the average binding over the array of HLA alleles tested with the single exception of 2231 MODs. As the 2231-O peptide did not bind well to begin with, modifications to 2231-O did not significantly reduce binding.
Fig. 2. Direct binding of C2 peptides to HLA alleles.

In 96-well plates, non-biotinylated test peptide at a concentration of 100 μM competes for binding to purified Class II molecules (50 nM). Blue bar is average across alleles. Dots represent percent inhibition of binding to HLA alleles: DRB1*0101, DRB1*0301, DRB1*0401, DRB1*0701 and DRB1*1501. The amino acid modifications were made to prevent binding across all HLA DR alleles; however, the predicted changes were not always universal and therefore some peptides could be expected to demonstrate residual binding to selected MHC.
It is perhaps not surprising that the different modifications had a variable effect on average HLA binding, and increasing the number of substitutions decreased the propensity of the peptide to bind. For example, 2191-M2, containing two substitutions, significantly reduced binding whereas the corresponding 2191-M1, containing only one, only moderately reduced binding (Fig. 2).
3.3 De novo immunogenicity of computationally predicted FVIII T-cell epitopes
To initially evaluate the immunodominant peptides predicted by our programs, E16 and DR transgenic mice were immunized with FVIII and then their LN cells stimulated in vitro with the identified ORG peptides in C2. Our results verified that FVIII could be processed to present the ORG peptides to the immune system of these mice to various degrees dependent on the MHC type of the host (data not shown); i.e. the peptides stimulated recall immune responses, indicating that the sequences are antigenic.
Next, to validate the de novo immunogenicity of ORG sequences, E16 and DR transgenic mice were immunized with all six ORG peptides and the ensuing immune response to the peptides evaluated in T cell proliferation assays using lymph node cells. As shown in Fig. 3A, only 2191-O stimulated proliferative responses in E16 mice. 2271-O had the highest response in DR3 mice (Fig. 3B). Further evidence of this selective immunodominance is revealed in DR3×E16 mice as T cell proliferation responses to both 2191-O and 2271-O peptides depended on the expression of H-2b as well as DR3 in the mice (Fig. 3C). Note that the response to 2271-O was independent of whether the DR3 mice were on a hemophilic background, suggesting that dissimilarity between the human FVIII epitope and the murine FVIII epitope may be a key driver of the immune response for this HLA allele. Indeed, there is a non-conservative sequence mismatch between mouse and human FVIII in a predicted DR3 epitope in 2271-O that could explain the immunogenicity of this peptide (Table 2). In DR4 mice, peptides 2231-O and 2310-O showed the highest responses (Fig. 3D). These four immunodominant epitopes (2191-O, 2271-O, 2231-O, 2310-O) were selected as candidates to study how modifications impact recall immune responses in mice immunized with ORG peptides (antigenicity) and de novo immune responses in mice exposed to MOD peptides (immunogenicity).
Fig. 3. De novo immunogenicity of C2 peptides in hemophilic and HLA DR transgenic mice.
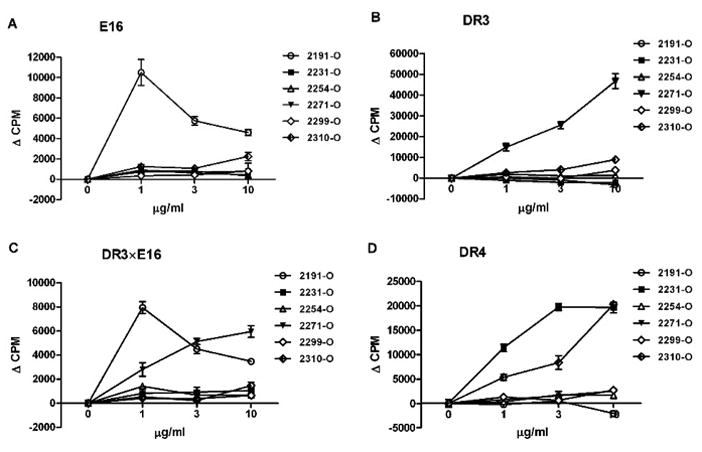
Six ORG peptides (2191-O, 2231-O, 2254-O, 2271-O, 2299-O, and 2310-O) were tested for their ability to stimulate T cells proliferation in different mouse strains: (A) E16, (B) DR3, (C) DR3×E16, and (D) DR4. Mice were immunized with mixed 6 ORG peptides (3 μg each), emulsified in CFA. After two weeks, T cell proliferation was determined using pooled lymph nodes (LN) cells from each mouse strain (n=2). Cells were primed with indicated ORG peptides at concentrations of 0, 1, 3 and 10 μg/ml. Values are means ± SEM of triplicate cultures, p < 0.05.
Table 2. Impact of FVIII C2 domain species-specific sequences on epitope immunogenicity.
Mouse and human FVIII sequences were aligned using ClustalW (SwissProt Accession Q06194 and P00451, respectively). EpiMatrix-predicted epitope cluster sequences in the human C2 domain are shown aligned with the corresponding mouse sequences and EpiMatrix hits for the MHC alleles tested in mice. Human sequence mismatches are colored red, DR3 hits as black bars, DR4 as gray and I-Ab as white. Epitope cluster immunogenicity is observed in HLA DR mice, which produce mouse FVIII, where non-conservative mismatches overlap with EpiMatrix hits. Figure 3 illustrates C2 ORG epitope immunogenicity.

|
To further understand these results, we explored the possibility that immunodominance observed in vivo was a function of peptide competition for MHC binding. 2191-O, which is immunodominant in E16 mice, was used to test this hypothesis. E16 mice were immunized with (i) 2191-O alone, (ii) a pool of ORG peptides including 2191-O or (iii) OVA with 2191-O. 2191-O-specific responses were then evaluated by proliferation assay in the presence of competitor peptide or OVA (Appendix, Figure 1). We found that 2191-O stimulated similar dose-dependent proliferation levels no matter whether mice were immunized with 2191-O alone (Appendix, Figure 1A) or with a pool of ORG peptides (Appendix, Figure 1B). Moreover, proliferation was not affected by the presence of competitor ORG peptides over a range of concentrations. E16 mice co-immunized with 2191-O and OVA mounted an immune response against both immunogens (Appendix, Figure 1C). Taken together, these results suggest that there was no competition among ORG peptides and presentation of different ORG peptides to T cells corresponded to their respective MHC binding affinities.
3.4 Modifications of immunodominant peptides lower their antigenicity and immunogenicity
To investigate the effects of epitope modification on the antigenicity and immunogenicity of immunodominant peptides, DR3×E16 mice and DR4 mice were immunized with either the ORG immunodominant peptides or the corresponding MOD epitopes. These studies were undertaken in consideration of the potential applications of de-immunized FVIII. Recall immune responses (antigenicity) measured in MOD peptide stimulations of LN cells taken from ORG peptide-immunized mice would correspond to the T cell response to de-immunized FVIII in hemophiliacs previously treated with wild type FVIII. De novo immune responses (immunogenicity) to MOD peptides in stimulations of LN cells taken from MOD peptide-immunized mice would represent the T cell response to de-immunized FVIII in FVIII-naïve hemophiliacs.
DR3×E16 mice immunized with 2191-O peptide, the H-2b dominant sequence, showed lower recall responses with 2191-M2 but not 2191-M1, indicating the importance of the F2200G mutation for diminished antigenicity (Fig. 4A, Table 1). Notably, the nearly identical response observed for 2191-O and 2191-M1 suggests that the F2196G mutation of 2191-M1 alone would not be sufficient to de-immunize this epitope in FVIII. Additional mutations would be required such as F2200G, which lowered antigenicity as shown for 2191-M2 (Figure 4A). Although this epitope is dominant in the context of mouse MHC, and not for the HLA alleles tested here, this result illustrates an important de-immunization principle: ORG-reactive T cells that may be present in FVIII-treated hemophiliacs could still produce undesired immune responses to a de-immunized FVIII bearing motifs for particular HLA alleles. Careful selection of modifications and testing of each of the MODs for potential immunogenicity, as done in this stepwise approach, would be necessary to produce an effectively de-immunized FVIII.
Fig. 4. Epitope modification of immunodominant C2 peptides lowers immune recall responses to original sequences.
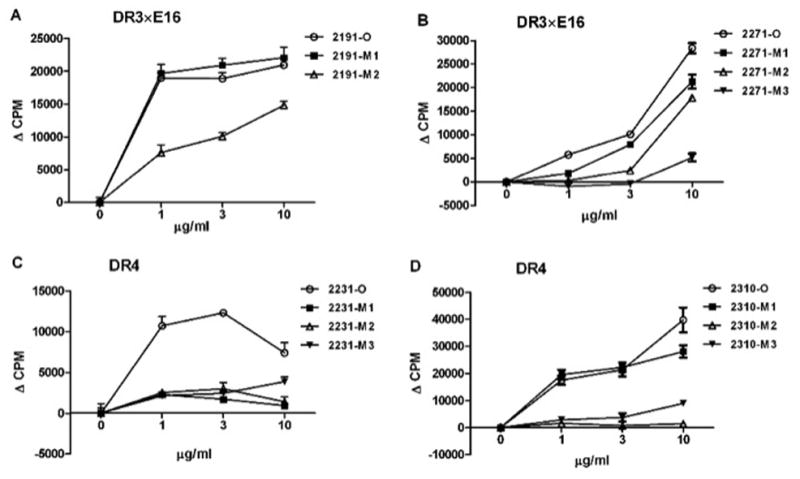
Corresponding HLA dominant peptides and their modifications were tested for their ability to lower antigenicity of protein in DR3×E16 and DR4 mice. A: DR3×E16 mice were immunized with 2191-O. B: DR3×E16 mice were immunized with 2271-O; C: DR4 mice were immunized with 2231-O. D: DR4 mice were immunized with 2310-O. 3 μg of each peptide was emulsified in CFA. Subcutaneous injection was given to mice via a hind footpad and the base of tail. Two weeks later, T cell proliferation was determined using pooled LN cells from immunized mice (n=2). Cells were primed with indicated ORG and MOD peptides at concentrations of 0, 1, 3 and 10 μg/ml. Values are means ± SEM of triplicate cultures, p < 0.05.
In DR3×E16 mice immunized with 2271-O peptide, the DR3 immunodominant epitope, all modifications lowered antigenicity in direct proportion to the number of strong anchor substitutions (Fig. 4B, Table 1). In DR4 mice immunized with 2231-O, 2231-M1 and -M2 showed the lowest antigenicity (Fig. 4C). The F2234G mutation present in 2231-M1 appears to be sufficient to reduce antigenicity as Q2246T does not further diminish the cellular response to 2231-M2 (Table 1).
We also found that in DR4 mice immunized with 2310-O, all modifications lowered antigenicity. While the W2313S mutation reduced proliferation only at the highest peptide concentration tested for 2310-O, the additional mutation L2319G and M2321S (M2) or L2324G (M2) reduced antigenicity with dose dependency (Fig. 4D and Table 1).
To measure immunogenicity of modified epitopes, DR3×E16 mice were immunized with either a mixture of 2191 (2191-M1, -M2) or 2271 MODs (2271-M1, -M2, -M3). We found that 2191-M2 was less immunogenic than 2191-M1; this was similar to the antigenicity results in 2191-O immunized mice (Fig. 5A). Notably, 2191-M1 was as immunogenic under these conditions as the 2191-O peptide. One explanation for the similarity may be that 2191-M1-reactive T cells cross-react with 2191-O, although this type of assay is not designed to test T cell cross-reactivity.
Fig. 5. Epitope modification of immunodominant C2 peptides reduces de novo immunogenicity.
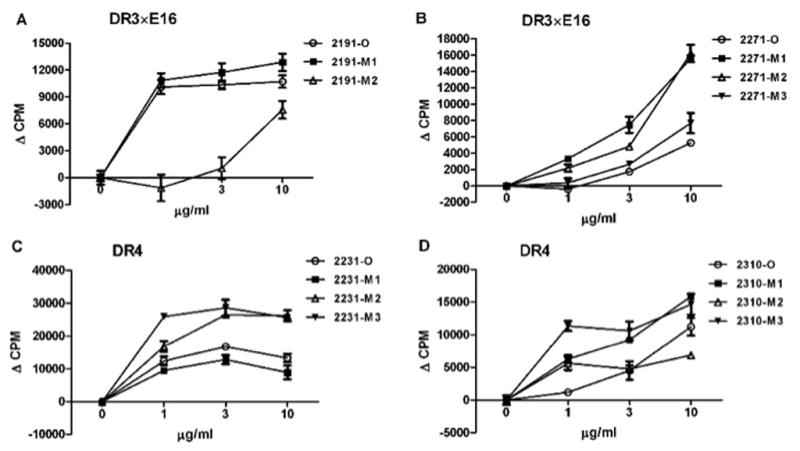
Corresponding HLA dominant peptides and their modifications were tested for their ability to lower immunogenicity of protein in DR3×E16 and DR4 mice. A: DR3×E16 mice were immunized with a mixture of 2191-M1 and 2191-M2; B: DR3×E16 mice were immunized with a mixture of 2271-M1, 2271-M2 and 2271-M3; C: DR4 mice were immunized with a mixture of 2231-M1, 2231-M2 and 2231-M3; D: DR4 mice were immunized with a mixture of 2310-M1, 2310-M2 and 2310-M3. 3 μg of each peptide was emulsified in CFA. Subcutaneous injection was given to mice via a hind footpad and the base of tail. Two weeks later, T cell proliferation was determined using pooled LN cells from immunized mice (N=2). Cells were primed with indicated ORG and MOD peptides at concentrations of 0, 1, 3 and 10 μg/ml. Values are means ± SEM of triplicate cultures, p < 0.05.
In DR3×E16 mice immunized with a mixture of 2271 MODs (2271-M1, -M2, M3), 2271-M3 was less immunogenic than the other MODs (Fig. 5B) as was observed in the antigenicity study. Notably, 2271-O cross-reacted to a lesser extent with MOD-reactive T cells than did 2291-O to T cells raised in response to immunization with its own MODs. Additionally, in DR4 mice immunized with a mixture of 2231 MODs (2231-M1, -M2, -M3), we found that 2231-M1 was less immunogenic than 2231-O and the other MODs of 2231 (Fig. 5C). 2310-M2 was less immunogenic, even at a high concentration in vitro, compared with the other MODs of 2310 in DR4 mice immunized with these MODs (Fig. 5D).
We conclude that modified sequences 2191-M2, 2271-M3, 2231-M1 and 2310-M2 successfully disrupt the ability of the peptides to bind to HLA molecules, thus leading to diminished T cell recognition. Where single and double amino acid substitutions were made, the double modifications were more likely to reduce antigenicity and immunogenicity. This stepwise approach to de-immunization of C2 resulted in the selection of peptides with diminished antigenicity and immunogenicity. As illustrated here, de-immunization involves 1) immunoinformatics, 2) in vitro validation of computational predictions using HLA binding assays and 3) in vivo validation studies that measure the impact of epitope modifications on antigenicity and immunogenicity. The approach may be useful to guide design of improved recombinant FVIII that elicits lower levels of FVIII neutralizing antibodies.
4. Discussion
The formation of anti-FVIII antibodies, also known as inhibitors, is a major obstacle to FVIII gene replacement therapy in hemophilia A patients. After intravenous administration of FVIII, the immune response mounted is dependent on CD4+ T helper cells, as has been demonstrated by numerous studies in mice and humans [8–14]. The essential role of CD4+ T cells in FVIII inhibitor development was confirmed in the case of hemophiliacs infected with HIV-1. Their inhibitor titers decreased as CD4+ T cells declined in the course of HIV disease, and returned when CD4 T cells were normalized following HIV treatment [13, 48]. Further evidence for the importance of CD4+ T help for the development inhibitory antibodies cells is the observation of somatic hypermutation, a known T cell dependent process [49–51]. More recently, interference with T-B cell interactions in hemophilic mice was shown to reduce inhibitor formation [10, 13].
If CD4 T cells play a central role in the development of inhibitors, then T cell epitopes are the key to reducing immunogenicity. T cell epitopes are peptides generated from exogenous antigens that bind to MHCII of antigen presenting cells (APC). Determination of immunodominant T cell epitopes is an important step in designing less immunogenic biotherapeutics, such as a de-immunized version of human FVIII [15, 18–20]. In this study, we demonstrated that immunogenic peptides in the C2 domain of FVIII could be modified to reduce MHCII binding and that these modified peptides also display reduced antigenicity in a hemophilic E16 mouse model (H-2b) and HLA-DR transgenic mice.
In the present study, we measured predicted epitope immunogenicity in two distinct mouse models that differ in FVIII and class II MHC expression: (i) a FVIII knockout mouse (E16) that expresses no C2 domain at any point in development and normally expresses mouse class II MHC and (ii) HLA DR transgenic models, which express whole FVIII protein throughout development, a single human class II MHC allele and no mouse class II MHC. The immunogenicity study results, in which mice were immunized with C2 domain ORG sequences, are consistent with predicted responses for the two different phenotypes in terms of epitope recognition. A genetic cross of the two mouse models, E16xDR3 mice, exhibited an intermediate immune response, as expected of mice that have endogenous FVIII and both mouse and human MHC.
To be specific, 2191-O immunogenicity in E16 mice is consistent with the absence of tolerance induction to this sequence (the mice do not have FVIII) and the presence of MHC (I-Ab) motifs in the sequence (Table 2). The lack of immunogenicity observed for the other ORG peptides, despite an expected lack of tolerance induction to these sequences as well, may be explained by the absence of I-Ab MHC binding motifs in their sequences, with the exception of 2231-O, which does have I-Ab motifs. 2231-O may not be immunogenic for other reasons, such as peptide processing.
With regard to the HLA DR transgenic models, peptides predicted to be HLA ligands were immunogenic when the sequence contained mismatches between the human FVIII sequence used in the immunization and the corresponding sequence in the native mouse FVIII protein (Table 2). To be immunogenic, the mismatches had to occur within 9-mer sequences that also contained HLA DR3 or DR4 binding motifs. As these 9-mers could be presented to the mouse T cell and would be ‘foreign’ to the mouse, they stimulate T cell proliferation. Peptides that were predicted to bind HLA DR3 and/or DR4 but did not stimulate immune responses were found to contain no mouse/human sequence mismatches. As these sequences do not appear ‘foreign’ to the mouse, no immune response is expected because tolerance induction mechanisms either limited or eliminated auto-reactive T cells. Notably, immunization with complete Freund’s adjuvant did not break tolerance, but was sufficient to tip the regulatory and effector T cell population balance in favor of proliferation only in response to the ‘foreign’ sequences.
Following selection of immunogenic ORG peptides in the E16 and DR transgenic mouse models, we set out to determine to what degree modification of these peptides would reduce their immune potential in terms of both antigenicity and de novo immunogenicity. In proliferation assays, MOD peptides were less antigenic than ORG peptides (Figure 4). The only exception to this finding was 2191-M1, which exhibited immunogenic properties similar to the parent ORG across a ten-fold range of peptide concentrations. In general, lower antigenicity was observed for those peptides that had two, rather than just one amino acid substitution. For example, in DR3×E16 mice, T cells showed the lowest responses to 2191-M2 and 2271-M3; in DR4 mice, T cells showed the lowest responses to 2231-M1, 2231-M2 and 2310-M2. With the exception of 2231-M1, these MODs have two or more amino acids substitutions; where only a single residue was mutated, the reduction in proliferative response was less pronounced. Similarly in terms of de novo immunogenicity, in general, the more mutations, the lower the observed proliferative response. While proliferative responses to 2191-M1 and 2271-M1 in DR3×E16 mice and 2231-M2 and 2310-M3 in DR4 mice were observed, 2191-M2, 2271-M3, 2231-M1 and 2310-M2 were significantly lower.
Despite the link between the number of mutations and decreased proliferation, the de-immunization process cannot be simply reduced to an accumulation of random mutations that would eventually abrogate HLA binding and reduce immunogenicity. Rather, it is a process that involves minimizing the number of targeted substitutions that will broadly minimize immunogenicity across genetically diverse HLA types while also minimizing their impact on bioactivity. Taken together, the data suggest that multiple and carefully selected amino acid substitutions were advantageous to disruption of HLA binding and reduction of immunogenicity, and thus, multiple, select modifications may contribute to better de-immunization of the C2 domain. We estimate that as few as one or two amino acid changes in a single immunodominant epitope in the C2 domain could be sufficient to lower the immunogenicity threshold enough to render a C2 domain protein non-immunogenic. However, it is likely that additional modifications would be required not only in C2 but also in other domains to fully de-immunize whole FVIII. Given that sequence modifications may also impact protein structure and/or function, it would be optimal to minimize the number of deimmunizing mutations to avoid detrimental effects on structural and/or functional integrity. Future studies using intact C2 domain and whole FVIII will address the critical balance between these competing factors that influence clinical success.
In summary, we identified immunodominant T cell epitopes in the C2 domain of FVIII and developed de-immunized versions by targeted sequence modification. We synthesized original and modified peptides to evaluate their abilities to stimulate HLA-restricted T cells. We found that while 2271-O was reactive in DR3 transgenic mice, 2231-O and 2310-O responses were mainly observed in DR4 mice. Peptide 2191-O was apparently H-2b-restricted, in that it caused T cell proliferation only in E16 and E16xDR3 mice. We also found that the MODs 2191-M2, 2231-M1, 2271-M2 and 2310-M2 showed lower HLA binding and proliferation responses compared with their respective ORGs. Although most modifications resulted in reduced immunogenicity, some modifications remained partially immunogenic.
This stepwise approach to reducing biotherapeutic immunogenicity promises to advance the design and production of a modified, yet functional, recombinant FVIII protein. It is anticipated that a T-cell epitope-modified FVIII would be useful for the treatment of existing and new hemophilia A patients. New patients would not develop T cells that are required for FVIII inhibitor production in the absence of immunogenic T-cell epitopes. Reduced FVIII-specific T cell activation in existing patients would prevent inhibitor production, too, as the association of T cells and inhibitor development in the case of HIV-infected hemophiliacs suggests: as CD4+ T cells decline in the course of disease, so do FVIII inhibitors; restoration of T cell function by treatment with antiretroviral therapy results in the return of inhibitors to previous levels [13,42]. Finally, the de-immunization approach applied to FVIII will serve as a guide for improvement of other immunogenic biologics.
Supplementary Material
Selected ORG peptides were tested for competition in the stimulation of T cells: (A) E16 mice were immunized with 2191-O peptides, and LN cells stimulated with 2191 in the presence of the other identified peptides in the sub-panels 1, 2 and 3. (B) E16 mice were immunized with a mixture of 2191-O, 2271-O, 2254-O, and 2310-O peptides, and LN cells stimulated with 2191 in the presence of the other identified peptides as in (A). (C) Similarly, E16 mice were immunized with a mixture of 2191-O and ovalbumin (OVA), and LN cells stimulated with OVA in the presence of the other identified peptides. Mixtures of peptide at 3 μg each were emulsified in CFA and two weeks later, T cell proliferation was determined using pooled LN cells from immunized mice (n=4). Cells were primed with indicated mixed ORG peptides or OVA at concentrations of 0, 1, 3 and 10 μg/ml. Values are means ± SEM of triplicate cultures, p < 0.05.
Highlights.
Resistance to Factor VIII therapy for hemophiliacs is attributed to immunogenicity.
We use a systematic approach to reduce immunogenicity by epitope modification.
Immunoinformatic tools find FVIII T-cell epitopes and de-immunizing modifications.
Pinpoint mutations reduce FVIII epitope immunogenicity in transgenic mouse models.
De-immunization can be broadly applied to lower the risk of immunogenic biologics.
Acknowledgments
This work was supported by NIH grant R43 HL088834-01 (ADG). We are grateful to Timothy Messitt and Patrick Adair for their careful review of the manuscript.
Footnotes
Conflict-of-interest
Two of the contributing authors, Anne S. De Groot and William D. Martin, are senior officers and majority shareholders at EpiVax, Inc., a privately owned biotechnology company located in Providence, RI. Leonard Moise and David Scott hold stock options in EpiVax. These authors acknowledge that there is a potential conflict of interest related to their relationship with EpiVax and attest that the work contained in this research report is free of any bias that might be associated with the commercial goals of the company.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lenting PJ, van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998;92:3983–3996. [PubMed] [Google Scholar]
- 2.Fay PJ, Koshibu K, Mastri M. The A1 and A2 subunits of factor VIIIa synergistically stimulate factor IXa catalytic activity. J Biol Chem. 1999;274:15401–15406. doi: 10.1074/jbc.274.22.15401. [DOI] [PubMed] [Google Scholar]
- 3.Fay PJ, Scandella D. Human inhibitor antibodies specific for the factor VIII A2 domain disrupt the interaction between the subunit and factor IXa. J Biol Chem. 1999;274:29826–29830. doi: 10.1074/jbc.274.42.29826. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien LM, Medved LV, Fay PJ. Localization of factor IXa and factor VIIIa interactive sites. J Biol Chem. 1995;270:27087–27092. doi: 10.1074/jbc.270.45.27087. [DOI] [PubMed] [Google Scholar]
- 5.Lusher JM, Lee CA, Kessler CM, Bedrosian CL. The safety and efficacy of B-domain deleted recombinant factor VIII concentrate in patients with severe haemophilia A. Haemophilia. 2003;9:38–49. doi: 10.1046/j.1365-2516.2003.00708.x. [DOI] [PubMed] [Google Scholar]
- 6.Lacroix-Desmazes S, Misra N, Bayry J, Artaud C, Drayton B, Kaveri SV, Kazatchkine MD. Pathophysiology of inhibitors to factor VIII in patients with haemophilia A. Haemophilia. 2002;8:273–279. doi: 10.1046/j.1365-2516.2002.00624.x. [DOI] [PubMed] [Google Scholar]
- 7.Saint-Remy JM, Lacroix-Desmazes S, Oldenburg J. Inhibitors in haemophilia: pathophysiology. Haemophilia. 2004;10(Suppl 4):146–151. doi: 10.1111/j.1365-2516.2004.01009.x. [DOI] [PubMed] [Google Scholar]
- 8.Ragni MV, Bontempo FA, Lewis JH. Disappearance of inhibitor to factor VIII in HIV-infected hemophiliacs with progression to AIDS or severe ARC. Transfusion. 1989;29:447–449. doi: 10.1046/j.1537-2995.1989.29589284147.x. [DOI] [PubMed] [Google Scholar]
- 9.Clark EA, Ledbetter JA. How B and T cells talk to each other. Nature. 1994;367:425–428. doi: 10.1038/367425a0. [DOI] [PubMed] [Google Scholar]
- 10.Reipert BM, Sasgary M, Ahmad RU, Auer W, Turecek PL, Schwarz HP. Blockade of CD40/CD40 ligand interactions prevents induction of factor VIII inhibitors in hemophilic mice but does not induce lasting immune tolerance. Thromb Haemost. 2001;86:1345–1352. [PubMed] [Google Scholar]
- 11.Holder MJ, Wang H, Milner AE, Casamayor M, Armitage R, Spriggs MK, Fanslow WC, MacLennan IC, Gregory CD, Gordon J, et al. Suppression of apoptosis in normal and neoplastic human B lymphocytes by CD40 ligand is independent of Bc1–2 induction. Eur J Immunol. 1993;23:2368–2371. doi: 10.1002/eji.1830230948. [DOI] [PubMed] [Google Scholar]
- 12.Klaus GG, Choi MS, Lam EW, Johnson-Leger C, Cliff J. CD40: a pivotal receptor in the determination of life/death decisions in B lymphocytes. Int Rev Immunol. 1997;15:5–31. doi: 10.3109/08830189709068169. [DOI] [PubMed] [Google Scholar]
- 13.Qian J, Burkly LC, Smith EP, Ferrant JL, Hoyer LW, Scott DW, Haudenschild CC. Role of CD154 in the secondary immune response: the reduction of pre-existing splenic germinal centers and anti-factor VIII inhibitor titer. Eur J Immunol. 2000;30:2548–2554. doi: 10.1002/1521-4141(200009)30:9<2548::AID-IMMU2548>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 14.Lavigne-Lissalde G, Schved JF, Granier C, Villard S. Anti-factor VIII antibodies: a 2005 update. Thromb Haemost. 2005;94:760–769. doi: 10.1160/TH05-02-0118. [DOI] [PubMed] [Google Scholar]
- 15.Pratt KP, Qian J, Ellaban E, Okita DK, Diethelm-Okita BM, Conti-Fine B, Scott DW. Immunodominant T-cell epitopes in the factor VIII C2 domain are located within an inhibitory antibody binding site. Thromb Haemost. 2004;92:522–528. doi: 10.1160/TH03-12-0755. [DOI] [PubMed] [Google Scholar]
- 16.Healey JF, Barrow RT, Tamim HM, Lubin IM, Shima M, Scandella D, Lollar P. Residues Glu2181-Val2243 contain a major determinant of the inhibitory epitope in the C2 domain of human factor VIII. Blood. 1998;92:3701–3709. [PubMed] [Google Scholar]
- 17.Scandella D, Gilbert GE, Shima M, Nakai H, Eagleson C, Felch M, Prescott R, Rajalakshmi KJ, Hoyer LW, Saenko E. Some factor VIII inhibitor antibodies recognize a common epitope corresponding to C2 domain amino acids 2248 through 2312, which overlap a phospholipid-binding site. Blood. 1995;86:1811–1819. [PubMed] [Google Scholar]
- 18.Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Human CD4+ T-cell epitope repertoire on the C2 domain of coagulation factor VIII. J Thromb Haemost. 2003;1:1777–1784. doi: 10.1046/j.1538-7836.2003.00251.x. [DOI] [PubMed] [Google Scholar]
- 19.Hu GL, Okita DK, Conti-Fine BM. T cell recognition of the A2 domain of coagulation factor VIII in hemophilia patients and healthy subjects. J Thromb Haemost. 2004;2:1908–1917. doi: 10.1111/j.1538-7836.2004.00918.x. [DOI] [PubMed] [Google Scholar]
- 20.Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. Epitope repertoire of human CD4(+) T cells on the A3 domain of coagulation factor VIII. J Thromb Haemost. 2004;2:1385–1394. doi: 10.1111/j.1538-7836.2004.00850.x. [DOI] [PubMed] [Google Scholar]
- 21.Skupsky J, Zhang AH, Su Y, Scott DW. A role for thrombin in the initiation of the immune response to therapeutic factor VIII. Blood. 2009;114:4741–4748. doi: 10.1182/blood-2008-10-186452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bray GL, Gomperts ED, Courter S, Gruppo R, Gordon EM, Manco-Johnson M, Shapiro A, Scheibel E, White G, 3rd, Lee M. A multicenter study of recombinant factor VIII (recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. The Recombinate Study Group. Blood. 1994;83:2428–2435. [PubMed] [Google Scholar]
- 23.Ehrenforth S, Kreuz W, Scharrer I, Linde R, Funk M, Gungor T, Krackhardt B, Kornhuber B. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet. 1992;339:594–598. doi: 10.1016/0140-6736(92)90874-3. [DOI] [PubMed] [Google Scholar]
- 24.Kreuz W, Ettingshausen CE, Zyschka A, Oldenburg J, Saguer IM, Ehrenforth S, Klingebiel T. Inhibitor development in previously untreated patients with hemophilia A: a prospective long-term follow-up comparing plasma-derived and recombinant products. Semin Thromb Hemost. 2002;28:285–290. doi: 10.1055/s-2002-32664. [DOI] [PubMed] [Google Scholar]
- 25.Lusher JM, Arkin S, Abildgaard CF, Schwartz RS. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. Kogenate Previously Untreated Patient Study Group. N Engl J Med. 1993;328:453–459. doi: 10.1056/NEJM199302183280701. [DOI] [PubMed] [Google Scholar]
- 26.Barrow RT, Healey JF, Gailani D, Scandella D, Lollar P. Reduction of the antigenicity of factor VIII toward complex inhibitory antibody plasmas using multiply-substituted hybrid human/porcine factor VIII molecules. Blood. 2000;95:564–568. [PubMed] [Google Scholar]
- 27.Nilsson IM, Berntorp E, Zettervall O, Dahlback B. Noncoagulation inhibitory factor VIII antibodies after induction of tolerance to factor VIII in hemophilia A patients. Blood. 1990;75:378–383. [PubMed] [Google Scholar]
- 28.Oldenburg J, Schwaab R, Brackmann HH. Induction of immune tolerance in haemophilia A inhibitor patients by the ‘Bonn Protocol’: predictive parameter for therapy duration and outcome. Vox Sang. 1999;77(Suppl 1):49–54. doi: 10.1159/000056717. [DOI] [PubMed] [Google Scholar]
- 29.De Groot AS, Moise L, McMurry JA, Wambre E, Van Overtvelt L, Moingeon P, Scott DW, Martin W. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes”. Blood. 2008;112:3303–3311. doi: 10.1182/blood-2008-02-138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moghimi B, Sack BK, Nayak S, Markusic DM, Mah CS, Herzog RW. Induction of tolerance to factor VIII by transient co-administration with rapamycin. J Thromb Haemost. 2011;9:1524–1533. doi: 10.1111/j.1538-7836.2011.04351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miao CH, Harmeling BR, Ziegler SF, Yen BC, Torgerson T, Chen L, Yau RJ, Peng B, Thompson AR, Ochs HD, Rawlings DJ. CD4+FOXP3+ regulatory T cells confer long-term regulation of factor VIII-specific immune responses in plasmid-mediated gene therapy-treated hemophilia mice. Blood. 2009;114:4034–4044. doi: 10.1182/blood-2009-06-228155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Groot AS, Knopp PM, Martin W. De-immunization of therapeutic proteins by T-cell epitope modification. Dev Biol (Basel) 2005;122:171–194. [PubMed] [Google Scholar]
- 33.De Groot AS, Moise L. Prediction of immunogenicity for therapeutic proteins: state of the art. Curr Opin Drug Discov Devel. 2007;10:332–340. [PubMed] [Google Scholar]
- 34.Warmerdam PA, Plaisance S, Vanderlick K, Vandervoort P, Brepoels K, Collen D, De Maeyer M. Elimination of a human T-cell region in staphylokinase by T-cell screening and computer modeling. Thromb Haemost. 2002;87:666–673. [PubMed] [Google Scholar]
- 35.Moise SJL, Tassone R, McMurry JA, Martin WD, De Groot AS, Scott DW. De-Immunization of Human Factor VIII: Identification of Epitopes in the C2 Domain. Blood (ASH Annual Meeting Abstracts) 2008;112:1030. [Google Scholar]
- 36.Schafer JR, Jesdale BM, George JA, Kouttab NM, De Groot AS. Prediction of well-conserved HIV-1 ligands using a matrix-based algorithm, EpiMatrix. Vaccine. 1998;16:1880–1884. doi: 10.1016/s0264-410x(98)00173-x. [DOI] [PubMed] [Google Scholar]
- 37.De Groot AS, Jesdale BM, Szu E, Schafer JR, Chicz RM, Deocampo G. An interactive Web site providing major histocompatibility ligand predictions: application to HIV research. AIDS Res Hum Retroviruses. 1997;13:529–531. doi: 10.1089/aid.1997.13.529. [DOI] [PubMed] [Google Scholar]
- 38.Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, Hoffman S, Kubo RT, Chesnut RW, Grey HM, Sette A. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol. 1998;160:3363–3373. [PubMed] [Google Scholar]
- 39.De Groot AS. Immunomics: discovering new targets for vaccines and therapeutics. Drug Discov Today. 2006;11:203–209. doi: 10.1016/S1359-6446(05)03720-7. [DOI] [PubMed] [Google Scholar]
- 40.Steere AC, Klitz W, Drouin EE, Falk BA, Kwok WW, Nepom GT, Baxter-Lowe LA. Antibiotic-refractory Lyme arthritis is associated with HLA-DR molecules that bind a Borrelia burgdorferi peptide. J Exp Med. 2006;203:961–971. doi: 10.1084/jem.20052471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10:119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 42.Qian J, Collins M, Sharpe AH, Hoyer LW. Prevention and treatment of factor VIII inhibitors in murine hemophilia A. Blood. 2000;95:1324–1329. [PubMed] [Google Scholar]
- 43.Cheng S, Smart M, Hanson J, David CS. Characterization of HLA DR2 and DQ8 transgenic mouse with a new engineered mouse class II deletion, which lacks all endogenous class II genes. J Autoimmun. 2003;21:195–199. doi: 10.1016/s0896-8411(03)00120-3. [DOI] [PubMed] [Google Scholar]
- 44.Mangalam A, Rodriguez M, David C. A new humanized HLA transgenic mouse model of multiple sclerosis expressing class II on mouse CD4 T cells. Ann N Y Acad Sci. 2007;1103:112–117. doi: 10.1196/annals.1394.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lei TC, Scott DW. Induction of tolerance to factor VIII inhibitors by gene therapy with immunodominant A2 and C2 domains presented by B cells as Ig fusion proteins. Blood. 2005;105:4865–4870. doi: 10.1182/blood-2004-11-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reding MT, Wu H, Krampf M, Okita DK, Diethelm-Okita BM, Christie BA, Key NS, Conti-Fine BM. Sensitization of CD4+ T cells to coagulation factor VIII: response in congenital and acquired hemophilia patients and in healthy subjects. Thromb Haemost. 2000;84:643–652. [PubMed] [Google Scholar]
- 47.Jacquemin M, Vantomme V, Buhot C, Lavend’homme R, Burny W, Demotte N, Chaux P, Peerlinck K, Vermylen J, Maillere B, van der Bruggen P, Saint-Remy JM. CD4+ T-cell clones specific for wild-type factor VIII: a molecular mechanism responsible for a higher incidence of inhibitor formation in mild/moderate hemophilia A. Blood. 2003;101:1351–1358. doi: 10.1182/blood-2002-05-1369. [DOI] [PubMed] [Google Scholar]
- 48.Bray GL, Kroner BL, Arkin S, Aledort LW, Hilgartner MW, Eyster ME, Ragni MV, Goedert JJ. Loss of high-responder inhibitors in patients with severe hemophilia A and human immunodeficiency virus type 1 infection: a report from the Multi-Center Hemophilia Cohort Study. Am J Hematol. 1993;42:375–379. doi: 10.1002/ajh.2830420408. [DOI] [PubMed] [Google Scholar]
- 49.Jacquemin MG, Desqueper BG, Benhida A, Vander Elst L, Hoylaerts MF, Bakkus M, Thielemans K, Arnout J, Peerlinck K, Gilles JG, Vermylen J, Saint-Remy JM. Mechanism and kinetics of factor VIII inactivation: study with an IgG4 monoclonal antibody derived from a hemophilia A patient with inhibitor. Blood. 1998;92:496–506. [PubMed] [Google Scholar]
- 50.van Den Brink EN, Turenhout EA, Davies J, Bovenschen N, Fijnvandraat K, Ouwehand WH, Peters M, Voorberg J. Human antibodies with specificity for the C2 domain of factor VIII are derived from VH1 germline genes. Blood. 2000;95:558–563. [PubMed] [Google Scholar]
- 51.van den Brink EN, Bril WS, Turenhout EA, Zuurveld M, Bovenschen N, Peters M, Yee TT, Mertens K, Lewis DA, Ortel TL, Lollar P, Scandella D, Voorberg J. Two classes of germline genes both derived from the V(H)1 family direct the formation of human antibodies that recognize distinct antigenic sites in the C2 domain of factor VIII. Blood. 2002;99:2828–2834. doi: 10.1182/blood.v99.8.2828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Selected ORG peptides were tested for competition in the stimulation of T cells: (A) E16 mice were immunized with 2191-O peptides, and LN cells stimulated with 2191 in the presence of the other identified peptides in the sub-panels 1, 2 and 3. (B) E16 mice were immunized with a mixture of 2191-O, 2271-O, 2254-O, and 2310-O peptides, and LN cells stimulated with 2191 in the presence of the other identified peptides as in (A). (C) Similarly, E16 mice were immunized with a mixture of 2191-O and ovalbumin (OVA), and LN cells stimulated with OVA in the presence of the other identified peptides. Mixtures of peptide at 3 μg each were emulsified in CFA and two weeks later, T cell proliferation was determined using pooled LN cells from immunized mice (n=4). Cells were primed with indicated mixed ORG peptides or OVA at concentrations of 0, 1, 3 and 10 μg/ml. Values are means ± SEM of triplicate cultures, p < 0.05.