

Abstract
Mechanistic understanding of RP105 has been confounded by the fact that this TLR homolog has appeared to have opposing, cell type-specific effects on TLR4 signaling. While RP105 inhibits TLR4-driven signaling in cell lines and myeloid cells, impaired LPS-driven proliferation by B cells from RP105−/− mice has suggested that RP105 facilitates TLR4 signaling in B cells. We show here that modulation of B cell proliferation by RP105 is not a function of B cell-intrinsic expression of RP105, and identify a mechanistic role for dysregulated BAFF expression in the proliferative abnormalities of B cells from RP105−/− mice: serum BAFF levels are elevated in RP105−/− mice, and partial BAFF neutralization rescues aberrant B cell proliferative responses in such mice. These data indicate that RP105 does not have dichotomous effects on TLR4 signaling, and emphasize the need for caution in interpreting the results of global genetic deletion.
Introduction
Immunobiological understanding of RP105 was shaped by its discovery and initial analysis in B cells. Ab-mediated cross-linking of RP105 leads to B cell activation and proliferation, providing protection against radiation- and steroid-induced apoptosis (1), but sensitization to apoptosis in response to BCR ligation (2). Anti-RP105-driven proliferative responses have been studied in detail; roles for the Lyn/CD19/Vav1 complex, Bruton's tyrosine kinase, protein kinase CβI/II and MEK have been found (1, 3–5).
Cloning revealed RP105 to be a TLR homolog, albeit one lacking a signaling TIR domain. Further, while LPS-driven TLR4 signaling depends on the association of TLR4 with MD-2, RP105 activity depends on its association with the MD-2 homolog, MD-1. Subsequent study revealed an apparent role for RP105 in TLR4 signaling in B cells. Whereas LPS-induced B cell proliferation is strictly dependent on TLR4, B cells from RP105−/− mice exhibit impaired LPS-driven proliferation in the face of normal proliferative responses to engagement of TLR9, IgM or CD40 (6, 7). Such data suggested that RP105 facilitates TLR4 signaling in B cells, although the underlying mechanisms have not been identified. The converse is not the case, however; B cell proliferation induced by Ab to RP105 is unimpaired in mice lacking TLR4 (4).
Further analysis revealed a broader pattern of expression. In addition to B cells, RP105 is expressed by diverse cell types expressing TLRs, including most APCs (8). Notably, RP105 was shown to inhibit TLR4 signaling in HEK293 cells, inhibit TLR4 signaling in dendritic cells, and restrain in vivo cytokine production in response to LPS (8). Recent solution of the crystal structure of RP105/MD-1 has been informative (9). The overall architecture of the 1:1 RP105/MD-1 complex is similar to that of TLR4/MD-2. However, RP105/MD-1 assembles into a unique 2:2 homodimer, with head-to-head dimerization of RP105's C-terminal leucine-rich repeats. This displaces the intracellular domains of RP105 to opposing sides of the complex; unlike the tail-to-tail dimerization of the N-terminal leucine-rich repeats observed in liganded TLR dimers which juxtaposes intracellular TIR domains, allowing signaling. Modeling of the interaction of RP105/MD-1 with TLR4/MD-2 based on their solved structures suggests that the TIR domains of TLR4 are prevented from apposition by the interaction of TLR4/MD-2 with RP105/MD-1 (9). Such modeling fails to suggest a mechanism by which RP105/MD-1 might facilitate TLR4 signaling in B cells.
In light of these paradoxical findings—with RP105 appearing to facilitate or inhibit TLR4 signaling depending on the cell type involved—we reinvestigated the B cell proliferative responses of RP105−/− mice.
Materials and Methods
Mice
RP105−/− mice (6) backcrossed ≥12 generations to a C57BL/6 background; C57BL/6 mice, μMT mice (C57BL/6; Jax); BAFF-Tg mice (C57BL/6; Biogen Idec) (10) and TACI−/− mice (C57BL/6) (11) were maintained in specific pathogen-free facilities. Age- and sex-matched mice were used in all experiments. Care was provided in accordance with NIH guidelines in studies approved by institutional IACUC committees.
In vitro assays and reagents
Splenic B cells were purified by negative selection with immunomagnetic beads (Miltenyi Biotech B cell Isolation Kit: CD43, CD4, Ter119 microbeads) to ≥ 98% purity. Purified B cells (1 × 106 cells/mL) or sorted MZ and FO B cells (0.45 × 106 cells/mL) were plated in triplicate, stimulated for 36 h with repurified E coli K235 LPS (S. Vogel), CpG DNA (Coley Pharm.) or PBS. Proliferation was quantified by thymidine incorporation over an additional 8 h. Single cell leukocyte suspensions were incubated with fluorochrome-labeled Ab for 30 min at 4° C. 100–150K events/data point were acquired on a LSR II flow cytometer and analyzed using FlowJo software. For B cell subset sorting, purified B cells were labeled with CD19, B220, CD21 and CD23, and sorted to ≥ 97% purity using a FACS Aria II. Except for CD24 Ab (Biolegend), all Ab were from eBioscience; appropriate isotype controls were used for all FACS. 7AAD was from eBioscience. BAFF was quantified by ELISA (Apotech); IL-6 and IL-10 by ELISA (BD OptEIA). huBCMA-Fc was from Biogen Idec; human Ig (IV-Ig, Gammaguard) from Baxter. Splenic mRNA was quantified by qRT–PCR (8); primers: BAFF 5' TGGATGCCGCCATTCTCAAC; 3' GCCTGTTTGCCTCACCACTATTTTG; APRIL 5' CCTTTCGGTTGCTCTTTGGTTGAG; 3' TGCTTCTTCTTGTGCTTCTGGGTG; β-actin 5' GTGACGTTGACATCCG; 3' CAGTAACAGTCCGCCT.
In vivo transfers and proliferation assays
Splenic B cells were purified by negative immunomagnetic separation (CD43, CD4, Ter119 microbeads; Miltenyi Biotech) and 30 – 40 × 106 were injected i.v. into μMT mice. Eight wk later, splenic B cells were similarly purified for in vitro proliferation assays. For in vivo proliferation, mice were challenged i.p. with 40 μg E coli K235 LPS, or pyrogen-free PBS, for 48 h. Mice were injected i.p. with 1 mg BrdU (BD Pharmingen) 24 and 12 h prior to sacrifice. Single cell leukocyte suspensions were fixed, permeabilized, treated with DNase, and stained with anti-BrdU APC (BD Biosciences); B cell (CD19+B220+) BrdU incorporation was quantified by FACS.
Results and Discussion
Impaired TLR4-driven proliferation is recapitulated in marginal zone B cells from RP105−/− mice
The B cell proliferative phenotype of RP105−/− mice has remained robust despite extensive crossing onto a C57BL/6 background: impaired LPS-driven proliferation, but normal proliferation to other stimuli (Suppl. Fig. 1a). This is not due to decreased B cell expression of TLR4/MD-2 ((7); data not shown). Of note, blunting of LPS-driven proliferation is not associated with increased B cell death (Suppl. Fig. 1b). The lack of genotype-specific effects on apoptosis, in the face of an apparent dose response relationship in the ability of TLR signaling to prevent apoptosis (no stimulation—LPS—CpG; Suppl. Figs. 1a and 1b), suggested that impaired LPS-driven proliferation might not be due to suppressed TLR4 signaling in RP105−/− B cells. Impaired LPS-driven proliferation was also seen in vivo: FACS analysis revealed that significantly fewer splenic B cells in RP105−/− mice, compared to B cells in wild type controls, underwent proliferation in response to LPS challenge (Fig. 1a).
Figure 1. The LPS-driven proliferative defect of B cells from RP105−/− mice is recapitulated in marginal zone B cells.
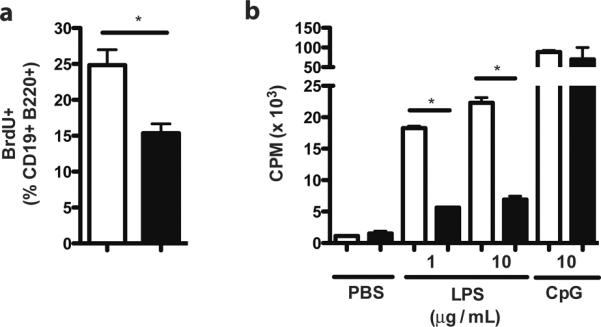
(a) Mice were challenged i.p. with LPS (40 μg), and splenic B cell proliferation was quantified by BrdU incorporation 48 h later. Means +/− SE are depicted; N=10 mice/genotype, pooled from 2 independent experiments. *P<0.005; unpaired, two-tailed t test. (b) FACS-sorted MZ B cells were stimulated as indicated, and proliferation was quantified by thymidine incorporation. Means +/− SE of triplicate cultures are depicted; N=4 pooled mice/genotype; representative of 2 separate experiments. *P<0.0001, unpaired, two-tailed t test.
There is marked variability in LPS proliferative responsiveness across splenic B cell populations. Despite the fact that they represent a minor subset numerically, marginal zone (MZ) B cells constitute the dominant such subpopulation, proliferating robustly after LPS stimulation. By contrast, LPS-stimulated proliferation by follicular (FO) B cells is much weaker, indeed nearly absent when sort purities are high (12). Transitional type 2 B cells exhibit intermediate sensitivity (13). While B cell development proceeds normally in RP105−/− mice (6), it remained possible that these proliferative abnormalities were a function of alterations in the development or survival of B cell subsets variably responsive to LPS-driven proliferation. The proportions of these subpopulations were, however, indistinguishable between RP105−/− and wild type mice (Suppl. Figure 1c).
We subsequently directly tested the ex vivo proliferative responses of highly purified B cell subpopulations. Notably, MZ B cells from RP105−/− mice recapitulated the blunted response to LPS observed in unfractionated splenic B cells, despite normal proliferative responsiveness to CpG DNA (Fig. 1b), data that underscore the fact that this phenotype is not due to altered B cell subset representation. As expected, FO B cells from RP105−/− and wild type mice exhibited little to no proliferation (and no differential proliferation) in response to LPS (Suppl. Fig. 1d). It should be noted that it is not possible to analyze LPS-driven proliferation of these subsets in vivo, as LPS stimulation alters expression of surface markers that can be used in their definition, including CD23, CD1d and IgD ((14); data not shown).
Despite proliferative differences, MZ and FO B cells exhibit similar changes in LPS-driven surface activation marker expression and signaling pathway activation (12). Similarly, we found that LPS-driven CD69 and MHC class II expression was comparable in MZ B cells (and FO B cells) purified from RP105−/− and wild type mice (Suppl. Figs. 1e and 1f; and data not shown). Further, no significant genotype-specific differences in LPS-driven B cell secretion of IL-10 or IL-6 were observed (Suppl. Figs. 1g and 1h). These findings of altered LPS-driven proliferation, in the absence of differences in other LPS-driven responses (including prevention of apoptosis; above), are difficult to reconcile with a fundamental difference in responsiveness at the level of the LPS receptor.
RP105-associated defects in LPS-driven B cell proliferation are not B cell autonomous
Of note, splenic size and leukocyte numbers were significantly increased in RP105−/− mice (Fig. 2a, 2b). That said, RP105−/− mice exhibited no disproportionate changes in cell composition in either B cell or non-B cell compartments (Suppl. Fig. 1c, 1i): all leukocyte populations were proportionately increased. Further, despite blunted LPS-driven in vivo B cell proliferation, basal in vivo B cell proliferation was significantly increased in RP105−/− mice (Fig. 2c). Increased BrdU incorporation was specifically observed in RP105−/− MZ B cells (Fig. 2d), indicating increased basal B cell proliferation in the RP105−/− MZ B compartment—the same compartment exhibiting blunted LPS-driven proliferation.
Figure 2. RP105−/− mice exhibit increased baseline spleen size, splenic leukocyte number and in vivo B cell proliferation.
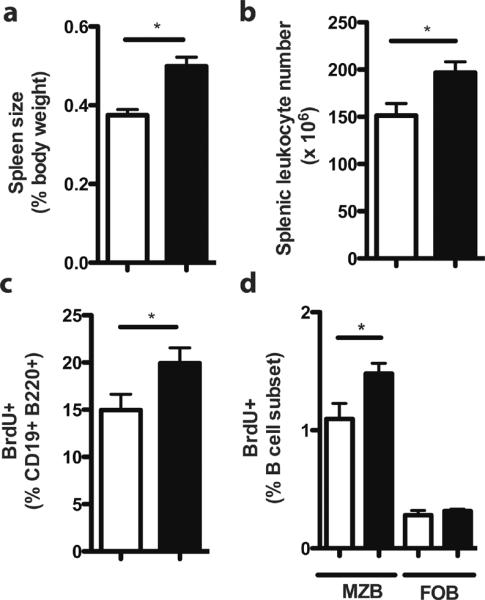
(a) Spleen size and (b) splenic leukocytes were quantified in wild type (white bars) and RP105−/− mice (black bars) mice. Means +/− SE from a single experiment are depicted. (a) N=28 mice/genotype; *P<0.0001, unpaired two-tailed t test. (b) N=12 mice/genotype; *P<0.05, unpaired, two-tailed t test. (c) In vivo baseline B cell proliferation was analyzed by flow cytometric quantification of BrdU incorporation, 48 h after i.p. injection of PBS. N=6 mice/genotype, representative of 3 independent experiments. *P<0.005, unpaired, two-tailed t test. (d) In vivo baseline B cell proliferation was quantified by flow cytometric analysis of BrdU incorporation in MZ (CD19+ B220+ CD21high CD23lo) and FO (CD19+ B220+CD21int CD23+) B cell subsets from wild type (white bars) and RP105-deficient (black bars) mice. Means +/− SE are depicted; N = 6 mice/genotype. *P<0.05 (unpaired, two-tailed t test).
Taken together, these data suggested that the proliferative differences between wild type and RP105−/− B cells might not be due to B cell expression (or not) of RP105. To test this, we purified splenic B cells from wild type and RP105−/− mice and transferred them into B cell-deficient, RP105-sufficient μMT mice. Eight wk later, splenic B cells were purified and stimulated in vitro. Given the contrasting proliferative responsiveness of MZ and FO B cells, we first quantified the engraftment of these subsets. Skewing towards MZ B cell engraftment, reported previously, was observed; however, no genotype-specific differences in the proportions of MZ and FO B cells were seen (Fig. 3a). Notably, as shown in Figure 3b, engraftment of RP105-deficient and -sufficient B cells in an RP105-sufficient environment obviated differences in LPS-driven B cell proliferative responses. These data strongly suggest that RP105-associated defects in LPS-driven B cell proliferation are not a function of B cell autonomous lack of expression of RP105.
Figure 3. RP105-deficient and -sufficient B cells have similar TLR4-driven proliferative responses after adoptive transfer into an RP105-sufficient environment.
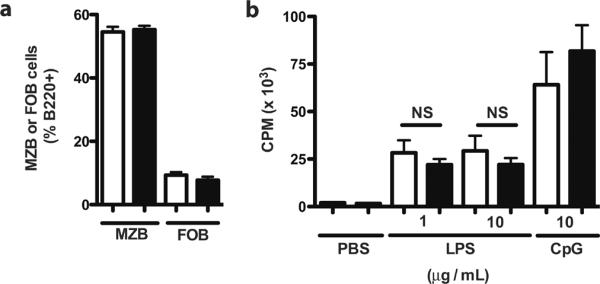
Splenic B cells were purified from wild type (white bars) and RP105−/− mice (black bars) mice, transferred into μMT (B cell deficient, RP105+/+) mice, and allowed to reconstitute for 8 wks. (a) Following engraftment, MZ B and FO B cell subsets were quantified by flow cytometry. (b) B cell proliferative responses were quantified by thymidine incorporation. Means +/− SE are depicted. N=12–15 mice/genotype, pooled from 3 independent experiments. NS (not significant; unpaired, two-tailed t test).
Integral role for BAFF in altered proliferative responses of B cells from RP105−/− mice
BAFF is essential for B cell maturation and survival. Overexpression drives B cell hyperplasia; neutralization leads to B cell death (15). As TLR signaling induces BAFF expression (15), we tested whether compromised control of TLR signaling in RP105−/− mice led to increased BAFF activity. Indeed, RP105−/− mice exhibited significantly increased basal serum BAFF levels compared to wild type controls, a difference exacerbated by LPS stimulation (Fig. 4a). No genotype-specific differences in splenic APRIL expression were observed (data not shown). B cell expression of 2 of the 3 BAFF receptors, TACI and BAFF-R, can also be upregulated by TLR stimulation (15). As might thereby be expected, B cells from RP105−/− mice exhibited significantly increased basal expression of TACI (Fig. 4b)–although genotype-specific differences in BAFF-R expression were not observed (data not shown). TACI expression is thought to limit the amount of soluble BAFF available for signaling (15). That said, no in vivo data bearing directly on the subject appear to have been published. TACI−/− mice did indeed have significantly increased serum BAFF concentrations at baseline—despite their expanded B cell compartment (11)—and after LPS stimulation (Fig. 4c). Increased basal TACI expression may thus lead to underestimation of basal BAFF production in RP105−/− mice. Consonant with this, qRT-PCR-mediated quantification of splenic BAFF mRNA revealed a mean 30% increase in basal expression levels in RP105−/− mice (data not shown).
Figure 4. Dysregulated BAFF expression is integral to the altered TLR4 proliferative responsiveness of RP105−/− B cells.

(a) Kinetic analysis of serum BAFF levels from wild type (white bars) and RP105−/− (black bars) mice in response to i.p. challenge with LPS (40 μg). Means +/− SE are depicted; N=6 mice/genotype. * P < 0.05, ** P<0.005, unpaired, two-tailed t test. (b) Splenic B cell expression of TACI, 48 h after i.p. injection of PBS in wild type (white bars) or RP105−/− (black bars) mice. Means +/− SE are depicted; N=9–12 mice/genotype, pooled from 2 independent experiments, *P<0.05, **P<0.0001, unpaired, two-tailed t test. (c) Kinetic analysis of serum BAFF levels from wild type (white bars) and TACI−/− (black bars) mice in response to i.p. challenge with LPS (40 μg). Means +/− SE are depicted; N=5 mice/genotype from a single experiment, * P<0.05, ** P<0.005, unpaired, two-tailed t test. (d–f) RP105−/− (black bars) and wild type (white bars) mice were treated i.p. with BCMA-Fc (300 ng) or IV-Ig (300 ng). Six days later, (d) splenic MZ B and FO B cell subsets were quantified by flow cytometry, and (e, f) splenic B cell proliferation was quantified by BrdU incorporation, 48 h after intraperitoneal challenge with (e) LPS (40 μg) or (f) PBS. Black bars, RP105−/− mice; white bars, wild type mice. Means +/− SE are depicted, of N=8–11 mice/group (e, f), pooled from 2 independent experiments. *P<0.05, **P<0.005, ANOVA/Bonferroni post-hoc test.
To define the relevance of BAFF overexpression to B cell proliferation in RP105−/− mice, we turned first to BAFF-Tg mice (10). Such mice also exhibit significantly increased basal B cell proliferation (data not shown). As the even more robust BAFF overexpression seen in BAFF-Tg mice is associated with a significant increase in the proportion of MZ B cells (10), we analyzed proliferation of purified MZ B cells. Similar to RP105−/− mice, MZ B cells from BAFF-Tg mice exhibited blunted LPS-driven, but intact CpG-driven, proliferation (Suppl. Fig. 2). These data suggested a possible role for BAFF overexpression in the B cell proliferative abnormalities of RP105−/− mice—mice with considerably less dramatic perturbations in immune homeostasis than BAFF-Tg mice. We thus used a huBCMA-Fc fusion protein (16) to test whether partial BAFF neutralization would rescue B cell proliferative responses in RP105−/− mice. Robust neutralization of BAFF by high doses of huBCMA-Fc leads to B cell depletion (16). We thus quantified in vivo B cell proliferation 6 d after treating mice with a low dose of huBCMA-Fc, defined by preliminary dose-titration experiments (300 ng), that altered neither B cell numbers nor B cell subset composition (Fig. 4d), nor the proliferative responsiveness of B cells in wild type mice (data not shown). Notably, such treatment reversed the LPS proliferative hypo-responsiveness of RP105−/− B cells (Fig. 4e). It also reversed their basal increase in B cell proliferation (Fig. 4f). Thus, BAFF concentrations are increased in RP105−/− mice, and partial neutralization of BAFF rescues aberrant B cell proliferation in such mice.
These data indicate that the B cell proliferative abnormalities of RP105−/− mice are not the result of B cell-autonomous (lack of) expression of RP105, and implicate dysregulated BAFF expression in the generation of these abnormalities. There may well be broader biological relevance for the pathways outlined here. In line with recent data showing BAFF over-expression in diverse human autoimmune diseases (15), it was reported almost 30 years ago that patients with lupus exhibit defective LPS-driven B cell proliferation (17). Whether these two findings are mechanistically related as suggested by the current data remains to be tested.
While TACI facilitates T cell-independent type II Ab responses, TACI negatively regulates B cell compartment size. Current data support the existence of both indirect (restriction of BAFF availability) and direct (B cell signaling) mechanisms for the latter activity (15). Given increased basal TACI expression by RP105−/− mice, it is tempting to speculate that the mechanisms underlying restriction of LPS-driven proliferation by BAFF overexpression in such mice involve TACI signaling. Concordant with this, despite the fact that TACI−/− mice exhibit increased serum BAFF levels, MZ B cells from TACI−/− mice (subset purification performed because of elevated percentages of MZ B cells in such mice (18)) do not exhibit significant blunting of LPS-driven proliferation (data not shown).
We note that we have focused here on B2 B cells; indeed, all in vitro studies employed a negative selection approach that depletes B1 cells. Whether the pathways defined here also affect B1 cell function (B1 cell numbers are unchanged in RP105−/− mice (6)) remains to be defined.
Our data resolve some of the apparent biological complexities associated with RP105. There is no need to postulate dichotomous, cell type-specific effects on TLR4 signaling. Generation of mice with cell type-specific deletions in RP105 expression should allow for clear definition of whether RP105 is a biologically relevant inhibitor of TLR4 signaling in B cells, as it is in myeloid cells. More broadly, the critical function(s) of B cell-intrinsic RP105 expression remain to be defined. In particular, it is not known whether the pathways of B cell activation and proliferation stimulated by Ab-mediated cross-linking of RP105 model the effects of any natural ligands for RP105. If so, identification of such ligands and mechanistic analysis of the roles of the pathways activated by such ligands in homeostasis and disease would be of obvious biological, and potentially therapeutic, interest.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants AI075159 (to C.L.K.), HD037091, HL075453 and AI071163 (to D.J.R.), T32AI055406, and a Department of Veterans Affairs Merit Award (to F.D.F.).
Abbreviations
- FO
follicular
- MZ
marginal zone
References
- 1.Miyake K, Yamashita Y, Hitoshi Y, Takatsu K, Kimoto M. Murine B cell proliferation and protection from apoptosis with an antibody against a 105-kD molecule: unresponsiveness of X-linked immunodeficient B cells. J. Exp. Med. 1994;180:1217–1224. doi: 10.1084/jem.180.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamashita Y, Miyake K, Miura Y, Kaneko Y, Yagita H, Suda T, Nagata S, Nomura J, Sakaguchi N, Kimoto M. Activation mediated by RP105 but not CD40 makes normal B cells susceptible to anti-IgM-induced apoptosis: a role for Fc receptor coligation. J. Exp. Med. 1996;184:113–120. doi: 10.1084/jem.184.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan VW, Mecklenbrauker I, Su I, Texido G, Leitges M, Carsetti R, Lowell CA, Rajewsky K, Miyake K, Tarakhovsky A. The molecular mechanism of B cell activation by toll-like receptor protein RP-105. J. Exp. Med. 1998;188:93–101. doi: 10.1084/jem.188.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yazawa N, Fujimoto M, Sato S, Miyake K, Asano N, Nagai Y, Takeuchi O, Takeda K, Okochi H, Akira S, Tedder TF, Tamaki K. CD19 regulates innate immunity by the toll-like receptor RP105 signaling in B lymphocytes. Blood. 2003;102:1374–1380. doi: 10.1182/blood-2002-11-3573. [DOI] [PubMed] [Google Scholar]
- 5.Hebeis B, Vigorito E, Kovesdi D, Turner M. Vav proteins are required for B-lymphocyte responses to LPS. Blood. 2005;106:635–640. doi: 10.1182/blood-2004-10-3919. [DOI] [PubMed] [Google Scholar]
- 6.Ogata H, Su I, Miyake K, Nagai Y, Akashi S, Mecklenbrauker I, Rajewsky K, Kimoto M, Tarakhovsky A. The toll-like receptor protein RP105 regulates lipopolysaccharide signaling in B cells. J. Exp. Med. 2000;192:23–29. doi: 10.1084/jem.192.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagai Y, Kobayashi T, Motoi Y, Ishiguro K, Akashi S, Saitoh S, Kusumoto Y, Kaisho T, Akira S, Matsumoto M, Takatsu K, Miyake K. The radioprotective 105/MD-1 complex links TLR2 and TLR4/MD-2 in antibody response to microbial membranes. J. Immunol. 2005;174:7043–7049. doi: 10.4049/jimmunol.174.11.7043. [DOI] [PubMed] [Google Scholar]
- 8.Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A, Finberg RW, Tarakhovsky A, Vogel SN, Belkaid Y, Kurt-Jones EA, Karp CL. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat. Immunol. 2005;6:571–578. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon SI, Hong M, Wilson IA. An unusual dimeric structure and assembly for TLR4 regulator RP105-MD-1. Nat. Struct. Mol. Biol. 2011;18:1028–1035. doi: 10.1038/nsmb.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, Browning JL. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J. Exp. Med. 1999;190:1697–1710. doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Bulow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity. 2001;14:573–582. doi: 10.1016/s1074-7613(01)00130-3. [DOI] [PubMed] [Google Scholar]
- 12.Meyer-Bahlburg A, Bandaranayake AD, Andrews SF, Rawlings DJ. Reduced c-myc expression levels limit follicular mature B cell cycling in response to TLR signals. J. Immunol. 2009;182:4065–4075. doi: 10.4049/jimmunol.0802961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei B, Su TT, Dalwadi H, Stephan RP, Fujiwara D, Huang TT, Brewer S, Chen L, Arditi M, Borneman J, Rawlings DJ, Braun J. Resident enteric microbiota and CD8+ T cells shape the abundance of marginal zone B cells. Eur. J. Immunol. 2008;38:3411–3425. doi: 10.1002/eji.200838432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson L, Cady CT, Cambier JC. TLR4-mediated signaling induces MMP9-dependent cleavage of B cell surface CD23. J Immunol. 2009;183:2585–2592. doi: 10.4049/jimmunol.0803660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackay F, Schneider P. Cracking the BAFF code. Nat. Rev. Immunol. 2009;9:491–502. doi: 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 16.Pelletier M, Thompson JS, Qian F, Bixler SA, Gong D, Cachero T, Gilbride K, Day E, Zafari M, Benjamin C, Gorelik L, Whitty A, Kalled SL, Ambrose C, Hsu YM. Comparison of soluble decoy IgG fusion proteins of BAFF-R and BCMA as antagonists for BAFF. J. Biol. Chem. 2003;278:33127–33133. doi: 10.1074/jbc.M305754200. [DOI] [PubMed] [Google Scholar]
- 17.Abe T, Toguchi T, Takeuchi T, Kiyotaki M, Homma M. Mitogenic responses to lipopolysaccharide by B lymphocytes from patients with systemic lupus erythematosus. Scand. J. Immunol. 1981;15:475–482. doi: 10.1111/j.1365-3083.1982.tb00673.x. [DOI] [PubMed] [Google Scholar]
- 18.Yan M, Wang H, Chan B, Roose-Girma M, Erickson S, Baker T, Tumas D, Grewal IS, Dixit VM. Activation and accumulation of B cells in TACI-deficient mice. Nat. Immunol. 2001;2:638–643. doi: 10.1038/89790. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.