

Abstract
Background
Drug-induced long QT syndrome (diLQTS) is an adverse drug effect that has an important impact on drug use, development, and regulation. Here, we tested the hypothesis that common variants in key genes controlling cardiac electrical properties modify the risk of diLQTS.
Methods and Results
In a case-control setting, we included 176 patients of European descent from North America and Europe with diLQTS, defined as documented torsades de pointes during treatment with a QT prolonging drug. Control samples were obtained from 207 patients of European ancestry who displayed <50 msec QT lengthening during initiation of therapy with a QT-prolonging drug, and 837 controls from the population based KORA study. Subjects were successfully genotyped at 1,424 single nucleotide polymorphisms (SNPs) in 18 candidate genes including 1,386 SNPs tagging common haplotype blocks, and 38 non-synonymous ion channel gene SNPs. For validation we used a set of cases (n=57) and population-based controls of European descent. The SNP KCNE1 D85N (rs1805128), known to modulate an important potassium current in the heart, predicted diLQTS with an odds ratio of 9.0 (95% confidence interval: 3.5–22.9). The variant allele was present in 8.6% of cases, 2.9% of drug-exposed controls, and 1.8% of population controls. In the validation cohort the variant allele was present in 3.5% of cases, and in 1.4% of controls.
Conclusions
This high-density candidate SNP approach identified a key potassium channel susceptibility allele that may be associated with the rare adverse drug reaction torsades de pointes.
Keywords: candidate genes; death, sudden; SNP; torsade de pointes; adverse drug events
Unexpected marked prolongation of the QT interval after initiation of drug therapy, associated with risk of a morphologically distinct pattern of polymorphic ventricular tachycardia (torsades de pointes) has been recognized since the 1960s.1–3 The most common class of drugs causing drug-induced long QT syndrome (diLQTS) is the membrane-active antiarrhythmic drug category, with an incidence approaching 8% in some reports.1–3 However, diLQTS can occur, albeit more rarely, during treatment with various drugs including many that are not prescribed for cardiovascular indications.1–3 DiLQTS has been a major cause for drug relabeling and withdrawal, creating obstacles for drug development and uncertainties during drug approval and regulatory processes.1–3
Clinical studies have identified risk factors for diLQTS such as hypokalemia4–6 or female sex,7 but the phenomenon remains unpredictable in a given individual. Moreover, the congenital long QT syndromes, usually caused by mutations in ion channel or associated genes, are characterized by the same pattern of torsades de pointes. Since the congenital LQTS may have variable penetrance,8 and some mutation carriers may display normal QT intervals, this finding has lead to the hypothesis that the drug-induced “acquired” LQT syndromes are conditions predisposed by heritable variants in QT-controlling genes. Indeed, screening the common disease genes associated with the congenital syndrome has identified mutations in 5–15% of subjects with diLQTS.9–14 One recent study reported congenital LQTS mutations in 8/20 (40%) Japanese cases.15
A second hypothesis is that common genetic variants modulate diLQTS risk, and small studies have identified such variants.12,14 In addition, one study reported that the extent of QT prolongation among first degree relatives of subjects with diLQTS was greater than that seen in first degree relatives of subjects who tolerated QT prolonging drugs without relevant QT prolongation.16 In the present study, we tested the second hypothesis, using a large cohort of cases drawn from multiple collaborating sites, and interrogating common variants across ion channel and other high priority candidate genes.
Methods
Subject sample
The 176 diLQTS patients that constituted the case cohort, defined as documented torsades de pointes during treatment with a drug having known QT prolonging potential, were from North America or Europe, and all were of self-identified European descent. The diagnosis was ascertained by cardiologists who specialize in clinical electrophysiology at 7 participating centers. The case definition required documented torsades de pointes, associated with any reversible QT prolongation during the drug exposure. Covariates sought included age, sex, self-reported ethnicity, arterial hypertension, history of atrial fibrillation, serum creatinine, and culprit drug, as well as information serum potassium concentrations (with particular emphasis on hypokalemia) at the time of the index arrhythmia. Written informed consent to participate in the study including the collection and use of DNA samples for genetic analysis was obtained with Institutional Review Board (IRB) approval at each center.
The drug-exposed controls consisted of 207 self-identified European ancestry subjects derived from a clinical study at Vanderbilt University Medical Center, under appropriate IRB-approved protocols. The study used electronic medical record-based surveillance to identify in-patients in whom QT prolonging antiarrhythmics were being initiated. Patients consenting to participate (91% of those approached) agreed to allow collection of clinical data, including drug-associated QT interval duration changes, as well as DNA samples. For this study, the drug-exposed control cohort was defined by the absence of qualifying arrhythmias, <50 msec increase in QTc interval using Bazett’s formula for heart rate correction, and no QTc interval exceeding 500 msec during drug treatment.
We also used control subjects from the general population. These were recruited from the population-based KORA Study (Cooperative Health Research in the Region of Augsburg),17 and were all of European descent. In brief, 4856 subjects in ten subsets of equal size, stratified according to sex and age, underwent a survey in 1994–95 (KORA S3), and again in 2004–2005 (KORA F3). From among the KORA population, we included 837 randomly selected subjects as controls, maintaining the original sex-distribution. The study was approved by the ethics committee of the Bavarian Medical Association.
Finally, we used cases with diLQTS from the Drug Induced Arrhythmia Risk Evaluation (DARE) Study, who met the inclusion criteria above, for a validation cohort.18 These validation cases were compared to healthy population controls recruited by DARE from the primary care practices of cases to achieve geographical matches, aiming for a 2:1 to 4:1 ratio of controls to cases. Controls were excluded if there was any prior history of ventricular arrhythmia, drug-induced arrhythmia, syncope or congenital long QT syndrome. All validation control subjects underwent digital 12 lead ECGs and were excluded if there was significant abnormality.
Genotyping
Eighteen candidate genes were studied. These included genes reported to be disease-causing in cardiac arrhythmia syndromes, functionally important subunits of these genes, and genes significantly associated with the QT interval in the general population based on the results of genome wide association studies (Table 1).19,20
Table 1.
Candidate gene based SNP selection
| Gene | chr | Gene position | Tagged region | tag SNPs | ns SNPs |
|---|---|---|---|---|---|
| AKAP9 Isoform 1 | 7 | 91,214,843–91,384,640 | 91,104,000–91,700,000 | 16 | 4 |
| ANK 2 Isoform 1 | 4 | 114,328,474–114,662,489 | 114,160,000–114,690,000 | 114 | 4 |
| CACNA1C | 12 | 2,032,725–2,672,368 | 1,900,000–2,820,000 | 173 | 3 |
| NOS1AP | 1 | 158,771,239–159,071,471 | 158,680,000–159,170,000 | 130 | - |
| CASQ2 | 1 | 115,954,685–116,023,312 | 115,810,000–116,130,000 | 66 | 4 |
| CAV3 | 3 | 8,750,496–8,763,450 | 8,700,000–8,840,000 | 62 | 1 |
| FKBP1B | 2 | 24,184,279–24,198,199 | 24,134,278–24,248,198 | 13 | - |
| GPD1L | 3 | 32,123,148–32,185,205 | 31,980,000–32,230,000 | 50 | - |
| KCNE1 | 21 | 34,740,858–34,805,483 | 34,640,000–34,930,000 | 68 | 3 |
| KCNE2 | 21 | 34,658,193–34,665,310 | 34,640,000–34,930,000 | 28 | - |
| KCNH2 / HERG | 7 | 150,079,697–150,112,662 | 149,930,000–150,200,000 | 65 | 2 |
| KCNJ2 | 17 | 65,677,271–65,687,776 | 65,610,000–65,765,000 | 42 | - |
| KCNQ1 | 11 | 2,422,797–2,826,915 | 2,260,000–2,900,000 | 150 | 1 |
| RYR2 | 1 | 233,531,743–234,323,329 | 233,427,218–234,427,217 | 232 | 8 |
| SCN1B | 19 | 40,213,374–40,223,192 | 40,140,000–40,270,000 | 24 | - |
| SCN4B | 11 | 117,509,302–117,528,745 | 117,350,000–117,620,000 | 47 | - |
| SCN4A | 17 | 59,369,646–59,404,010 | 59,220,000–59,520,000 | 24 | 1 |
| SCN5A | 3 | 38,564,557–38,666,167 | 33,843,000–38,790,000 | 82 | 7 |
| Subtotal | 1386 | 38 | |||
| Total | 1424 | ||||
Basic information on the 18 genes under investigation and the number of investigated SNPs. All data based on human genome built hg 17, NCBI built 35. chr – chromosome; tag SNP – haplotype tagging SNP; ns SNP – non-synonymous coding SNP (for detailed information per SNP, see Supplemental Table 1).
Haplotype tagging SNPs (tagSNPs) were selected to systematically analyze each gene using Tagger21 based on HapMap22 data release #20 / phase II using the March 2006 NCBI B35 genome assembly and dbSNP build 125 data. The following criteria were applied for tagSNP selection: HapMap CEU population, pairwise only tagging with r2 ≥ 0.8 and a minor allele frequency (MAF) ≥ 10%. To account for genetic variation in regions surrounding each gene, we considered up- and downstream genetic data in the tagging procedure. Included regions were defined by linkage-disequilibrium (LD), blocks as previously reported.23 We included at least 50 kb of both up- and downstream information. A total of 1,386 SNPs were selected for genotyping using this method. In addition, we included all 38 non-synonymous SNPs with minor allele frequencies ≥1% previously reported in the 18 candidate genes. A summary of all 1,424 SNPs chosen for study is presented in Table 1, and detailed information including a complete SNP-list is presented in Supplemental Table 1.
Genotyping of all cases, drug-exposed controls, and KORA population-based controls was performed using a custom Illumina GoldenGate assay (Illumina OPA GS0007558) according to the manufacturer’s recommendations using the Illumina Beadstation 500GX (Illumina Inc., SanDiego, USA) in a single laboratory (Genome Analysis Center, Helmholtz Zentrum München, Germany). The Illumina BeadStudio software clustering algorithm was used for initial data analysis. Thereafter, intensity plots of all variants were examined individually and manual genotype calling was performed if necessary.
Monomorphic SNPs or those with a minor allele frequency <1%, and SNPs, as well as samples with call rates <90%, were excluded from the analysis. A total of 70 SNPs were removed prior to analysis. Hardy Weinberg Equilibrium (HWE) was calculated for the remaining SNPs, and deviations from HWE expectations were noted but not excluded from analyses (Supplemental Table 1). After filtering, the overall genotyping call rate was 97.1%, and a total of 1,354 SNPs had sufficient quality for analysis. Details of SNPs as well as their clustering relative to the 18 selected genes can be found in Table 1. Clusterplots for SNPs selected for replication are shown in Supplemental Figure 1.
For validation of rs1805128, 57 additional diLQTS cases from the DARE cohort and 211 controls were genotyped for KCNE1 D85N (rs1805128) using standard direct Sanger sequencing of the PCR amplicon in both directions. We also used the DARE set to examine whether an initial finding with rs7295250 would replicate. For this experiment, samples were genotyped using the Illumina Infinium Human CVD 50K Bead Array24 analyzed with the Illumina platform 500GX (the Biomics Centre, St. George’s University of London). Of 138 individuals, two had >10% missing genotypes and were excluded. Another three control subjects were excluded having an overall genotype pattern suggestive of non-European descent despite self-report. Thereafter, 47 cases and 86 controls were available with 100% genotyping call rate for rs7295250.
Statistical analysis
All subjects included in the study were of European descent. We attempted the STRUCTURE25,26 and EIGENSTRAT27 methods to account for population stratification, but no relevant stratification was identified. In particular, the first five principal components were nonsignificant.
All tests of association were performed using PLINK.28 Each SNP was tested for association to diLQTS between cases and controls using logistic regression adjusted for age and sex in two different analyses: diLQTS cases vs. drug-exposed controls, and diLQTS cases vs. population controls. In addition, we attempted sex-stratified analyses adjusted for age in each analysis group. Bonferroni correction for multiple comparisons would have required an uncorrected p-value of 3×10−5. Given our sample size and the fact that ours presumably represents already one of the largest collections of diLQTS cases, we applied less stringent criteria for identifying SNPs to be evaluated by replication. We planned to take forward to the DARE follow-up set any SNP with a P value of <0.01 for both drug-drug exposed and drugcontrol population comparisons.
The validation stage associations in the DARE cohort for rs1805128 and rs7295250 were assessed among cases and controls only assuming an additive genetic model adjusted for age and sex.
Results
The clinical characteristics of the cohorts are presented in Table 2. Among the 176 diLQTS cases, 128 (73%) were female, and the mean age was 63 ± 17 years. The single most common drug associated with torsades de pointes was sotalol (n=52); other common culprit antiarrhythmic drugs included quinidine (n=18) and amiodarone (n=17). Cumulatively, antiarrhythmics (n=107), antipsychotics (n=14) and antibiotics (n=12) were associated with diLQTS in 133 (76%) of the cases. In 28 cases, combinations of two or more culprit drugs were implicated.
Table 2.
Baseline characteristics of the study sample
| diLQTS cases (n=176) |
drug-exposed controls (n=207) |
p-value (diLQTS cases vs. controls) |
Population controls (n=837) |
p-value (diLQTS cases vs. population controls) |
DARE diLQTS cases (n=57)* |
DARE population controls (n=211)* |
p-value (DARE cases vs. controls) |
|
|---|---|---|---|---|---|---|---|---|
| Clinical characteristics | ||||||||
| Male sex, n (%) | 48 (27.3%) | 137 (66.2%) | <0.001 | 417 (49.8%) | <0.001 | 19 (33.3%) | 101 (47.9%) | 0.053 |
| Age (at event) (years) | 63.0 ± 16.7 | 61.4 ± 14.0 | 0.32 | 60.7 ± 10.2 | 0.10 | 63.4 ± 14.2 | 68.6 ± 11.3 | 0.005 |
| Arterial Hypertension n (%) | 82 (46.6%) | 124 (59.9%) | 0.73 | 485 (57.9%) | 0.39 | 33 (64.7%) | 87 (42.7%) | 0.007 |
| Atrial fibrillation, n (%) | 94 (53.4%) | 184 (88.9%) | <0.001 | 14 (1.7) | <0.001 | 28 (49.0%) | - | - |
| Hypokalemia at event n (%) | 34 (19.3%) | - | - | 12 (21.0%) | - | - | ||
| ECG characteristics | ||||||||
| QT interval, ms | 442.7 ± 91.8 | 381.1 ± 47.2 | <0.001 | 403.2 ± 30.4 | <0.001 | 416.8 ± 51.7 | 407.8 ± 29.4 | 0.107 |
| RR interval, ms | 910.8 ± 228.7 | 792.5 ± 212.1 | <0.001 | 964.7 ± 165.2 | 0.009 | 900.0 ± 230.0 | 950.0 ± 150.0 | 0.048 |
| QTc Bazett, ms | 468.3 ± 77.2 | 433.8 ± 31.9 | <0.001 | 413.0 ± 23.4 | <0.001 | 444.7 ± 25.3 | 420.2 ± 20.1 | <0.001 |
| QTc Fridericia, ms | 458.1 ± 77.0 | 414.6 ± 28.7 | <0.001 | 409.3 ± 20.1 | <0.001 | 434.4 ± 26.2 | 416.7 ± 33.6 | 0.001 |
| QTc Framingham, ms | 448.8 ± 75.6 | 409.8 ± 27.7 | <0.001 | 403.6 ± 19.6 | <0.001 | 432.8 ± 25.5 | 415.5 ± 34.3 | 0.001 |
| QTc RAS, ms | 447.1 ± 73.8 | 411.2 ± 27.4 | <0.001 | 405.4 ± 19.9 | <0.001 | - | - | - |
The table provides baseline clinical characteristics and ECG measures prior to the event or at steady state after the event after culprit drug withdrawal. QT corrections: QTc Bazett = QT / RR^1/2; QTc Friderica = QT / RR^1/3; QTc Framingham = QT + 0.154*(1-RR). Continuous parameters are expressed as mean ± standard deviation and are compared using a two-sample t-test. Discrete variables are displayed as frequency and percentage, and are compared between groups using the Fisher’s Exact Test.
The DARE sub-sample of 47 cases and 86 controls was a subset of the entire DARE cohort showing similar demographic characteristics, and is not presented separately.
Serum potassium was available in 119 cases at the time of the event and was below the laboratory specific reference range in 34 (19.3%) cases. Resting heart rate was different between cases (70.0 ± 17.4 /min), drug-exposed controls (80.7 ± 23.9 /min) and population controls (69.0 ± 11.2 /min). Atrial fibrillation was more prevalent in the drug-exposed control group (88.9%) compared to the population control group (1.7%) and to the diLQTS cases (53.4%). Quantitatively analyzable 12-lead resting ECG measurements were available in 135 cases, 205 drug-exposed controls, and 824 population controls. ECG measurements were included prior to the event or at steady state post event and after culprit drug withdrawal. Baseline QT intervals as well as corrected QT intervals using multiple formulae was prolonged at baseline in cases compared to both control groups.
Single SNP tests of association assuming an additive genetic model were performed for 1,354 variants across all 18 genes of interest between diLQTS cases and drug-exposed controls (Figure 1, black symbols). The association results for all SNPs at a significance threshold of p≤0.01 are shown in Table 3, and the results for all associations are shown in Supplemental Table 1. All tests of association used a logistic regression model adjusted for age and sex. SNP rs7295250, a variant on chromosome 12 in the genomic region of the L-type calcium channel gene CACNA1C, was associated with the smallest p-value among all tests of association. This variant conferred an odds ratio of 1.88 (95% confidence interval (CI) 1.30 – 2.71, p=7.62 × 10−4). The strongest effect size within this dataset was detected for rs1805128, which is a non-synonymous coding (missense) SNP in exon 1 of the cardiac ion channel gene KCNE1. This variant leads to an amino acid substitution from aspartic acid to asparagine at position 85 of the translated protein (Asp 85 Asn; D85N). The variant was significantly associated with diLQTS with an odds ratio of 9.92 (95% CI 2.36 – 41.80, p=1.77 × 10−3). The SNP showed a marked difference in allele frequency between diLQTS cases and drug-exposed controls: 8.6% vs. 2.9%.
Figure 1.
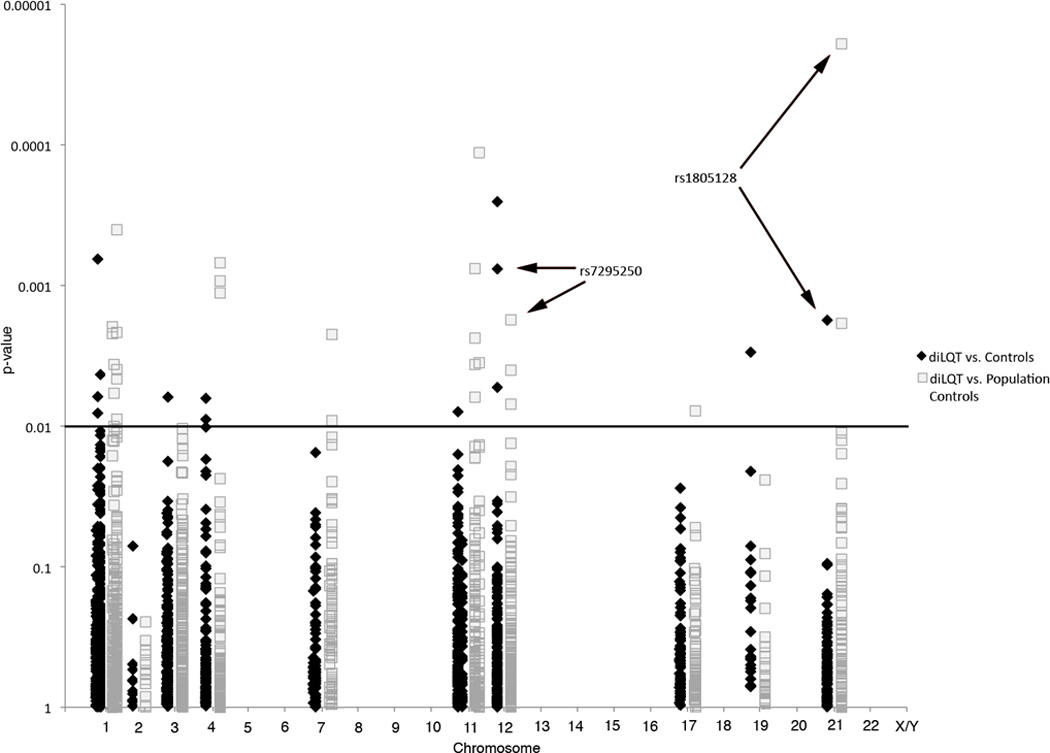
Manhattan-plot illustrating the genotyping results.–log10 p-values plotted against the SNPs’ chromosomal location. diLQTS cases vs. drug-exposed controls in black; diLQTS cases vs. population controls in gray. The horizontal line indicates threshold of p=0.01 for SNP replication. Arrows mark associations with p≤0.01 in both comparisons.
Table 3.
SNPs with significant associations between diLQTS cases to controls or population controls
| diLQT cases (n=176) vs. drug-exposed controls (n=207) |
diLQT cases (n=176) vs. population controls (n=837) |
DARE cases (n=57) vs. DARE controls (n=211) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | chr | Gene | Coded Allele |
Non- coded Allele |
MAF | Function | OR(95%CI) | p-value | OR(95%CI) | p-value | OR(95%CI) | p-value |
| rs2238018 | 12 | CACNA1C | C | T | 0.20 | Intron | 2.22(1.45–3.41) | 2.53×10−04 | 1.32(0.97–1.79) | 7.94×10−02 | ||
| rs164124 | 1 | CAPON | A | G | 0.33 | Intron | 0.55(0.39–0.78) | 6.49×10−04 | 0.8(0.6–1.06) | 1.16×10−01 | ||
| rs7295250 | 12 | CACNA1C | C | T | 0.22 | Intron | 1.88(1.3–2.71) | 7.62×10−04 | 1.56(1.18–2.07) | 1.77×10−03 | 0.95(0.54–1.67) | 8.7×10−1 |
| rs1805128 | 21 | KCNE1 | A | G | 0.02 | D85N | 9.92(2.36–41.8) | 1.77×10−03 | 8.88(3.26–24.17) | 1.95×10−05 | 1.51(0.35–6.57) | 5.8×10−1 |
| rs8103371 | 19 | SCN1B | C | T | 0.12 | 2.06(1.28–3.32) | 2.98×10−03 | 1.53(1.06–2.21) | 2.43×10−02 | |||
| rs2249287 | 1 | RYR2 | C | T | 0.21 | Intron | 0.54(0.36–0.83) | 4.30×10−03 | 0.67(0.47–0.96) | 2.86×10−02 | ||
| rs2238017 | 12 | CACNA1C | A | G | 0.35 | Intron | 1.62(1.16–2.28) | 5.31×10−03 | 1.37(1.05–1.78) | 2.22×10−02 | ||
| rs10800465 | 1 | CAPON | C | T | 0.26 | Intron | 0.6(0.42–0.87) | 6.17×10−03 | 0.83(0.62–1.12) | 2.28×10−01 | ||
| rs7427106 | 3 | SCN5A | C | G | 0.13 | Intron | 1.98(1.21–3.22) | 6.22×10−03 | 1.13(0.79–1.61) | 4.95×10−01 | ||
| rs1395112 | 4 | ANK2 | A | G | 0.34 | 0.62(0.44–0.87) | 6.35×10−03 | 0.72(0.54–0.96) | 2.38×10−02 | |||
| rs10741726 | 11 | KCNQ1 | G | T | 0.45 | Intron | 1.55(1.12–2.15) | 7.91×10−03 | 1.18(0.9–1.55) | 2.22×10−01 | ||
| rs12026931 | 1 | CAPON | C | T | 0.08 | Intron | 2.36(1.25–4.45) | 8.09×10−03 | 1.28(0.81–2.02) | 2.96×10−01 | ||
| rs10155204 | 4 | ANK2 | C | T | 0.13 | 0.48(0.27–0.83) | 8.96×10−03 | 0.62(0.39–0.99) | 4.43×10−02 | |||
| rs10216051 | 7 | KCNH2 | A | G | 0.37 | 1.49(1.08–2.05) | 1.54×10−02 | 1.51(1.16–1.96) | 2.25×10−03 | |||
| rs1032913 | 4 | ANK2 | C | T | 0.21 | 0.61(0.4–0.91) | 1.72×10−02 | 0.55(0.38–0.79) | 1.14×10−03 | |||
| rs3766884 | 1 | RYR2 | C | T | 0.18 | Intron | 1.59(1.07–2.38) | 2.30×10−02 | 1.78(1.29–2.45) | 4.06×10−04 | ||
| rs16928297 | 11 | KCNQ1 | G | T | 0.23 | Intron | 1.52(1.05–2.21) | 2.78×10−02 | 1.69(1.25–2.3) | 7.66×10−04 | ||
| rs596502 | 1 | RYR2 | C | T | 0.21 | Intron | 1.5(1.03–2.17) | 3.51×10−02 | 1.54(1.14–2.08) | 4.65×10−03 | ||
| rs11062288 | 12 | CACNA1C | C | T | 0.27 | Intron | 1.46(1.03–2.09) | 3.57×10−02 | 1.53(1.15–2.04) | 4.04×10−03 | ||
| rs1532876 | 4 | ANK2 | C | G | 0.15 | 0.57(0.33–0.97) | 3.91×10−02 | 0.46(0.3–0.72) | 6.97×10−04 | |||
| rs3733617 | 4 | ANK2 | C | T | 0.05 | P2835S | 1.97(1–3.88) | 4.89×10−02 | 2.6(1.48–4.59) | 9.38×10−04 | ||
| rs689264 | 11 | SCN4B | C | T | 0.08 | Intron | 1.68(0.96–2.92) | 6.93×10−02 | 2.38(1.53–3.7) | 1.15×10−04 | ||
| rs16847548 | 1 | CAPON | C | T | 0.22 | Promoter | 1.41(0.97–2.06) | 7.57×10−02 | 1.55(1.14–2.12) | 5.88×10−03 | ||
| rs757086 | 11 | KCNQ1 | C | T | 0.08 | Intron | 1.39(0.96–2) | 7.78×10−02 | 1.59(1.18–2.14) | 2.39×10−03 | ||
| rs1786176 | 11 | SCN4B | C | T | 0.09 | 1.53(0.9–2.57) | 1.13×10−01 | 1.82(1.22–2.72) | 3.57×10−03 | |||
| rs2237898 | 11 | KCNQ1 | A | C | 0.07 | Intron | 1.59(0.88–2.85) | 1.21×10−01 | 1.84(1.19–2.86) | 6.27×10−03 | ||
| rs4817639 | 21 | KCNE2 | C | T | 0.21 | 1.31(0.9–1.91) | 1.64×10−01 | 1.63(1.2–2.21) | 1.88×10−03 | |||
| rs2238092 | 12 | CACNA1C | A | G | 0.25 | Intron | 1.29(0.9–1.86) | 1.68×10−01 | 1.52(1.12–2.06) | 7.03×10−03 | ||
| rs2819774 | 1 | RYR2 | A | T | 0.34 | Intron | 1.22(0.88–1.7) | 2.28×10−01 | 1.48(1.13–1.94) | 4.00×10−03 | ||
| rs11022922 | 11 | KCNQ1 | C | T | 0.22 | Intron | 1.24(0.87–1.78) | 2.39×10−01 | 1.6(1.17–2.19) | 3.67×10−03 | ||
| rs4427395 | 1 | RYR2 | A | G | 0.34 | Intron | 1.17(0.84–1.63) | 3.43×10−01 | 1.54(1.17–2.03) | 2.18×10−03 | ||
| rs4564142 | 1 | CASQ2 | C | T | 0.16 | 1.18(0.78–1.8) | 4.38×10−01 | 1.7(1.21–2.38) | 2.22×10−03 | |||
| rs7522269 | 1 | CAPON | C | T | 0.22 | Intron | 1.16(0.8–1.66) | 4.38×10−01 | 1.6(1.17–2.2) | 3.67×10−03 | ||
| rs3860222 | 1 | CASQ2 | A | G | 0.23 | 1.15(0.8–1.67) | 4.50×10−01 | 1.63(1.2–2.22) | 1.98×10−03 | |||
| rs1985961 | 17 | SCN4A | C | T | 0.40 | 1.13(0.82–1.56) | 4.68×10−01 | 1.44(1.1–1.89) | 7.85×10−03 | |||
| rs2805468 | 1 | RYR2 | A | G | 0.32 | Intron | 0.92(0.66–1.3) | 6.47×10−01 | 1.44(1.1–1.89) | 8.98×10−03 | ||
| rs10240738 | 7 | KCNH2 | C | T | 0.46 | Promoter | 1.07(0.78–1.46) | 6.66×10−01 | 1.42(1.09–1.85) | 9.2010−03 | ||
The table presents association results for all SNPs that reached a significance level of p ≤ 0.01 either comparing diLQTS cases vs. drug-exposed controls or diLQTS cases vs. population controls. The two SNPs fulfilling these criteria are printed in bold. MAF – minor allele frequency.
In parallel, we performed an analysis comparing diLQTS cases with population control subjects (Figure 1, grey symbols). Tests of association results with p≤0.01 are shown in Table 3 and the complete results are shown in Supplemental Table 1. The most significantly associated variant at this stage was the previously detected rs1805128 / D85N, with an odds ratio of 8.88 (95% CI 3.26 – 24.17, p=1.95 × 10−5). The difference in allele frequencies between cases and population-based controls was 8.6% versus 1.8%. SNP rs7295250 showed weaker, but significant association in the analysis versus population controls subjects: odds ratio 1.56 (95% CI 1.18 – 2.07, p=1.77 × 10−3). Other SNPs showed strong associations, but were not (or not as significantly) associated in the parallel analysis comparing diLQTS cases and drug-exposed controls.
To validate the association between diLQTS and SNPs rs1805128 / D85N and rs7295250, we assessed the relationship in the independent DARE sample consisting of 57 diLQTS cases and 211 population-based, matched control subjects. Clinical characteristics of this validation sample are provided in Table 2. Cases displayed a higher proportion of cardiovascular disease than controls, and hypokalemia at the time of the index arrhythmia was identified in 25% of those cases for whom admission potassium levels were available. Amiodarone (51%) and sotalol (26%) were the two most common culprit drugs. Six patients (11%) were receiving diuretics, usually in combination with another drug. In 12 patients (21%) more than one culprit drug was implicated.
The effect of rs1805128 / D85N in the validation analysis showed a trend in the same direction as observed in the discovery analysis (Table 3). We identified 1 homozygote and 2 heterozygotes among diLQTS cases, and 6 heterozygotes among controls. The minor allele frequencies of cases and controls in this dataset were 3.5% and 1.4%, respectively, which translated into an odds ratio of 1.5 (95% CI 0.35 – 6.57, p=0.58).
In an analysis of rs1805128 stratified by sex, we found similar effect estimates and significance levels for both men and women suggesting no major effect modification by sex. In each subgroup, the results were comparable to the main age- and sex-adjusted analysis (data not shown). SNP rs7295250 failed to replicate in the DARE cohort with a non-significant effect estimate in the opposite direction (Table 3).
Discussion
In this study, we tested the hypothesis that common genetic variants modulate the risk for diLQTS. We observed that the KCNE1 variant rs1805128, resulting in D85N, confers substantially increased risk for drug-induced torsades de pointes. The key resources that allowed us to identify this variant as a risk allele were the accumulation of a large set of cases through an international collaboration, large control sets of subjects with drug response phenotypes, and availability of population controls.
The D85N variant has been implicated previously as a modulator of QT interval19,29,30 and T wave morphology.31 In addition, D85N had been described as a rare variant in a small study of 32 diLQTS cases.12 One recent study reported that co-expression in heterologous cells of KCNE1-D85N with genes encoding potassium channel pore-forming subunits (KCNQ1 and KCNH2) impaired IKr and IKs, two key repolarizing potassium currents, and suggested it contributed to the variable penetrance of the congenital long QT syndrome.32
Life-threatening adverse drug reactions are rare by their nature, since high-risk drugs are not generally widely prescribed. Further, many such adverse reactions occur in an unpredictable fashion, and sudden death due to this mechanism may not be recognized clinically. Thus, the impact of culprit drugs on the balance between risk and benefit of drug therapy in an individual patient has not been predictable a priori. Contemporary genomic approaches to analyze human phenotypes, such as common diseases, rely on discovery and replication cohorts, often accumulating tens of thousands of subjects to validate the predictive power of variants with odds ratios <2. Accumulating such large numbers of cases is by definition impossible for rare adverse drug effects.33,34 Hence, the expectation with the use of either an intensive candidate gene interrogation or even a genome-wide approach is that common contributory variants can be identified only if they confer relatively high odds ratios. Further, demonstration of biologic plausibility, as in this study, bolsters an argument that a variant identified in this fashion contributes to the phenotype under study. Other examples of rare adverse drug effects analyzed using genomic approaches include statin-related myopathy35 and abacavir-related skin reactions.36 In both cases, individual genetic variants with strong effects on risk and with biologic plausibility arguments have been identified.
We studied cases of diLQTS caused by a wide range of drugs and drug classes, although the majority was antiarrhythmic-related. Atrial fibrillation was most prevalent in the drug-exposed control group compared to the population control group and to the diLQTS cases. The reason for this distribution pattern was the fact that many diLQTS cases and drug-exposed controls received culprit drugs for atrial fibrillation treatment. It is widely accepted that culprit drugs all share a common proximate mechanism, which is inhibition of the rapid component of the cardiac delayed rectifier potassium current, IKr.1–3 This common mechanism justifies the inclusion in this analysis of multiple culprit drugs. High concentrations of offending drugs are also clinical risk factors for diLQTS, although mechanisms producing such elevations are drug-specific. Thus the analysis we report did not consider variability in drug concentrations. While the phenotype we studied here required documented torsades de pointes, this arrhythmia has the potential to degenerate to ventricular fibrillation, and thus cause sudden cardiac death. Indeed, use of QT-prolonging antipsychotics37 and of other high potency IKr blockers38 has been associated with an increased risk of SCD.
Electrophysiological experiments using a range of approaches, from whole hearts39–41 to single mammalian cells42 to computational simulations,43 support the concept that variability in the extent to which IKr block prolongs QT interval is determined not only by this specific current but also by multiple other currents whose integrated activity generates normal cardiac repolarization. A framework for analyzing diLQTS susceptibility suggests that multiple mechanisms control normal cardiac repolarization. Thus, loss of a single component in this system is unlikely to cause marked QT derangement; this redundancy has been referred to as “repolarization reserve”.44 As a consequence, individuals with a reduced repolarization reserve, due to common genetic variants or other risk factors such as sex, may display entirely normal QT intervals until drug challenge eliminates the remaining reserve, resulting in marked QT prolongation and torsades de pointes. Recent studies have implicated a second key repolarizing current, IKs, as a major contributor to repolarization reserve.41–43 Our finding, that a variant in KCNE1 is a risk factor for diLQTS, is entirely consistent with this view, and supports the idea of using such a “system” approach45 to analyze the physiologic underpinnings of other rare, previously unpredictable adverse drug effects. Our diLQTS cases presented with significantly prolonged QTc measurements compared to controls. We attribute this finding in part to the fact that many ECGs in cases unavoidably were collected when the patients were not entirely off the QT prolonging drugs. Our findings demonstrate the power both of multi-institutional collaborations to study rare adverse drug effects, and of contemporary genomic approaches as a first step to definitive studies to directly test the value of “pre-prescription genotyping” to optimize drug therapy. Further studies will be required to determine whether pre-prescription genotyping at this site can reduce the incidence of diLQTS.
Limitations and conclusions
Restricting our study to participants of European descent does not allow us to make any conclusion with regard to other ethnicities. Although we did not detect relevant population stratification in our cohort, diLQTS cases were of European descent, but all population controls were Germans only. Current standards in genetic association studies generally require rigorous standards regarding adjustment for multiple comparisons and independent replication. As outlined above, the rarity of our phenotype prevented us from collecting higher numbers of participants to meet this standard. However, the clinical importance of the phenotype justified the acceptance of a trade-off in requirements. In addition, we aimed to minimize false-positive results by using two control groups different in nature, and an independent validation step, which showed consistent but not significant results. As particularly the sample size of the case group was small, we attribute this finding to a limitation of statistical power. In addition to the sample size, also the low frequency of the D85N risk allele is of importance when interpreting the nonsignificant validation. However, the D85N variant is known to modulate potassium current and prolong repolarization. Data attributing risk to this variant in our study and previous work is creating a portfolio of evidence that supports the idea of genotyping subjects at risk prior to drug exposure. At present, such an option might be still distant, however, will become more interesting once further supporting replication evidence is available. How much evidence is required to act on a specific variant in this fashion remains controversial, and is likely to evolve as genotyping costs and very large databases evolve.46 We submit that the clarity of our findings would have benefitted from additionally adjusted models including ECG measurements like the QTc interval, or other clinical covariates like potassium levels or concomitant diseases. Due to an excess of missing data in our case samples, we were restricted to a limited adjustment as presented. Future investigations will be required to gain further insight regarding the effect of other factors on the association between D85N and diLQTS.
Supplementary Material
Acknowledgments
These data have been deposited at Pharmacogenetics and Pharmacogenomics Knowledge Base / PS209206. (pharmgkb.org), and are being deposited at dbGaP, The database of Genotypes and Phenotypes (http://www.ncbi.nlm.nih.gov/gap).
Funding Sources: This study was supported by U01HL65962, the Pharmacogenomics of Arrhythmia Therapy site of the Pharmacogenetics Research Network and by a grant from the Fondation Leducq (Trans-Atlantic Network of Excellence “Alliance Against Sudden Cardiac Death”, 05 CVD 01). Additional support was provided from Bundesministerium für Bildung und Forschung (BMBF) in the context of the German National Genome Research Network NGFNplus 01GS0838, the LMU Excellence Initiative, and within the Munich Center of Health Sciences (MC Health) as part of LMUinnovativ. The KORA research platform and the MONICA Augsburg studies were initiated and financed by the Helmholtz Zentrum München, German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research and by the State of Bavaria. Additional support for the collection and genotyping of the Miami cohort was provided by the Florida Heart Research Institute. The DARE study was funded by the British Heart Foundation under special project grant SP/02/001. Dr. Sinner is supported by the German Heart Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosures: A.A.M.W is a member of the scientific advisory board of Transgenomics (formerly PGxHealth). R.J.M. reports consultancy for Sanofi-Aventis, GSK, and Boston Scientific. He reports various expert testimonies not related to the present study. He was paid for lectures by Sanofi-Aventis and Boston Scientific. Travel and accommodations have been paid by Sanofi-Aventis. He is chair of DSMB of a multicenter study co-supported by the NHLBI, and Medtronic, GE, and Zoll. A.J.C.has acted as a consultant for Sanofi, Servier, Daiichi, Boehringer Ingelheim, Bayer, Novartis, Gilead and Menarini. He is a member of DSMB for studies funded by Servier, Novartis, BMs and Pfizer. A.L.G. reports additional funding to his institution by Gilead Sciences to study proprietary compounds, but unrelated to the present study. Together with D.M.R. he holds U.S. Letters Patent No. 6,458,542, issued October 1, 2002 for “Method of Screening for Susceptibility to Drug-Induced Cardiac Arrhythmia”. D.M.R. in addition reports consultancy for Merck, Novartis, Dai-Ichi, Astellas, Avanir, Pfizer, ARCA, and Vitae Pharm.
References
- 1.Fenichel RR, Malik M, Antzelevitch C, Sanguinetti M, Roden DM, Priori SG, et al. Drug-induced torsades de pointes and implications for drug development. J Cardiovasc Electrophysiol. 2004;15:475–495. doi: 10.1046/j.1540-8167.2004.03534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Antzelevitch C, et al. The potential for qt prolongation and proarrhythmia by non-antiarrhythmic drugs: Clinical and regulatory implications. Report on a policy conference of the european society of cardiology. Eur Heart J. 2000;21:1216–1231. doi: 10.1053/euhj.2000.2249. [DOI] [PubMed] [Google Scholar]
- 3.Roden DM. Drug-induced prolongation of the qt interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 4.Bens JL, Quiret JC, Lesbre JP. Spike torsades with syncopal expression caused by hypokalemia. Coeur Med Interne. 1972;11:293–307. [PubMed] [Google Scholar]
- 5.Prandota J, Iwanczak F. Long q-t syndrome precipitated by atropine and hypokalemia. Dev Pharmacol Ther. 1983;6:356–364. doi: 10.1159/000457336. [DOI] [PubMed] [Google Scholar]
- 6.Roden DM, Woosley RL, Primm RK. Incidence and clinical features of the quinidine-associated long qt syndrome: Implications for patient care. Am Heart J. 1986;111:1088–1093. doi: 10.1016/0002-8703(86)90010-4. [DOI] [PubMed] [Google Scholar]
- 7.Makkar RR, Fromm BS, Steinman RT, Meissner MD, Lehmann MH. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA. 1993;270:2590–2597. doi: 10.1001/jama.270.21.2590. [DOI] [PubMed] [Google Scholar]
- 8.Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-qt syndrome: Clinical impact. Circulation. 1999;99:529–533. doi: 10.1161/01.cir.99.4.529. [DOI] [PubMed] [Google Scholar]
- 9.Donger C, Denjoy I, Berthet M, Neyroud N, Cruaud C, Bennaceur M, et al. Kvlqt1 c-terminal missense mutation causes a forme fruste long-qt syndrome. Circulation. 1997;96:2778–2781. doi: 10.1161/01.cir.96.9.2778. [DOI] [PubMed] [Google Scholar]
- 10.Lehtonen A, Fodstad H, Laitinen-Forsblom P, Toivonen L, Kontula K, Swan H. Further evidence of inherited long qt syndrome gene mutations in antiarrhythmic drug-associated torsades de pointes. Heart Rhythm. 2007;4:603–607. doi: 10.1016/j.hrthm.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 11.Napolitano C, Schwartz PJ, Brown AM, Ronchetti E, Bianchi L, Pinnavaia A, et al. Evidence for a cardiac ion channel mutation underlying drug-induced qt prolongation and life-threatening arrhythmias. J Cardiovasc Electrophysiol. 2000;11:691–696. doi: 10.1111/j.1540-8167.2000.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 12.Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, et al. Genetic variations of kcnq1, kcnh2, scn5a, kcne1, and kcne2 in drug-induced long qt syndrome patients. J Mol Med. 2004;82:182–188. doi: 10.1007/s00109-003-0522-z. [DOI] [PubMed] [Google Scholar]
- 13.Schulze-Bahr E, Haverkamp W, Hordt M. Do mutations in cardiac ion channel genes predispose to drug-induced (acquired) long-qt syndrome? Circulation. 1997;96 I–211. [Google Scholar]
- 14.Yang P, Kanki H, Drolet B, Yang T, Wei J, Viswanathan PC, et al. Allelic variants in long-qt disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 15.Itoh H, Sakaguchi T, Ding WG, Watanabe E, Watanabe I, Nishio Y, et al. Latent genetic backgrounds and molecular pathogenesis in drug-induced long-qt syndrome. Circ Arrhythm Electrophysiol. 2009;2:511–523. doi: 10.1161/CIRCEP.109.862649. [DOI] [PubMed] [Google Scholar]
- 16.Kannankeril PJ, Roden DM, Norris KJ, Whalen SP, George AL, Jr, Murray KT. Genetic susceptibility to acquired long qt syndrome: Pharmacologic challenge in first-degree relatives. Heart Rhythm. 2005;2:134–140. doi: 10.1016/j.hrthm.2004.10.039. [DOI] [PubMed] [Google Scholar]
- 17.Holle R, Happich M, Lowel H, Wichmann HE. Kora--a research platform for population based health research. Gesundheitswesen. 2005;67 Suppl 1:S19–S25. doi: 10.1055/s-2005-858235. [DOI] [PubMed] [Google Scholar]
- 18.Dare: The drug-induced arrhythmia risk evaluation study. Psychiatric Bulletin. 2004;28:431. [Google Scholar]
- 19.Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, et al. Common variants at ten loci influence qt interval duration in the qtgen study. Nat Genet. 2009;41:399–406. doi: 10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeufer A, Sanna S, Arking DE, Muller M, Gateva V, Fuchsberger C, et al. Common variants at ten loci modulate the qt interval duration in the qtscd study. Nat Genet. 2009;41:407–414. doi: 10.1038/ng.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Bakker PI, Yelensky R, Pe'er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 22.A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 24.Keating BJ, Tischfield S, Murray SS, Bhangale T, Price TS, Glessner JT, et al. Concept, design and implementation of a cardiovascular gene-centric 50 k snp array for large-scale genomic association studies. PLoS One. 2008;3:e3583. doi: 10.1371/journal.pone.0003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 28.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marjamaa A, Newton-Cheh C, Porthan K, Reunanen A, Lahermo P, Vaananen H, et al. Common candidate gene variants are associated with qt interval duration in the general population. J Intern Med. 2009;265:448–458. doi: 10.1111/j.1365-2796.2008.02026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gouas L, Nicaud V, Chaouch S, Berthet M, Forhan A, Tichet J, et al. Confirmation of associations between ion channel gene snps and qtc interval duration in healthy subjects. Eur J Hum Genet. 2007;15:974–979. doi: 10.1038/sj.ejhg.5201866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Porthan K, Marjamaa A, Viitasalo M, Vaananen H, Jula A, Toivonen L, et al. Relationship of common candidate gene variants to electrocardiographic t-wave peak to t-wave end interval and t-wave morphology parameters. Heart Rhythm. 2010;7:898–903. doi: 10.1016/j.hrthm.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, et al. D85n, a kcne1 polymorphism, is a disease-causing gene variant in long qt syndrome. J Am Coll Cardiol. 2009;54:812–819. doi: 10.1016/j.jacc.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Giacomini KM, Krauss RM, Roden DM, Eichelbaum M, Hayden MR, Nakamura Y. When good drugs go bad. Nature. 2007;446:975–977. doi: 10.1038/446975a. [DOI] [PubMed] [Google Scholar]
- 34.Wilke RA, Lin DW, Roden DM, Watkins PB, Flockhart D, Zineh I, et al. Identifying genetic risk factors for serious adverse drug reactions: Current progress and challenges. Nat Rev Drug Discov. 2007;6:904–916. doi: 10.1038/nrd2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, et al. Slco1b1 variants and statin-induced myopathy--a genomewide study. N Engl J Med. 2008;359:789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 36.Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic J, et al. Hla-b*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568–579. doi: 10.1056/NEJMoa0706135. [DOI] [PubMed] [Google Scholar]
- 37.Reilly JG, Ayis SA, Ferrier IN, Jones SJ, Thomas SH. Qtc-interval abnormalities and psychotropic drug therapy in psychiatric patients. Lancet. 2000;355:1048–1052. doi: 10.1016/s0140-6736(00)02035-3. [DOI] [PubMed] [Google Scholar]
- 38.van Noord C, Sturkenboom MC, Straus SM, Witteman JC, Stricker BH. Non-cardiovascular drugs that inhibit herg-encoded potassium channels and risk of sudden cardiac death. Heart. 2011;97:215–220. doi: 10.1136/hrt.2009.188367. [DOI] [PubMed] [Google Scholar]
- 39.Akar FG. The perfect storm: Defective calcium cycling in insulated fibers with reduced repolarization reserve. Circ Res. 2007;101:968–970. doi: 10.1161/CIRCRESAHA.107.164426. [DOI] [PubMed] [Google Scholar]
- 40.Wu Y, Anderson ME. Reduced repolarization reserve in ventricular myocytes from female mice. Cardiovasc Res. 2002;53:763–769. doi: 10.1016/s0008-6363(01)00387-x. [DOI] [PubMed] [Google Scholar]
- 41.Xiao L, Xiao J, Luo X, Lin H, Wang Z, Nattel S. Feedback remodeling of cardiac potassium current expression: A novel potential mechanism for control of repolarization reserve. Circulation. 2008;118:983–992. doi: 10.1161/CIRCULATIONAHA.107.758672. [DOI] [PubMed] [Google Scholar]
- 42.Jost N, Virag L, Bitay M, Takacs J, Lengyel C, Biliczki P, et al. Restricting excessive cardiac action potential and qt prolongation: A vital role for iks in human ventricular muscle. Circulation. 2005;112:1392–1399. doi: 10.1161/CIRCULATIONAHA.105.550111. [DOI] [PubMed] [Google Scholar]
- 43.Silva J, Rudy Y. Subunit interaction determines iks participation in cardiac repolarization and repolarization reserve. Circulation. 2005;112:1384–1391. doi: 10.1161/CIRCULATIONAHA.105.543306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roden DM. Taking the "idio" out of "idiosyncratic": Predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 45.Berger SI, Ma'ayan A, Iyengar R. Systems pharmacology of arrhythmias. Sci Signal. 2010;3:ra30. doi: 10.1126/scisignal.2000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roden DM, Wilke RA, Kroemer HK, Stein CM. Pharmacogenomics: The genetics of variable drug responses. Circulation. 2011;123:1661–1670. doi: 10.1161/CIRCULATIONAHA.109.914820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.