

Abstract
Mutations in the PHEX gene cause X-linked hypophosphatemia (XLH). Hypophosphatemia in XLH results from increased circulating levels of a phosphaturic hormone, fibroblast growth factor 23 (FGF23), which inhibits renal phosphate reabsorption and 1,25-dihydroxyvitamin D (calcitriol) synthesis. The current standard therapy for XLH – high dose phosphate and calcitriol – further increases FGF23 concentrations, suggesting that patients with XLH may have an altered response to extracellular phosphate. To test for the presence of abnormal phosphate responsiveness, we compared serum biochemistries and femoral Fgf23 mRNA expression between wild-type mice, murine models of XLH (PhexK496X) and hyperphosphatemic tumoral calcinosis (Galnt3 -/-), and Galnt3/Phex double mutant mice. Phex mutant mice had not only increased Fgf23 expression, but also reduced proteolytic cleavage of intact Fgf23 protein, resulting in markedly elevated intact Fgf23 levels and consequent hypophosphatemia. In contrast, despite markedly increased Fgf23 expression, Galnt3 knockout mice had significantly high proteolytic cleavage of Fgf23 protein, leading to low intact Fgf23 concentrations and hyperphosphatemia. Galnt3/Phex double mutant mice had an intermediate biochemical phenotype between wild-type and Phex mutant mice, including slightly elevated intact Fgf23 concentrations with milder hypophosphatemia. Despite the hypophosphatemia, double mutant mice attempted to reduce serum phosphate back to the level of Phex mutant mice by up-regulating Fgf23 expression as much as 24 fold higher than Phex mutant mice. These data suggest that Phex mutations alter the responsiveness of bone cells to extracellular phosphate concentrations and may create a lower set point for “normal” phosphate levels.
Keywords: Fgf23, Galnt3, Phex, Phosphate, X-linked hypophosphatemia (XLH)
Introduction
X-linked hypophosphatemia (XLH) is the most common genetic disorder of phosphate homeostasis. It is caused by inactivating mutations in the PHEX gene (1-4), which encodes a phosphate-regulating endopeptidase homolog (4,5). Biochemical hallmarks of this disorder include hypophosphatemia and inappropriately low or normal serum 1,25-dihydroxyvitamin D [1,25(OH)2D, calcitriol] concentrations, resulting in rickets and osteomalacia.
The primary cause of the hypophosphatemia in XLH is elevated levels of a circulating phosphaturic hormone, fibroblast growth factor 23 (FGF23), which inhibits renal tubular phosphate reabsorption and 1,25(OH)2D production (6). Although the majority of patients with XLH have increased circulating FGF23 concentrations, some patients have normal FGF23 levels, which are inappropriate in light of the hypophosphatemia (7-9). Both PHEX and FGF23 are expressed in osteoblasts, osteocytes, and odontoblasts (10-14); however, the mechanism by which PHEX deficiency results in overproduction of FGF23 is currently unclear.
In contrast to XLH, persistently low levels of intact FGF23 causes hyperphosphatemia in familial tumoral calcinosis, which is characterized by ectopic calcifications in soft tissues (15-19). Although inactivating mutations in FGF23 (15,19-21) and Klotho (KL) (22) can cause tumoral calcinosis, most of the patients have mutations in GALNT3 (17), which encodes a Golgi-associated glycosyltransferase, UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 3 (ppGalNAc-T3). This enzyme adds O-glycosylation in the subtilisin-like proprotein convertase recognition site in FGF23 protein, thereby protecting FGF23 from proteolytic cleavage and allowing intact FGF23 to be secreted (23,24). Thus, in the absence of this enzyme, most FGF23 is processed into inactive fragments before secretion, leading to low or undetectable levels of biologically active intact FGF23. The low intact Fgf23 and subsequent hyperphosphatemia make tumoral calcinosis a biochemical mirror image of XLH.
The current standard therapy for XLH is high dose phosphate and calcitriol (25). Although the therapy is effective in treating osteomalacia, it does not ameliorate the main cause of hypophosphatemia – i.e. increased FGF23 concentrations – because phosphate and calcitriol are known stimulators of FGF23 expression (6,26-29). Furthermore, recent studies indicate that standard therapy increases FGF23 concentrations in patients with XLH (30,31). These observations suggest that FGF23-producing cells (mainly osteoblasts and osteocytes) in XLH sense the phosphate level to be too high even when it nears normal and this abnormal responsiveness to phosphate may be the underlying cause of increased FGF23 production in XLH. In this study, we tested this hypothesis using murine models of XLH and tumoral calcinosis.
Materials and Methods
Generation and maintenance of experimental animals
A new murine model of XLH carries a nonsense mutation (K496X) in exon 14 of the Phex gene. The PhexK496X mouse line was created and kindly provided by Drs. Jane E. Aubin and Frieda Chen at the University of Toronto, Canada and Dr. Ann M. Flenniken, Celeste Owen and other members of the Centre for Modeling Human Disease, Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, Ontario, Canada. A detailed phenotype of this animal will be described elsewhere (32). Creation and initial characterization of Galnt3 knockout mice, the murine model of familial tumoral calcinosis, was described previously (33). To eliminate the effect of strain background, both Phex and Galnt3 mutant mice were backcrossed to C57/BL6J at least 10 generations. These congenic lines were used for all experiments.
Galnt3/Phex double mutant mice and single mutant mice were generated by crossing the two congenic lines. Since Galnt3 knockout males (-/-) were infertile (33), homozygous females were crossed to affected Phex males (-/Y) to generate F1 generations. F1 offspring were subsequently intercrossed to generate F2 experimental groups. Animals were fed Teklad Global 18% Protein Extruded Rodent Diet containing 1.01% calcium, 0.65% phosphorus, and 2.05 IU/g vitamin D3 (2018SX, Harlan, Indianapolis, Indiana) and tap water ad libitum. All animal studies were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee.
Serum biochemistry measurements
Mice were anesthetized with a ketamine/xylazine mix, and blood was drawn by cardiac puncture and stored at -80 °C until analysis. Serum alkaline phosphatase, BUN, calcium, creatinine, and phosphorus were measured, using Roche COBAS Mira S (Roche Diagnostics, Indianapolis, Indiana). 1,25(OH)2D concentrations were measured, using 1,25-Dihydroxy Vitamin D EIA (Immunodiagnostic Systems Ltd, Fountain Hills, Arizona). Intact Fgf23 levels were measured using FGF-23 ELISA kit, which detects only intact Fgf23 (Kainos Laboratories, Inc., Tokyo, Japan) (9). Fgf23 levels were also measured using mouse FGF-23 (C-Term) ELISA Kit, which detects both intact and C-terminal fragments of Fgf23 (Immutopics International, San Clemente, California). Fgf23 concentrations in Galnt3 knockout mice, hemizygous male and heterozygous female Phex mutant mice, and Galnt3/Phex double mutant mice were measured in serums diluted at 1:10 with diluents suggested by the manufacturers.
Fgf23 mRNA quantification
After the blood draw, femurs were collected and stored in RNAlater RNA Stabilization Reagent (QIAGEN Inc., Valencia, California). Total RNA was extracted from the whole femurs, using TRIzol Plus RNA Purification System (Invitrogen, Carlsbad, California) and used for first-strand cDNA synthesis, using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, California). The cDNA was subsequently used for quantification of Fgf23 expression by probe-based quantitative PCR, using TaqMan® Gene Expression Master Mix in the 7900HT Fast Real-Time PCR System (Applied Biosystems). To identify stably expressed genes in femurs, ten different “housekeeping” genes (18s, Actb, B2m, Gapdh, Gusb, Hmbs, Hprt, Sdha, Tbp, and Tfrc) were amplified in eight cDNA samples each of male and female wild-type, Phex mutant, Galnt3 knockout, and Galnt3/Phex double mutant mice (both 4 and 12 weeks old). The most stable gene, Tbp (TATA box binding protein), determined by NormFinder (34), was used to normalize the expression of Fgf23. Relative gene expression was determined by analyzing the data using the relative standard curve method. Probe-based quantitative real-time PCR assays were designed using Integrated DNA Technologies SciTools (Coralville, Iowa).
The forward primer, probe, and reverse sequences were: GGTGATAACAGGAGCCATGAC, TCAGCCCAGAGAATTGCAAGTTCCG, and TGCTTCTGCGACAAGTAGAC for Fgf23 (NCBI Reference Sequence: NM_022657) and AAGAAAGGGAGAATCATGGACC, CCTGAGCATAAGGTGGAAGGCTGTT, and GAGTAAGTCCTGTGCCGTAAG for Tbp (NCBI Reference Sequence: NM_013684). Sequences for other genes are available upon request.
Statistical analysis
Means, standard deviations, and standard errors were calculated for all outcome measures by genotype. Differences between the twelve genotypic groups were tested for significance using analysis of variance (ANOVA). When the ANOVA p-values were significant, differences between two groups were tested for significance using unpaired student's t-test. Due to significant differences between sexes and ages, the between-genotype comparisons were made for each sex and age, separately. P-values less than 0.05 were considered significant for all analyses.
Results
Galnt3/Phex double mutant mice have an intermediate phenotype between wild-type and Phex mutant mice
To produce Galnt3/Phex double mutant mice, Phex mutant mice carrying a nonsense mutation, K496X, were crossed to Galnt3 knockout mice, a murine model of hyperphosphatemic tumoral calcinosis (33). The double mutant mice were born at the expected ratio (χ2 test p-value = 0.52), and the presence of both Galnt3 and Phex mutations had no effect on the survival of these mice up to 12 weeks. The presence of Galnt3 heterozygosity by itself had no major effects on the phenotype of the mice, and the mean values for various measurements were comparable between wild-type littermates and Galnt3 heterozygotes (Supplemental Figures 1-3). Therefore, phenotypic comparisons were limited to four main groups within each sex: wild type, Galnt3 knockout (Galnt3 -/-), Phex mutant (Phex -/Y and +/-) and double mutant (Galnt3 -/-, Phex -/Y or Galnt3 -/-, Phex +/-). Phex mutant mice had characteristic short, stubby tails (Figure 1A). The tails of double mutant mice were less stubby than Phex mutant littermates, but shorter and thicker than wild-type and Galnt3 knockout littermates. Growth of Galnt3 knockout mice was comparable to that of wild-type littermates; however, Phex mutant mice (in Galnt3 wild-type background) had significant growth retardation from before weaning at three weeks old (Figure 1B). Both male and female Galnt3/Phex double mutant mice were also smaller than their respective wild-type controls. Sizes of double mutant and Phex mutant mice were comparable at weaning, but diverged after weaning, resulting in double mutants having an intermediate body weight between wild-type and Phex mutant mice. Interestingly, body weight of female double mutants was closer to that of wild-type, whereas male double mutant mice were more similar to Phex mutant mice.
Figure 1.
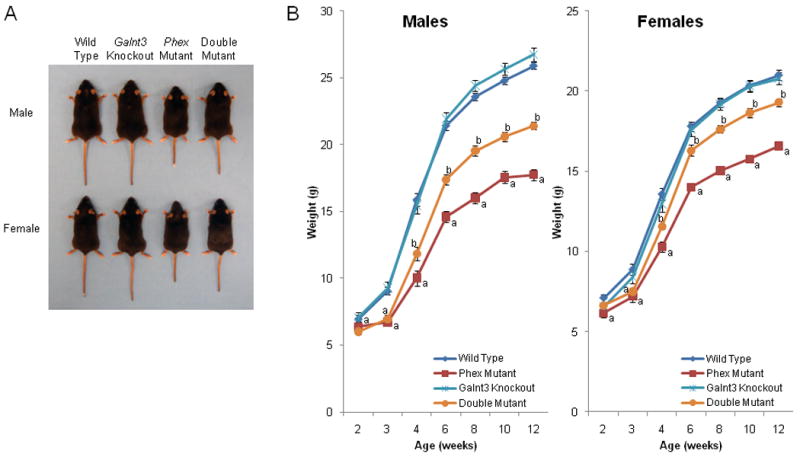
Gross phenotype of Galnt3/Phex double mutant mice. (A) Gross appearance of representative 12-week-old mice. Note that Phex mutant mice (Phex hemizygous male and heterozygous female) had characteristic short tails, whereas Galnt3/Phex double mutant mice have an intermediate tail length. (B) Growth curves. Male and female groups of comparable genotypes are indicated by same colors and symbols. Data are presented as mean ± SEM. Number of animals at any given time point = 8-20 per group. P-value < 0.05: a, compared to same-sex wild-type control mice; b, compared to both wild-type mice and Phex mutant mice. There were no significant differences between wild-type and Galnt3 knockout mice.
Serum biochemistries were measured in four- and twelve-week-old mice. Phex mutant mice, which were significantly smaller than wild-type and Galnt3 knockout mice, had lower creatinine and blood urine nitrogen (BUN) levels, particularly at 12 weeks (Figure 2). Similar to other murine models of XLH (35,36), Phex mutant mice used in this study had hypocalcemia, hypophosphatemia, and high alkaline phosphatase levels (Figure 2). 1,25(OH)2D level was only elevated in male Phex mutant mice. As previously described (33), Galnt3 knockout mice had an opposite biochemical phenotype including hyperphosphatemia, mild hypercalcemia, and low alkaline phosphatase concentrations. Although Galnt3 knockout mice had higher BUN and creatinine levels than Phex mutant mice, these levels were mostly normal compared to wild-type littermates (Figure 2). Similar to Phex mutant mice, Galnt3/Phex double mutant mice were hypophosphatemic with increased alkaline phosphatase levels; however, the phenotype of the double mutant was less severe than that of the Phex mutant (Figure 2). Serum phosphorus in 12-week-old double mutant mice were only 16% less than wild-type mice, compared to 43% in Phex mutants. At four weeks, the degree of the reduction was similar with 49% in Phex mutant and 40% in double mutant mice; however, the difference between the two groups was still significant. Double mutant mice also exhibited hypocalcemia observed in Phex mutant mice, but only in four-week-olds.
Figure 2.
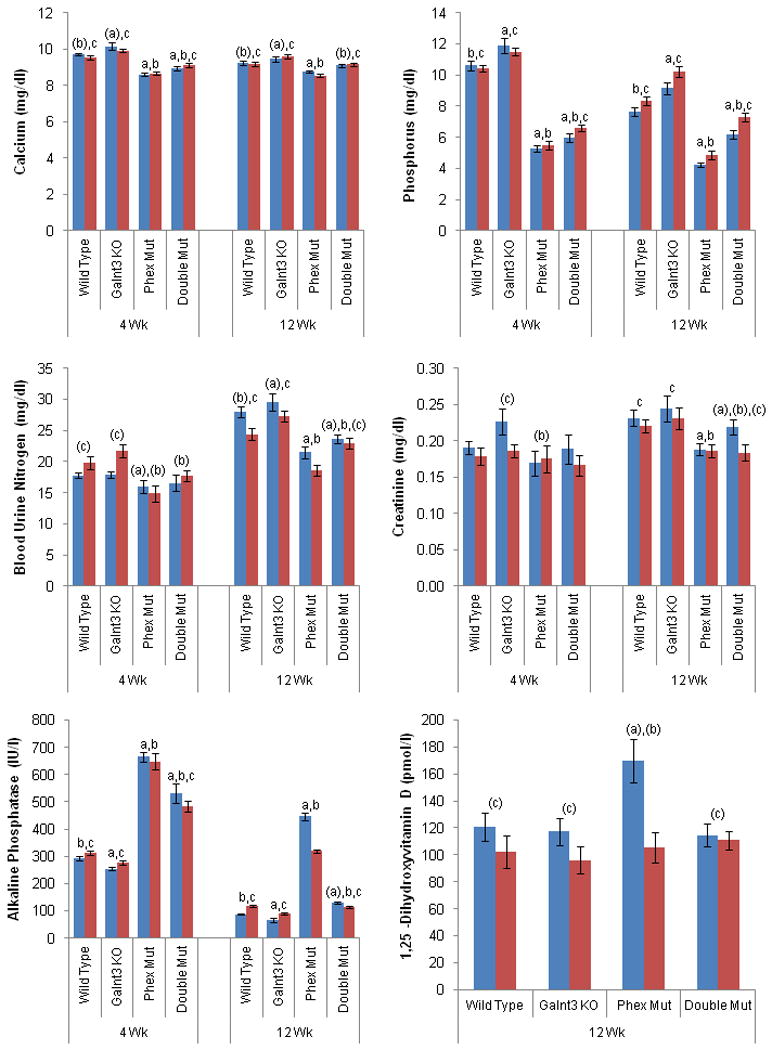
Biochemical analysis of mouse serums. Bars: Males (blue) and females (red). WT, wild-type; Mut, mutant (either hemizygote or heterozygote for Phex); KO, knockout (homozygote). Data are presented as mean ± SEM. Number of animals = 9-15 (4 weeks old) and 9-20 (12 weeks old) per group except 9-13 for 1,25(OH)2D. P-value < 0.05: a, compared to same-sex, same-age wild-type mice; b, Phex mutant mice; and c, Galnt3 knockout mice. a, b, and c, when both males and females of comparable genotypes had significant differences in the comparisons; (a), (b), and (c), when only one sex group had significant differences.
Although same-age males and females of the comparable genotypes had statistically significant differences in some biochemical parameters – most notably alkaline phosphatase for 12-week-old mice, overall trends between genotypes were essentially the same for male or female groups.
Galnt3/Phex double mutant mice have higher Fgf23 gene expression than Phex mutant mice, despite less severe hypophosphatemia
To test for the presence of altered phosphate responsiveness in XLH, we compared serum biochemistries and femoral Fgf23 mRNA expression between wild-type, Phex mutant, Galnt3 knockout, as well as Galnt3/Phex double mutant mice. Phex mutant mice had increased femoral Fgf23 expression, resulting in elevated serum intact Fgf23 levels (Figure 3) and consequent hypophosphatemia. Consistent with our previous study (33), Galnt3 knockout mice had significantly increased Fgf23 expression, but lower circulating concentrations of intact Fgf23 than wild-type littermates (Figure 3). As a result of the low intact Fgf23 concentration, Galnt3 knockout mice had hyperphosphatemia (Figure 2).
Figure 3.
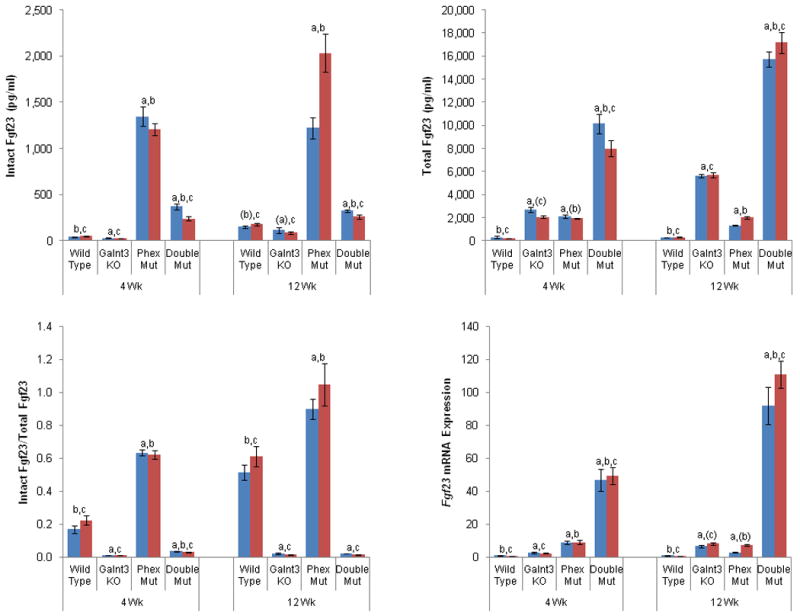
Femoral Fgf23 mRNA expression and circulating Fgf23 concentrations in mutant mice. Total Fgf23 concentrations include intact Fgf23 protein and C-terminal Fgf23 fragments. Fgf23 mRNA expression is normalized to internal control, Tbp, and values are presented by arbitrarily setting wild-type males as 1.0. Bars: Males (blue) and females (red). WT, wild-type; Mut, mutant (either hemizygote or heterozygote for Phex); KO, knockout (homozygote). A subset of the animals used for biochemical analysis (Figure 2) was used in all measurements. Data are presented as mean ± SEM. Number of animals = 5-12 (4 weeks old) and 9-20 (12 weeks old) per group. P-value < 0.05: a, compared to same-sex, same-age wild-type mice; b, Phex mutant mice; and c, Galnt3 knockout mice. a, b, and c, when both males and females of comparable genotypes had significant differences in the comparisons; (a), (b), and (c), when only one sex group had significant differences.
Although the Galnt3/Phex double mutant mice were still hypophosphatemic, their serum phosphorus level was significantly higher than Phex mutant mice and closer to normal because the double mutant had lower intact Fgf23 concentrations compared to the Phex mutant (Figures 2 and 3). In the absence of the altered phosphate responsiveness, Fgf23 mRNA expression in the double mutant should remain similar to Phex mutant mice. Instead, despite the presence of the improved, but still low phosphorus levels, the double mutant mice up-regulated Fgf23 gene expression even higher than Phex mutant mice: on average, 5 and 24 fold higher than 4- and 12-week-old Phex mutant mice, respectively. Of note, these levels were 57 or 113 fold higher than wild-type controls (Figure 3).
Post-translational processing of Fgf23 is increased in Galnt3 mutant mice, but reduced in Phex mutant mice
Circulating Fgf23 levels were also measured using a newly developed sandwich ELISA. Since this assay measures both biologically active intact Fgf23 protein and inactive C-terminal Fgf23 fragments, this measurement represents total amounts of Fgf23 produced in the bone. Thus, the observed patterns between the four genotypic groups were similar to those found in the Fgf23 expression in the bone (Figure 3). Galnt3 knockout mice had significantly higher levels of total Fgf23; total Fgf23 levels were about 9 and 19 fold higher than 4- and 12-week-old normal mice, respectively. However, when the proportion of intact Fgf23 in circulation was determined in these mice, only less than 2% of Fgf23 were intact (Figure 3).
Although 4- and 12-week-old Phex mutant mice had on average 27 and 10 fold higher intact Fgf23 than wild-type mice, the increase was less pronounced in total Fgf23 measured by the new assay – only 7 and 6 fold higher than normal littermates (Figure 3). To corroborate this finding, the proportion of intact Fgf23 in total Fgf23 was more than 40% higher in Phex mutant mice than wild-type mice. In four-week-old Phex mutants, 63% of Fgf23 exist as intact, whereas almost all Fgf23 (97%) in 12-week-old Phex mutant mice were intact.
Consistent with the Fgf23 expression data, double mutant mice had significantly higher total Fgf23 levels than Phex mutant and Galnt3 knockout mice. Double mutant mice at 4 and 12 weeks increased overall production of Fgf23 to 32 and 55 fold higher than sex-matched, wild-type controls. Furthermore, the intact-to-total Fgf23 ratio revealed that compared to Galnt3 knockout mice, there was a small, but significant increase in the proportion of intact Fgf23 in double mutant mice at four weeks.
Discussion
In light of data suggesting that the current therapy for XLH (i.e. high dose phosphate and calcitriol (25)) may actually aggravate FGF23 production (30,31), we investigated whether bone cells in XLH have an abnormal response to circulating phosphate concentrations, using two murine models of disorders of phosphate homeostasis. Compared to Phex mutant mice, Galnt3/Phex double mutant mice had phosphate levels that were closer to normal levels, although still hypophosphatemic. However, despite the fact that double mutant mice had improved serum phosphate levels, they attempted to bring phosphate concentrations down to the level of Phex mutant mice by increasing Fgf23 expression and, to some extent, reducing proteolytic cleavage of intact Fgf23. This suggests that the mutation in the Phex gene alters the responsiveness of bone cells to extracellular phosphate concentrations. Although the improved phosphorus levels were likely the major stimulus for the increased Fgf23 expression, it is possible that the up-regulation of Fgf23 expression was due in part to inappropriately normal 1,25(OH)2D in double mutant mice. Furthermore, the increased Fgf23 expression could result from the abundant C-terminal Fgf23 fragments in these mice, which have been implicated as an inhibitor of intact FGF23 that increases circulating phosphate levels (37). An alternative mechanism for the increased Fgf23 expression in double mutant mice is that hypophosphatemia acts as a “brake” on the system such that as the phosphate level increases, the “brake” suppressing Fgf23 expression is released. However, given the magnitude of the observed increase in Fgf23 expression, this mechanism is less likely.
The altered phosphate responsiveness in Phex mutant mice can be explained by at least three potential mechanisms. First, Fgf23-producing bone cells have a phosphate sensor (or sensing mechanism), which is analogous to the calcium-sensing receptor that controls production of parathyroid hormone (38). Phex mutations may directly affect the phosphate sensing ability of this sensor and lower the set point for extracellular phosphate concentrations. Thus, normalization of phosphate levels in Phex mutant mice would lead to enhanced Fgf23 production. Second, Fgf23 synthesis may be a secondary gene response to chronic changes in intracellular phosphate concentrations. Bone cells in Phex mutant mice may maintain intracellular levels of phosphate higher than normal even when extracellular concentration is low, leading to increased Fgf23 expression. Third, Phex itself may have a direct role in detecting changes in extracellular levels of phosphate. In this scenario, bone cells lacking normal Phex function may become more sensitive to extracellular phosphate and produce more Fgf23. Determining the exact mechanism by which Fgf23 is regulated in bone cells is important for our understanding of the pathogenesis of XLH, as well as other FGF23-mediated disorders.
The increased FGF23 levels in XLH (and Phex mutant mice) were believed to be a result of FGF23 up-regulation. However, our data indicate that Phex mutant mice not only overexpressed the Fgf23 gene, but also secreted more intact Fgf23 protein, likely due to reduced proteolytic cleavage of Fgf23. In agreement with our data, inactive Fgf23 fragments were also undetectable by western blot in another murine model of XLH (Hyp) (39). These data indicate that decreased cleavage of Fgf23 is not dependent on the type of Phex mutation and is part of the pathophysiology in Phex mutant mice and, most likely, XLH patients as well. Furthermore, assays that detect both intact FGF23 and inactive fragments may underestimate the severity of FGF23 elevations in XLH patients because they are making a greater percentage of biologically active FGF23 compared to normal individuals.
The levels of mRNA expression and “total” Fgf23 protein had similar overall trends. However, the increase in Fgf23 mRNA in double mutant mice was markedly higher than that of total Fgf23 protein measured by the new ELISA. This observation suggests that Fgf23 mRNA expression is so high that not all mRNA can be translated into protein. Alternatively, Fgf23 fragments may be degraded quickly such that these cannot be detected by the ELISA used in this study. However, this finding may simply reflect an inherent detection limit and sensitivity of quantitative PCR and ELISA. It should also be noted that the levels of both intact and total Fgf23, as well as the proportion of intact Fgf23 in circulation, change between the two ages studied as Fgf23 was generally higher in the 12-week-old mice.
The finding that Phex mutations alter responsiveness of bone cells to extracellular phosphate has major clinical implications. Although the current therapy of high dose phosphate and calcitriol is effective in treatment of osteomalacia, our data suggest that in XLH the bone is actively trying to reduce serum phosphate levels by producing more FGF23. Thus, the current therapy may increase FGF23 production in XLH not only because both phosphate and calcitriol can stimulate FGF23 production in normal and pathological conditions (6,26-29), but also because PHEX mutations cause the intrinsic defect in phosphate responsiveness of bone cells. Therefore, long-term use of the current therapy can lead to marked increases in FGF23 concentrations as suggested in recent studies (30,31) and potentially reduce the effectiveness of this therapy.
A few observations on the growth and serum biochemistries are worth noting. First, our initial characterization of Galnt3 knockout mice showed that heterozygous and homozygous males were smaller than wild-type littermates (33). However, in this study the presence of Galnt3 mutant alleles did not affect growth of the mice up to 12 weeks. This discrepancy may be attributable to the strain difference used in these two studies. The original report used F2 generations in mixed 129SvEv and C57BL/6J background, whereas the present study used F2 offspring from two congenic C57BL/6J strains. Second, Galnt3/Phex double mutant mice had an intermediate phenotype between wild-type and Phex mutant mice. However, since O-glycosylation of Fgf23 by ppGalNAc-T3 plays a critical role in stability of Fgf23, it was conceivable that lack of Galnt3 would prevent secretion of most intact Fgf23 and cause hyperphosphatemia in double mutant mice. Instead, the double mutant mice had an opposite biochemical phenotype with increased intact Fgf23 concentration (although less than Phex mutant mice). This finding suggests that if Galnt3 knockout mice produce large amounts of Fgf23, they can override Galnt3 deficiency and produce more intact Fgf23 protein.
In conclusion, Phex mutations lead to intrinsic defects in the responsiveness of Fgf23-producing cells (osteoblasts and osteocytes) to phosphate levels. The impaired responsiveness in turn results in overexpression of the Fgf23 gene, as well as reduction in proteolytic cleavage of intact Fgf23. Therefore, elucidation of the mechanism for the abnormal phosphate responsiveness in these bone cells may open up a new therapeutic approach to control elevated FGF23 concentrations in XLH.
Supplementary Material
Acknowledgments
We thank Dr. Erik A. Imel at Indiana University for helpful suggestions on intact Fgf23 measurements and Jeffrey Lavigne at Immutopics International for providing some mouse FGF-23 (C-Term) ELISA Kits. This study was supported in part by Indiana University School of Medicine Biomedical Research Grant (to SI), Showalter Research Trust Fund Grant (to SI), and National Institutes of Health Grant R01 AR042228 (to MJE). The preparation of this report was partially supported by a career development award to SI from the Indiana Clinical and Translational Sciences Institute funded in part by National Institutes of Health Grant RR025760.
Footnotes
Disclosure: Michael J. Econs receives royalties from and is a consultant for Kyowa Hakko Kirin Co. Ltd. All other authors state that they have no conflicts of interest.
References
- 1.Francis F, Strom TM, Hennig S, Boddrich A, Lorenz B, Brandau O, Mohnike KL, Cagnoli M, Steffens C, Klages S, Borzym K, Pohl T, Oudet C, Econs MJ, Rowe PS, Reinhardt R, Meitinger T, Lehrach H. Genomic organization of the human PEX gene mutated in X-linked dominant hypophosphatemic rickets. Genome Res. 1997;7(6):573–85. doi: 10.1101/gr.7.6.573. [DOI] [PubMed] [Google Scholar]
- 2.Holm IA, Huang X, Kunkel LM. Mutational analysis of the PEX gene in patients with X-linked hypophosphatemic rickets. Am J Hum Genet. 1997;60(4):790–7. [PMC free article] [PubMed] [Google Scholar]
- 3.Rowe PS, Oudet CL, Francis F, Sinding C, Pannetier S, Econs MJ, Strom TM, Meitinger T, Garabedian M, David A, Macher MA, Questiaux E, Popowska E, Pronicka E, Read AP, Mokrzycki A, Glorieux FH, Drezner MK, Hanauer A, Lehrach H, Goulding JN, O'Riordan JL. Distribution of mutations in the PEX gene in families with X-linked hypophosphataemic rickets (HYP) Hum Mol Genet. 1997;6(4):539–49. doi: 10.1093/hmg/6.4.539. [DOI] [PubMed] [Google Scholar]
- 4.The_HYP_Consortium. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995;11(2):130–6. doi: 10.1038/ng1095-130. [DOI] [PubMed] [Google Scholar]
- 5.Du L, Desbarats M, Viel J, Glorieux FH, Cawthorn C, Ecarot B. cDNA cloning of the murine Pex gene implicated in X-linked hypophosphatemia and evidence for expression in bone. Genomics. 1996;36(1):22–8. doi: 10.1006/geno.1996.0421. [DOI] [PubMed] [Google Scholar]
- 6.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19(3):429–35. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 7.Endo I, Fukumoto S, Ozono K, Namba N, Tanaka H, Inoue D, Minagawa M, Sugimoto T, Yamauchi M, Michigami T, Matsumoto T. Clinical usefulness of measurement of fibroblast growth factor 23 (FGF23) in hypophosphatemic patients: proposal of diagnostic criteria using FGF23 measurement. Bone. 2008;42(6):1235–9. doi: 10.1016/j.bone.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 8.Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348(17):1656–63. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 9.Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87(11):4957–60. doi: 10.1210/jc.2002-021105. [DOI] [PubMed] [Google Scholar]
- 10.Thompson DL, Sabbagh Y, Tenenhouse HS, Roche PC, Drezner MK, Salisbury JL, Grande JP, Poeschla EM, Kumar R. Ontogeny of Phex/PHEX protein expression in mouse embryo and subcellular localization in osteoblasts. J Bone Miner Res. 2002;17(2):311–20. doi: 10.1359/jbmr.2002.17.2.311. [DOI] [PubMed] [Google Scholar]
- 11.Ruchon AF, Marcinkiewicz M, Siegfried G, Tenenhouse HS, DesGroseillers L, Crine P, Boileau G. Pex mRNA is localized in developing mouse osteoblasts and odontoblasts. J Histochem Cytochem. 1998;46(4):459–68. doi: 10.1177/002215549804600405. [DOI] [PubMed] [Google Scholar]
- 12.Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291(1):E38–49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- 13.Ruchon AF, Tenenhouse HS, Marcinkiewicz M, Siegfried G, Aubin JE, DesGroseillers L, Crine P, Boileau G. Developmental expression and tissue distribution of Phex protein: effect of the Hyp mutation and relationship to bone markers. J Bone Miner Res. 2000;15(8):1440–50. doi: 10.1359/jbmr.2000.15.8.1440. [DOI] [PubMed] [Google Scholar]
- 14.Miao D, Bai X, Panda D, McKee M, Karaplis A, Goltzman D. Osteomalacia in hyp mice is associated with abnormal phex expression and with altered bone matrix protein expression and deposition. Endocrinology. 2001;142(2):926–39. doi: 10.1210/endo.142.2.7976. [DOI] [PubMed] [Google Scholar]
- 15.Araya K, Fukumoto S, Backenroth R, Takeuchi Y, Nakayama K, Ito N, Yoshii N, Yamazaki Y, Yamashita T, Silver J, Igarashi T, Fujita T. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab. 2005;90(10):5523–7. doi: 10.1210/jc.2005-0301. [DOI] [PubMed] [Google Scholar]
- 16.Garringer HJ, Fisher C, Larsson TE, Davis SI, Koller DL, Cullen MJ, Draman MS, Conlon N, Jain A, Fedarko NS, Dasgupta B, White KE. The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J Clin Endocrinol Metab. 2006;91(10):4037–42. doi: 10.1210/jc.2006-0305. [DOI] [PubMed] [Google Scholar]
- 17.Ichikawa S, Baujat G, Seyahi A, Garoufali AG, Imel EA, Padgett LR, Austin AM, Sorenson AH, Pejin Z, Topouchian V, Quartier P, Cormier-Daire V, Dechaux M, Malandrinou F, Singhellakis PN, Le Merrer M, Econs MJ. Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. Am J Med Genet A. 2010;152A(4):896–903. doi: 10.1002/ajmg.a.33337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichikawa S, Imel EA, Sorenson AH, Severe R, Knudson P, Harris GJ, Shaker JL, Econs MJ. Tumoral calcinosis presenting with eyelid calcifications due to novel missense mutations in the glycosyl transferase domain of the GALNT3 gene. J Clin Endocrinol Metab. 2006;91(11):4472–5. doi: 10.1210/jc.2006-1247. [DOI] [PubMed] [Google Scholar]
- 19.Larsson T, Yu X, Davis SI, Draman MS, Mooney SD, Cullen MJ, White KE. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab. 2005;90(4):2424–7. doi: 10.1210/jc.2004-2238. [DOI] [PubMed] [Google Scholar]
- 20.Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14(3):385–90. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 21.Chefetz I, Heller R, Galli-Tsinopoulou A, Richard G, Wollnik B, Indelman M, Koerber F, Topaz O, Bergman R, Sprecher E, Schoenau E. A novel homozygous missense mutation in FGF23 causes Familial Tumoral Calcinosis associated with disseminated visceral calcification. Hum Genet. 2005;118(2):261–6. doi: 10.1007/s00439-005-0026-8. [DOI] [PubMed] [Google Scholar]
- 22.Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117(9):2684–91. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frishberg Y, Ito N, Rinat C, Yamazaki Y, Feinstein S, Urakawa I, Navon-Elkan P, Becker-Cohen R, Yamashita T, Araya K, Igarashi T, Fujita T, Fukumoto S. Hyperostosis-hyperphosphatemia syndrome: a congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J Bone Miner Res. 2007;22(2):235–42. doi: 10.1359/jbmr.061105. [DOI] [PubMed] [Google Scholar]
- 24.Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem. 2006;281(27):18370–7. doi: 10.1074/jbc.M602469200. [DOI] [PubMed] [Google Scholar]
- 25.Harrell RM, Lyles KW, Harrelson JM, Friedman NE, Drezner MK. Healing of bone disease in X-linked hypophosphatemic rickets/osteomalacia. Induction and maintenance with phosphorus and calcitriol. J Clin Invest. 1985;75(6):1858–68. doi: 10.1172/JCI111900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146(12):5358–64. doi: 10.1210/en.2005-0777. [DOI] [PubMed] [Google Scholar]
- 27.Burnett-Bowie SM, Henao MP, Dere ME, Lee H, Leder BZ. Effects of hPTH(1-34) infusion on circulating serum phosphate, 1,25-dihydroxyvitamin D, and FGF23 levels in healthy men. J Bone Miner Res. 2009;24(10):1681–5. doi: 10.1359/JBMR.090406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimada T, Yamazaki Y, Takahashi M, Hasegawa H, Urakawa I, Oshima T, Ono K, Kakitani M, Tomizuka K, Fujita T, Fukumoto S, Yamashita T. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol. 2005;289(5):F1088–95. doi: 10.1152/ajprenal.00474.2004. [DOI] [PubMed] [Google Scholar]
- 29.Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, Kato S, Ogata E, Segawa H, Miyamoto K, Fukushima N. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005;280(4):2543–9. doi: 10.1074/jbc.M408903200. [DOI] [PubMed] [Google Scholar]
- 30.Alon US, Levy-Olomucki R, Moore WV, Stubbs J, Liu S, Quarles LD. Calcimimetics as an adjuvant treatment for familial hypophosphatemic rickets. Clin J Am Soc Nephrol. 2008;3(3):658–64. doi: 10.2215/CJN.04981107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imel EA, DiMeglio LA, Hui SL, Carpenter TO, Econs MJ. Treatment of X-linked hypophosphatemia with calcitriol and phosphate increases circulating fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab. 2010;95(4):1846–50. doi: 10.1210/jc.2009-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Owen C, Chen F, Flenniken AM, Osborne LR, Adamson SL, Rossant J, Aubin JE. A novel Phex mutation in a new mouse model of hypophosphatemic rickets (In preparation) doi: 10.1002/jcb.24115. [DOI] [PubMed] [Google Scholar]
- 33.Ichikawa S, Sorenson AH, Austin AM, Mackenzie DS, Fritz TA, Moh A, Hui SL, Econs MJ. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology. 2009;150(6):2543–50. doi: 10.1210/en.2008-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64(15):5245–50. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 35.Lorenz-Depiereux B, Guido VE, Johnson KR, Zheng QY, Gagnon LH, Bauschatz JD, Davisson MT, Washburn LL, Donahue LR, Strom TM, Eicher EM. New intragenic deletions in the Phex gene clarify X-linked hypophosphatemia-related abnormalities in mice. Mamm Genome. 2004;15(3):151–61. doi: 10.1007/s00335-003-2310-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carpinelli MR, Wicks IP, Sims NA, O'Donnell K, Hanzinikolas K, Burt R, Foote SJ, Bahlo M, Alexander WS, Hilton DJ. An ethyl-nitrosourea-induced point mutation in phex causes exon skipping, x-linked hypophosphatemia, and rickets. Am J Pathol. 2002;161(5):1925–33. doi: 10.1016/S0002-9440(10)64468-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, Kuro-o M, Mohammadi M. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U S A. 107(1):407–12. doi: 10.1073/pnas.0902006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar R, Thompson JR. The regulation of parathyroid hormone secretion and synthesis. J Am Soc Nephrol. 22(2):216–24. doi: 10.1681/ASN.2010020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamazaki Y, Tamada T, Kasai N, Urakawa I, Aono Y, Hasegawa H, Fujita T, Kuroki R, Yamashita T, Fukumoto S, Shimada T. Anti-FGF23 neutralizing antibodies show the physiological role and structural features of FGF23. J Bone Miner Res. 2008;23(9):1509–18. doi: 10.1359/jbmr.080417. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.