

Abstract
During normal pregnancy, dramatically increased placental blood flow is critical for fetal growth and survival as well as neonatal birth weights and survivability. This increased blood flow results from angiogenesis, vasodilatation, and vascular remodeling. Locally produced growth factors including fibroblast growth factor2 (FGF2) and vascular endothelial growth factor (VEGFA) are key regulators of placental endothelial functions including cell proliferation, migration, and vasodilatation. However, the precise signaling mechanisms underlying such regulation in fetoplacental endothelium are less well defined, specifically with regard to the interactions amongst protein kinases, protein phosphatase, and nitric oxide (NO). Recently we and others researchers have obtained solid evidence showing that different signaling mechanisms participate in FGF2- and VEGFA-regulated fetoplacental endothelial cell proliferation and migration as well as NO production. This review will briefly summarize currently available data on signaling mediating fetoplacental angiogenesis with a specific emphasis on protein kinases, ERK1/2, AKT1, and p38 MAPK and protein phosphatases, PPP2 and PPP3.
INTRODUCTION
During normal pregnancy, dramatically increased feto- and uteo-placental blood flows are highly correlated with fetal growth and survival as well as neonatal birth weights and survivability (Alexander 1974, Rosenfeld et al. 1974, Reynolds & Redmer 1995, Reynolds et al. 2005). These increased blood flows are primarily caused by angiogenesis, vasodilatation, and vascular remodeling (Barcroft & Barron 1946, Magness & Zheng 1996, Osol & Mandala 2009). Two potent angiogenic factors, fibroblast growth factor 2 (FGF2) and vascular endothelial growth factor (VEGFA), are key factors regulating placental vascular growth, vasodilatation, and vascular remodeling (Brigstock 1991, Klagsbrun & D’Amore 1991, Ferrara et al. 2003, Osol & Mandala 2009). Local endothelial production of vasodilators such as nitric oxide (NO) is increased during pregnancy in temporal association with increases in the production of placental angiogenic factors and may play an active role in the integral regulation of placental angiogenesis and vasodilatation (Myatt 1992, Magness & Zheng 1996, Sladek et al. 1997, Magness, 1998, Szukiewicz et al. 2005, Brownbill et al. 2007, Sprague et al. 2010).
FGF2 is one of the most extensively studied members in FGF family, which consists of at least nine structurally related polypeptides (Klein et al. 1997, Powers et al. 2000). Not only FGF2 is expressed in endothelial cells but it is thought to act as an autocrine factor. However, it is still not fully understood how FGF2 is released from the cells. The cellular response to FGF2 is mediated by binding and activating its high affinity receptors that have cytoplasmic tyrosine kinase domains. Similar to FGF2, VEGFA is also a member of a family of structurally homologous growth factors with a potent angiogenic activity for vascular endothelial cells (Ferrara et al. 2003). Biological actions of VEGFA are initiated upon binding to its high affinity receptors including VEGFR1 (Flt1) and VEGFR2 (Flk1/KDR) (Gille et al. 2000, Ferrara et al. 2003). VEGFR2 is the major signal transducer of VEGFA, responsible for mediating VEGFA-stimulated major steps of angiogenesis (endothelial cell proliferation and migration) and vasodilatation, whereas VEGFR1 may inhibit VEGFR2-mediated endothelial functions (Gille et al. 2000, Ferrara et al. 2003). However, knocking down either of these receptors in the mouse impairs vascular growth and development growth during the early embryonic stage, ultimately leading to embryonic death, indicating that both VEGFR1 and VEGFR2 are important for vascular formation and growth during early embryonic stage (Fong et al. 1995, Shalaby et al. 1995).
Over the past decade, it has become clear that in addition to its vasodilatory activity, NO, serves as a downstream signaling mediates FGF2- and VEGFA-stimulated angiogenesis (Ziche et al. 1993, 1994, 1997, Arnal et al. 1996, Noiri et al. 1997, Babaei et al. 1998, Murohara et al. 1998, Fulton et al. 1999, Morales-Ruiz et al. 2000). However, it is still not well defined what the signaling pathways are participating in FGF2- and VEGFA-promoted placental angiogenesis and how NO modulates placental angiogenesis.
In Vitro Cell Models
As significant improvements in methodology have occurred over the past two decades (Manjunath et al. 2009, Morris 2009), it has become possible to analyze the roles of individual signaling molecule in vivo. For example, it is now possible to specifically knock down an individual signaling component in a selected tissue or cell type beyond the embryonic stage using lentivirus-mediated RNA interference in vivo. However, owing to the complexity of signaling networks and technical difficulties, it is still extremely challenging to dissect individual signaling induced by a single factor in tissues and cells in vivo, specifically since the same signaling pathway could be activated simultaneously by a number of humoral (i.e., steroid hormone and peptide growth factors) (Klein et al. 1997, Powers et al. 2000) and physical factors (i.e., shear stress and circumferential stress) (Li et al. 2003, 2004, Osol & Mandala 2009, Sprague et al. 2009, 2010). Hence, most of our current knowledge on signaling mechanisms governing placental endothelial cell functions has been built on in vitro cell models.
It is well established that under normal physiological states, angiogenesis in vivo primarily occurs in microvasculature consisting of arterioles, capillaries, and venules, suggesting that endothelial cells isolated from these microvascular beds are the best cell models for studying placental angiogenesis. Indeed, over the last two decades, great efforts have been made to obtain microvascular endothelial cells from human placentas (Challier et al. 1995, Kacemi et al. 1997, Ugele & Lange 2001, Lang et al. 2003, Wang et al. 2003, Dye et al. 2004). These placental microvascular and macrovascular (i.e., human umbilical and chorionic plate arteries and veins) endothelial cells differ in their embryological origins. For example, the villous capillary endothelia are de novo generated from mesodermally-derived haemangioblastic cells in early placental villi and the chorionic plate artery endothelia are derived from allantoic vessels in the connecting stack during early embryonic stage (Benirschke et al. 2006a, b, Wang & Zhao 2010). Additionally, these placental microvascular endothelial cells could significantly differ from those generated from macrovessels (i.e., human umbilical veins) in their morphologies and in their responses to certain growth factors such as placental growth factor, but not to others such as FGF2 and VEGFA (Lang et al. 2003). These placental microvascular endothelial cells reportedly were enriched either from digesting enzyme effluents perfused through placentas via a chorionic artery (Lang et al. 2003) or from enzyme solutions applied to digest placental tissues or terminal villous vessels (Challier et al. 1995, Kacemi et al. 1997, Ugele & Lange 2001, Wang et al. 2003, Dye et al. 2004). These cell preparations, however, were inevitably composed of mixed populations of endothelial cells from both macro- and microvaculatures, possibly including all three types of microvasculatures (arterioles, capillaries, and venules). Specifically, the exact origin of these endothelial cells was hard to confirm due to the lack of specific markers for identifying or separating macro- or micro-vascular endothelium or each individual microvascular endothelium. Given the fact that different origins of endothelia are highly heterogeneous in global gene expression profiles, possibly leading to different cell phenotypes including morphologies, growth rates, and responses to stimuli (Chi et al. 2003, Lang et al. 2003, 2008, Aitsebaomo et al. 2008, Rocha & Adams 2009, dela Paz NG & D’Amore PA 2009), it is still questionable whether these mixed populations of cells can closely represent overall phenotypes of placental endothelial cells in vivo, especially after extensive expansion in vitro, during which cells derived from a specific locus are likely to become dominant.
On the other hand, endothelial cells isolated from relatively larger vessels, particularly placental arteries and veins (Zheng et al. 2005, Lang et al. 2008, Wang et al. 2009) as well as umbilical cord vessels (Chi et al. 2003) have been widely used for studying human endothelial functions because of their technical feasibility for obtaining a large number of cells with a high purity from a single type of vessel (Baudin et al. 2007). Since the cell proliferative responses to FGF2 and VEGFA were similar between endothelial cells isolated from the placental microvasculature and human umbilical veins (Lang et al. 2003), endothelial cells from those large vessels could be used as cell models for studying placental endothelial functions at least in regard with actions of FGF2 and VEGFA. Indeed, much of our current knowledge on regulation of human endothelial functions and signaling mechanisms has been obtained from these endothelial cell models.
Protein Kinases
The cellular responses to FGF2 and VEGFA are mediated by activating their specific receptors that have cytoplasmic tyrosine kinase domains. Upon activation, these receptor-tyrosine kinases initiate a cascade of cellular protein phosphorylation by protein kinases, including ERK1/2, AKT1, and p38 MAPK (Cobb 1999, Powers et al. 2000, Gille et al. 2000, Boilly et al. 2000, Cross et al. 2003). ERK1/2, a threonine and tyrosine kinase is phosphorylated and activated predominantly by MEK1/2 in the cytosol, translocates to the nucleus, and subsequently stimulates transcription of early response genes (Davis 1993, Blenis 1993, Blumer & Johnson 1994). AKT1 (also referred to as protein kinase B [PKB]) is a serine and threonine kinase, which is one major downstream target of PI3K. The MEK1/2/ERK1/2 and the PI3K/AKT1 signaling pathway are heavily involved in regulation of cell survival, proliferation, and migration (Rousseau et al. 1997, Cobb 1999, Powers et al. 2000, Gille et al. 2000, Boilly et al. 2000, Matsumoto & Claesson-Welsh 2001, Vivanco & Sawyers 2002,). p38 MAPK is also a serine and threonine kinase comprising at least four isoforms, α, β, γ, and δ, and is activated predominantly by MEK3/6. p38 MAPKα, β, and δ isoforms are ubiquitously expressed, whereas p38γ appears to be specially expressed in skeletal muscle. Activation of p38 MAPK was initially considered to be induced by environmental stress and inflammatory cytokines (Kyriakis et al. 1996); however, increasing evidence has shown that p38 MAPK also participates in growth factor-regulated cell functions including growth and migration (Boilly et al. 2000, Cross et al. 2003).
Activation of the ERK1/2, AKT1, and p38 MAPK is well known to play a critical role in FGF2- and VEGFA-stimulated endothelial cell proliferation and differentiation (Sa et al. 1995, D’Angelo et al. 1995, Matsumoto et al. 2002). However, the integration of these different kinases is extremely complicated in endothelium. It has been shown that activation of both ERK1/2 and PI3K is required for inducing a complete cell proliferation in response to FGF2, but not VEGFA in bovine choroidal endothelial cells (Zubilewicz et al. 2001). Similarly, activation of both ERK1/2 and p38 MAPK was also needed to induce FGF2-stimulated cell proliferation and migration in mouse spleen endothelial cells (Tanaka et al. 1999). These data suggest important mediation of parallel activation of both ERK1/2/PI3K and ERK1/2/p38 MAPK in growth factor-induced angiogenesis. On the other hand, inhibition of p38 MAPK has been show to enhance VEGFA-induced angiogenesis, accompanied by prolonged ERK1/2 activation in human lung-derived microvascular endothelial (HLDME) cells (Issbrucker et al. 2003). Inhibition of p38 MAPK also promoted VEGFA-promoted endothelial cell survival, partially via enhancing activation of the PI3K/AKT1 pathway in bovine aortic endothelial (BAE) and human umbilical cord vein (HUVE) cells (Gratton et al. 2001). Thus, an antagonistic regulation between p38 MAPK and ERK1/2 or AKT1 may also be important for endothelial functions including angiogenesis.
In ovine fetal placental artery endothelial (OFPAE) cells which were derived from secondary and tertiary branches of umbilical cords of late pregnant ewes (Zheng et al. 2005), both FGF2 and VEGFA robustly induced activation of the ERK1/2, AKT1, and p38 MAPK pathways, which at least partially mediated FGF2- and VEGFA-stimulated cell proliferation and migration (Zheng et al. 1999, 2008, Wang et al. 2008, 2009, Song et al. 2009, Liao et al. 2009, 2010, Figs. 1 and 2). Intriguingly, the MEK1/2/ERK1/2 and/or PI3K/AKT1 pathways differentially mediated the FGF2- and VEGFA-stimulated OFPAE cell proliferation and migration (Zheng et al. 2008, Figs. 1 and 2). For example, inhibition of either of the MEK1/2/ERK1/2 or PI3K/AKT1 pathways alone only partially attenuated the FGF2-stimulated cell proliferation, whereas it completely blocked the VEGFA-stimulated cell proliferation as well as the VEGFA- and FGF2-stimulated cell migration (Zheng et al. 2008, Fig. 1). Similarly, inhibition of p38 MAPK moderately suppressed the FGF2-stimulated cell proliferation and migration (Fig. 2), whereas it did not alter VEGFA-stimulated cell proliferation (Fig. 2) and migration (Liao et al. 2009, 2010). Additionally, inhibition of MEK1/2 by PD98059 (a selective MEK inhibitor) significantly decreased FGF2- and VEGFA-induced p38 MAPK, but not AKT1 phosphorylation, whereas LY294002 (a selective PI3K inhibitor) and SB203580 (a selective p38 MAPK inhibitor) did not appear to alter ERK1/2 phosphorylation (Fig. 3). These observations suggest that activation of either MEK/1/2/ERK1/2 or PI3K/AKT1 pathway only partially mediates FGF2-stimulated cell proliferation, but is sufficient to mediate the VEGFA-stimulated complete cell proliferation as well as FGF2- and VEGFA-stimulated complete cell migration in OFPAE cells (Zheng et al. 2008). Moreover, activation of the p38 MAPK pathway critically mediated FGF2-stimulated cell proliferation and migration, whereas was not sufficient for mediating VEGFA-induced cell proliferation and migration in OFPAE cells (Zheng et al. 2006, Liao et al 2010). These data imply that unlike the antagonistic roles of p38 MAPK in HLDME, BAE, and HUVE cells (Gratton et al. 2001, Issbrucker et al. 2003), p38 MAPK plays a positive role in regulating FGF2-, but not VEGFA-stimulated angiogenic activities of OFPAE cells. It is noteworthy that distinct signaling pathways might differentially mediate endothelial cell responses to FGF2 and VEGFA in different origins of placental endothelial cells. For example, we have reported that the PI3K/AKT1, but not MEK/ERK1/2 pathway mediates FGF2-stimulated cell proliferation, whereas the MEK1/2/ERK1/2, but not the PI3K/AKT1 pathway mediates VEGFA-stimulated cell proliferation in human placental artery endothelial cells (Wang et al. 2009). Thus, a complex signaling network may mediate placental angiogenesis via a parallel, synergistic and/or antagonistic manner (Yashima et al. 2001), possibly depending on signaling pathways and other vascular beds.
Figure 1.

Effects of PD98059 and LY294002 on FGF2- and VEGFA-Stimulated OFPAE Cell Migration. Cell migration was measured using a Multiwell BD Falcon FluoroBlok Insert System (8.0-um pores, BD Biosciences). Cells were treated with 10 ng/ml of FGF2 or VEGFA in the absence or presence of PD98059 or LY294002 (1 hr of pretreatment). Cells were counted after 16 hr of treatment. A) Representative images are shown, in which concentrations of PD98059 and LY294002 were at 40 and 6 μM, respectively. Bar = 100 μm. B) Data are expressed as means ± SEM % of the controls. All data were analyzed using one-way ANOVA. When an F test was significant, data were compared to the control by Bonferroni’s multiple comparisons or Student’s t test. *differ from the control (n = 5; p ≤ 0.05).
Figure 2.


Effects of SB203580 on FGF2- and VEGFA-Stimulated Cell Proliferation and FGF2-Stimulated Cell Migration in OFPAE Cells. A) A time course for FGF2- and VEGFA-induced phosphorylation of p38 MAPK. Note: 1) Both FGF2 and VEGFA (10 ng/ml) rapidly (~ 10 min) induced (p ≤ 0.05) overall p38 MAPK phosphorylation; 2) FGF2 appeared to be much more potent in inducing p38 MAPK phosphorylation as compared to VEGFA; and 3) FGF2 and VEGFA did not significantly alter protein levels of total p38 MAPK (p38). Data shown were collected after 10 min of FGF2 and VEGFA stimulation, pooled from all three p38 MAPK isoforms, and analyzed as described above. Data normalized to total p38 are expressed as means ± SEM fold of the controls. *differ from the control (n = 5; p ≤ 0.05). B) Effects of SB203580 on OFPAE cell proliferation and migration. Cell proliferation assay was carried out using a crystal violet method. Cells were treated with 10 ng/ml of FGF2 or VEGFA in the absence or presence of SB203580 (1 hr pretreatment). Cells were counted after 72 or 16 hr of treatments, respectively for cell proliferation and migration assays.
Figure 3.
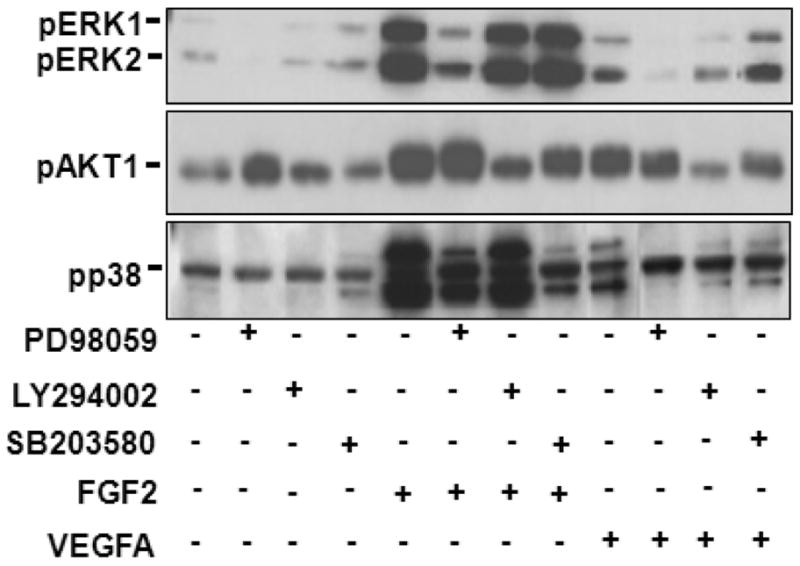
Effects of PD98059, LY294002, and SB203580 on Phosphorylation of ERK1/2, AKT1 and p38 MAPK Induced by FGF2 and VEGFA in OFPAE Cells. After 16 hr of serum starvation, cells were treated with 10 ng/ml of FGF2 or VEGFA for 10 min in the absence or presence of PD 98059 (20 μM), LY294002 (5 μM), or SB203580 (10 μM) (1 hr pretreatment). Proteins (20μg/lane) were subjected to Western blotting using antibody against phospho-specific ERK1/2 (pERK1/2; 1:2000), AKT1 (pAKT1; 1:1000) or p38 MAPK (pp38; 1:1000).
Protein Phosphatases: PPP2 and PPP3
After activation, protein kinases must undergo inactivation, returning to a status ready for the next stimulus. One of such mechanisms to inactivate protein kinases is to dephosphorylate these kinases by protein phosphatases. Reflecting the huge diversity and breadth of functions regulated by protein dephosphorylation, higher eukaryotes encode ~1000 protein phosphatase genes which can be classified into at least three families (Barford et al. 1998). Within each family, the catalytic domains are highly conserved, with functional diversity endowed by regulatory subunits. These protein phosphatases can also be cataloged into two major classes: tyrosine phosphatases and serine/threonine phosphatases, depending on their substrates (Liu et al. 2007). Serine/threonine phospho-protein phosphatases (PPP), which specifically dephosphorylate phosphoserine and phosphothreonine residues, include PPP1, PPP2 (formally termed as PP2A), PPP3 (PP2B or calcineurin), and PPM1B (PP2C) (Aramburu et al. 2004, Wilkins & Molkentin 2004).
PPP2, one of the most extensively studied members of PPP, is believed to make up most of total serine/threonine phosphatase activity in cells (Lechward et al. 2001, Sontag 2001). PPP2 consists of catalytic (C) and scaffolding (A) subunits, which can bind to at least another eighteen regulatory (B) subunits to form a trimeric holoenzyme (see Lechward et al. 2001, Sontag 2001, Cho et al. 2007, Shi 2009 for detail PPP2 structures). In mammals, the C subunit of PPP2 has two major isoforms, α (PPP2CA) and β (PPP2CB), between which PPP2CA is the most abundant isoform with the mRNA level ~ 10-fold more than PPP2CB in most tissues (Lechward et al. 2001, Sontag 2001). It is still unclear if these two catalytic subunits have distinct functions. However, PPP2CB cannot completely compensate for the absence of PPP2CA in early embryonic development, as knockdown of PPP2CA alone in the mouse lead to early embryonic death, mostly owing to impaired mesoderm formation (Gotz et al. 1998). PPP2 mediates a variety of essential cellular processes such as cell growth, protein synthesis, and metabolism (Lechward et al. 2001, Sontag 2001), which could be partially a result of dephosphorylating and inactivating protein kinases ERK1/2, AKT1, and p38 MAPK (Alessi et al. 1995, Camps et al. 2000, Janssens & Goris 2001, Silverstein et al. 2002, Lee et al. 2003, Liao & Hung 2004, Van Kanegan et al. 2005).
Similar to PPP2, PPP3 is also composed of catalytic and regulatory subunits, including three catalytic (α, β, and λ) and two regulatory (B1 and B2) subunits (Perrino et al. 2002, Aramburu et al. 2004, Wilkins & Molkentin 2004), among which α, β, and B1 subunits are widely expressed in mammalian tissues, whereas λ and B2 subunits are primarily found in the testis and brain (Perrino et al. 2004). The PPP3 catalytic subunit α (PPP3CA) appears to account for the majority (70–80%) of total phosphatase activity of PPP3, while the catalytic β subunit (PPP3CB) constitutes ~ 20–30% (Im & Rao 2004). Like PPP2, PPP3 also participates in many cellular functions (e.g., immune responses, and cardiac hypertrophy), partially via directly dephosphorylating members of nuclear factor of activated T cell (NFAT) transcriptional factors, which in turn could be modulated by ERK1/2 (Aramburu et al. 2004, Wilkins & Molkentin 2004). A positive role of PPP3 in mediating vascular development has been proposed as disruption of Ppp3r1, Nfatc3, and Nfatc4 genes in the mouse is embryonic lethal, due to impaired vascular development (Graef et al. 2001). This premise is further supported by the reports that suppression of PPP3 activity by its pharmacological inhibitor cyclosporin A (CsA) attenuated VEGFA-induced angiogenesis in HUVE cells (Hernandez et al. 2001) and intestinal microvascular endothelial cells (Rafiee et al. 2004). This inhibition by CsA was partially mediated via suppressing ERK1/2 and p38 MAPK activation (Rafiee et al. 2004, Farivar et al. 2005). Similar reciprocal relationships between PPP3 and ERK1/2 also have been reported in cardiomyocytes (Molkentin 2004) and in B cells (Gary-Gouy et al. 2006). However, in other types of cells, suppression of PPP3 activity could lead to completely opposite effects. For example, CsA has been shown to enhance ERK1/2 activation in human trophoblast cells (Du et al. 2007) and canine kidney epithelial cells (Kiely et al. 2003). Little is known about the mediation of PPP3 in activation of the PI3K/AKT1 cascade although it has been shown that PPP3 inhibition does not alter activation of the PI3K/AKT cascade in A549 cells (Wen et al. 2003).
The involvement of protein phosphatases in regulating endothelial functions has received much less attention as compared to the protein kinases, particularly regarding the roles of PPP2/PPP3 in placental angiogenesis. Recently, we have identified the expression of PPP2CA and PPP3CA in OFPAE cells (Wang et al. 2008, Song et al. 2009). Nonetheless, suppression of PPP2CA protein expression by its specific siRNA did not significantly affect VEGFA- and FGF2-stimulated OFPAE cell proliferation (Song et al. 2009). On the other hand, knockdown of PPP3CA protein by its specific siRNA only moderately enhanced VEGFA-stimulated (~20% increase), but not FGF2-stimulated cell proliferation. Thus, PPP3CA alone has a critical role in modulating VEGFA-stimulated cell proliferation, whereas PPP2CA alone does not have such a role in modulating both VEGFA- and FGF2-stimulated cell proliferation in OFPAE cells.
Failure of PPP2CA suppression to alter VEGFA- and FGF2-stimulated OFPAE cell proliferation is in disagreement with the previous reports showing that inhibition of PPP2 activity promoted cell motility in bovine aortic endothelial cells and primary human mouth endothelial cells (Gabel et al. 1999, Young et al. 2002) and cell proliferation in bovine aortic endothelial cells (Murata et al. 1996). Additionally, although participation of PPP3 in VEGFA-, but not FGF2-stimulated OFPAE cell proliferation was consistent with the previous reports using other endothelial cell types (Hernandez et al. 2001, Rafiee et al. 2004), the PPP3CA knockdown-enhanced VEGFA-stimulated OFPAE cell proliferation also contrasted with these previous reports (Hernandez et al. 2001, Rafiee et al. 2004). It is currently unknown whether different origins of endothelial cells cause these different or complete opposite actions of PPP3CA in modulating cell proliferation. Nonetheless, in those previous studies (Murata et al. 1996, Gabel et al. 1999, Hernandez et al. 2001, Young et al. 2002, Rafiee et al. 2004), inhibition of PPP2 and PPP3 activity was carried out by their pharmacological inhibitors (okadiac acid or cantharidin for PPP2 or CsA for PPP3). Given the specificity of these pharmacological inhibitors is highly dependent on the doses and relatively high doses of pharmacological PPP2 inhibitors were used in these studies, the effects of PPP2 and PPP3 inhibition on endothelial responses by their pharmacological inhibitors might result from suppression of multiple protein phosphatases. Moreover, unlike these pharmacological inhibitors which attenuate/block all catalytic subunits of PPP2 and PPP3, the siRNA used in OFPAE cells targeted only on catalytic subunit α of PPP2 and PPP3. This specific knockdown of a single catalytic subunit by the siRNA might cause upregulation of PPP2CB and PPP3CB to compensate the loss of phosphatase activity after PPP2CA and PPP3CA suppression, leading to differential modulation of different cell responses. Conversely, the different duration of PPP2 and PPP3 inhibition before growth factor stimulation can also contribute to such different cell responses caused by these pharmacological inhibitors (30 min to 1 hr) and siRNA (at least 16 hr). This supposition is supported by a recent observation that chronic and acute inhibition of PPP2 induced the opposite regulation of ERK1/2 and AKT1 activation since the PPP2 siRNA induced chronic ERK1/2 and AKT1 hyperphosphorylation, downregulating signaling molecules upstream of Ras in response to growth factors including FGF2 (Van Kanegan et al. 2005). In OFPAE cells, suppression of PPP2CA and PPP3CA by their special siRNA did attenuate FGF2-induced ERK1/2 and AKT1 activation (Wang et al. 2008, Song et al. 2009). Thus, a negative feedback mechanism might be involved in PPP2CA and PPP3CA modulation of ERK1/2 and AKT1 activation induced by FGF2 in OFPAE cells.
The roles of protein phosphatases in the modulation of placental angiogenesis and underlying signaling are much more complicated than originally thought, as PPP2 and PPP3 differentially modulated the VEGFA- and FGF2-stimulated cell proliferation and signaling cascades in OFPAE cells (Wang et al. 2008, Song et al. 2009, Fig. 4). Moreover, protein phosphatases other than PPP2 and PPP3 such as MAPK phosphatase and Phosphatase and TENsin (PTEN) homolog could play a more important role in differential modulation of ERK1/2, AKT1, and p38 MAPK activation in placental endothelial cells, ultimately regulating placental angiogenesis.
Figure 4.

A Proposed Model of the Signal Transduction Pathways for FGF2- and VEGFA-Stimulated Proliferation and Migration in OFPAE Cells. In this working model, we propose that FGF2 and VEGFA activate ERK1/2, AKT1, and p38 MAPK, which in turn increase eNOS protein expression and/or eNOS activity, increasing NO production. This increased NO as an intracellular signaling modulates FGF2- and VEGFA-stimulated cell proliferation and migration. Inhibition of PPP3CA, but not PPP2CA enhances VEGFA-, but not FGF2-stimulated cell proliferation, while failing to affect FGF2- and VEGFA-induced activation of ERK1/2 and AKT1, suggesting that yet to be identified signaling molecules play an important role in FGF2- and VEGFA-stimulated cell proliferation after knockdown of PPP3CA in OFPAE cells.
Angiogenesis and Nitric Oxide
Over the past two decades, it has become clear that apart from being a potent vasodilator (Dulak & Jozkowicz 2003), NO is also a key mediator of angiogenesis (Pipili-Synetos et al. 1993, Ziche et al. 1993, 1994, 1997a,b, Noiri et al. 1997, Parenti et al. 1998, Babaei et al. 1998, Murohara et al. 1998, Bussolati et al. 2001, Hida et al. 2004). The participation of NO in mediating angiogenesis was first reported by Pipili-Synetos et al. (1993) using the chick embryo chorioallantoic membrane (CAM) model, in which NO was believed to act as an anti-angiogenic mediator. However, Ziche and colleagues have subsequently provided several lines of evidence showing that NO functions as a positive mediator of angiogenesis (Ziche et al. 1993, 1994, 1997a,b, Parenti et al. 1998). They reported that both exogenous and endogenous NO stimulated cellular DNA synthesis, proliferation and migration in bovine post capillary venule endothelial cells in vitro (Ziche et al. 1993, 1994) and that exogenous NO potentiated angiogenesis in the rabbit “cornea pocket assay”, in which VEGFA-induced angiogenesis was also completely inhibited by the NOS inhibitor, L-NAME (Ziche et al. 1997a). Using the same in vivo assay, they further proposed that VEGFA-, but not FGF2-induced angiogenesis was mediated by NOS via the NO/cyclic guanylate monophosphate pathway (Ziche et al. 1997a). Positive involvement of endogenous NO as a downstream signal of VEGFA-induced angiogenesis was confirmed by in vivo observations showing significantly improved angiogenesis in response to dietary supplementation L-arginine in the rabbit (Murohara et al. 1998) and in the rat (eNOS overexpression, Namba et al. 2003) ischemia models, as well as the considerably limited angiogenesis in eNOS knockout mice (Fukumura et al. 2001). Bussolati and colleagues et al. (2001) have also proposed that VEGFR-1 promoted formation of capillary networks in HUVE cells via NO, while inhibiting VEGFR-2-mediated cell proliferation. Exogenous NO can also act as a crucial signal in the angiogenic response, in which NO promotes FGF2-induced endothelial cell differentiation into capillary tubes, while terminating the proliferative actions in both HUVE and calf pulmonary artery endothelial cells (Babaei et al. 1998). Thus, NO differentially regulate FGF2- and VEGFA-induced angiogenesis at different steps (i.e., proliferation/migration vs. capillary tube formation).
Expression of eNOS and iNOS has been identified in the placenta of human, rhesus monkey, rat, and sheep (Myatt et al. 1993, Conrad et al. 1993, Zarlingo et al. 1997). In association with robust fetoplacental angiogenesis (Reynolds LP & Redmer DA 1995, Magness RR & Zheng 1996), the NO level was increased in maternal circulation as pregnancy progresses in sheep (Vonnahme et al. 2005) and in late human pregnancy (Williams et al., 1997). In ovine cotyledons (fetal side of the placentome) during late pregnancy, eNOS, but not iNOS was present in the fetal component of the placentome, primarily in microvascular endothelial cells in the villous core (Zheng et al. 2000), similar to the findings reported in the term placentae of rhesus monkeys, baboon, guinea-pig, rat and sheep (Zarlingo et al. 1997). Together with the observation that increased expression of eNOS, but not iNOS protein run parallel to increased total NO (nitrate and nitrite) production (Zheng et al. 2000), eNOS seems to be a predominant isoform of NOS responsible for the NO production in the fetal component of the placentome during late ovine pregnancy. Additionally, these increases in eNOS protein expression and NO production in the fetal component of the placentome are temporally associated with increased placental vascular density and expression of FGF2 (Magness & Zheng 1996, Zheng et al. 1997), supporting a critical role NO in modulate fetoplacental angiogenesis.
Indeed, similar to the NO-mediated angiogenic responses in those endothelial cells reported (Ziche et al. 1993, 1994, 1997a,b, Parenti et al. 1998), exogenous NO (sodium nitroprusside [SNP], a potent NO donor) alone promoted OFPAE and human placental artery endothelial (HPAE) cell proliferation (Zheng et al. 2006) and OFPAE migration (Liao et al. 2010). Of note, NO mediation of angiogenesis is highly dependent on NO levels. At relatively lower levels, NO might be pro-angiogenic as shown in OFPAE cells (Zheng et al. 2006, Liao et al. 2010) and in other endothelial cells (Fukuo et al. 1995, Hida et al. 2004), while at relatively high levels NO could act as a pro-apoptotic or anti-angiogenic factor (Kimura & Esumi 2003, Zheng et al. 2006). These seemingly contradictory observations are likely due to the reaction of NO with super-oxidants, which in turn forms peroxynitrite, causing cytostasis and apoptosis (Pacher et al. 2007, Frey et al. 2009). Interestingly, the stimulatory actions of exogenous NO on OFPAE cell proliferation and migration (Zheng et al. 2006, Liao et al. 2010) were not associated with increased mRNA expression of FGF2, VEGFA, or their major receptors (VEGFR1, VEGFR2, NP1, NP2, and FGFR1) (Zheng et al. 2006). This is contradictory to previous reports demonstrating existence of the VEGFA/FGF2-NO reciprocal regulation between endothelial cells and the surrounding non-endothelial cells, including vascular smooth muscle cells, macrophages, keratinocytes, and tumor cells as described (Tuder et al. 1995, Tsurumi et al. 1997, Dembinska-Kiec et al. 1997, Ziche et al. 1997b, Frank et al. 1999, Namba et al. 2003, Zhang et al. 2003, Dulak J & Jozkowicz A 2003). In these studies, NO either promoted angiogenesis via increasing VEGFA expression in the rat ischemic hindlimb (Namba et al. 2003) and brain (Zhang et al. 2003) or FGF2 expression in bovine coronary venular endothelium (Ziche et al. 1997b) or suppressing mRNA expression of VEGFA and/or its receptors (VEGFR1 and VEGFR2) in rat vascular smooth muscle (Tsurumi et al. 1997), in rat lung tissue (Tuder et al. 1995), and in rat renal mesangial cells (Frank et al. 1999). Thus, the observation that SNP-derived NO failed to alter expression of FGF2, VEGFA, and their major receptors in OFPAE cells cultured under standard cell culture conditions (37° C, 5% CO2, 95% air) (Zheng et al. 2006) implicates that the VEGF/FGF2-NO reciprocal regulation may not occur within placental endothelia, although such regulation may exist between placental vascular endothelia and smooth muscle cells. It is noteworthy that this discrepancy in such NO regulation could also be attributed to different NO donors used since they are known to differentially mediate the cellular responses, possibly due to differences in the amount and duration of NO generation (Dulak & Jozkowicz 2003). Moreover, in our studies, even after stimulated with VEGFA and FGF2 at physiological concentrations, OFPAE and HUVE cells under standard culture conditions produced much lower levels of NO (unpublished data) as compared to those released from SNP at doses (1–10 μM) which stimulated OFPAE cell proliferation (Zheng et al. 2006). This suggests that under physiological conditions, NO generated by these endothelial cells might never reach to a dangerous level, even when one assumes the existence of a positive feedback loop for VEGFA/FGF2 and NO between placental vascular endothelial and smooth muscle cells. This phenomenon could also be attributed to the fact that NO itself can negatively regulate eNOS dimerization, expression and/or activity in endothelial cells, possibly via S-nitrosylation, thereby decreasing NO production (Sheehy et al. 1998, Black et al. 1999, Ravi et al. 2004, Kopincova et al. 2011) and potentially preventing apoptosis.
What the downstream signaling is for exogenous NO-mediated angiogenesis is still poorly defined. We have found that an NO donor might activate different signaling pathways, depending on the origin of the cells utilized. For example, SNP induced activation of ERK1/2, but not AKT1 in OFPAE cells, whereas activated both kinases in HPAE cells (Zheng et al. 2006). Additionally, a previous report has also shown NO donors (S-nitosol-L-glutathion and S-nitroso-N-penicillamine) promote migration and angiogenesis of human and bovine endothelial cells via activation of the soluble GC (sGC)/cGMP/PI3K/AKT1 pathway (Kawasaki et al. 2003). However, OFPAE cells used (Zheng et al. 2006, Liao et al. 2010) did not have detectable sGC activity and did not produce cGMP in response to SNP (Itoh et al. 1999). Thus, exogenous NO-stimulated angiogenic responses and NO-induced ERK1/2 activation (Zheng et al. 2006, Liao et al. 2010) are unlikely coupled to the sGC/cGMP pathway in OFPAE cells.
It is well recognized that FGF2 and VEGFA promote NO production by endothelial cells isolated from either placental (Zheng et al. 2008, Liao et al. 2010) or other non-placental tissues (Cuevas et al. 1991, Kadota et al. 1995, Kostyk et al. 1995, Rousseau et al. 1997, 2000, Babaei et al. 1998, Murohara et al. 1998, Dimmeler et al. 1999, Fulton et al. 1999, 2001, Michell et al. 1999, Morales-Ruiz et al. 2000, Zubilewicz et al. 2001, Fukumura et al. 2001, Namba et al. 2003). In OFPAE cells, FGF2 and VEGFA stimulated NO production via increasing eNOS protein expression (FGF2) or directly enhancing enzymatic activity of eNOS (FGF2/VEGFA) (Zheng et al. 2008, Mata-Greenwood et al. 2008, 2010, Liao et al. 2010), in which FGF2-increased eNOS expression and NO production was mediated primarily by AP-1-dependent transcription involving JunB and Fra1 up-regulation (Mata-Greenwood et al. 2010). After generation, endogenous NO positively mediated both FGF2- and VEGFA-stimulated cell proliferation and VEGFA-stimulated migration in OFPAE cells, primarily via an intracellular mechanism (Zheng et al. 2008, Liao et al 2010). However, in contrast to the previous studies showing that NO acted as an upstream signaling of ERK1/2 in VEGFA-stimulated cell proliferation and migration in non-placental endothelial cells (Ziche et al. 1997, Parenti et al. 1998), we found that NO lay downstream of ERK1/2 and AKT1 in OFPAE cells (Li et al. 2004, Zheng et al. 2008), consistent with the observations made in some other types of endothelial cells (Fulton 1999, Dimmeler et al. 1999, Souttou et al. 2001).
Activation of eNOS can be tightly regulated by multiple processes including phosphorylation and nitrosylation (Fulton et al. 2001, Boo & Jo 2003, Kopincova et al. 2011), the former of which has been recognized as critical for eNOS activation (Fulton et al. 2001, Boo & Jo 2003). Human eNOS can be phosphorylated on at least three residues: serine 116 (Ser116) and 1177 (Ser1177; Ser1179 in ovine and bovine) and threonine 495 (Thr495; Thr497 in ovine and bovine). Upon phosphorylation of these residues by the kinases, eNOS activity could be either enhanced (at Ser1177) or attenuated (at Thr495 and Ser116) (Boo & Jo 2003). To date, one of the best-studied signaling pathways which mediate eNOS phosphorylation is PI3K/AKT1. It has been reported that VEGFA-activated PI3K/AKT directly phosphorylates eNOS Ser1179, leading to stimulating NO production, and endothelial migration, proliferation, and capillary-like structure formation (Dimmeler et al. 1999, Fulton et al. 1999, Michell et al. 1999 Morales-Ruiz et al. 2000, Souttou et al. 2001). In OFPAE cells, shear stress-elevated NO production was associated with an increased NOS Ser1179 phosphorylation, which was blocked by PI3K inhibitors Wortmannin and LY294002, but not the MEK inhibitor UO126, suggesting that PI3K/Akt-eNOS Ser1179 is also major signaling pathways for activating eNOS activity in OFPAE cells (Li et al. 2003, 2004). In contrast to Ser1177, the VEGFA-induced eNOS Ser116 phosphorylation was completely blocked by the protein kinase C inhibitor calphostin, but not by either Wortmannin or U0126 (Kou et al. 2002). Thus, these data indicate that eNOS phosphorylation at different sites is controlled by a variety of protein kinases, coordinating eNOS activity and NO production.
Little is known about signaling pathways modulating eNOS dephosphorylation by protein phosphatases. It has been shown that PPP2 preferentially dephosphorylates eNOS at Thr497 and Ser1177, but not at Ser116, leading to deactivation of eNOS and impaired angiogenesis (Michell et al. 2001, Greif et al. 2002, Urbich et al. 2002, Leidi et al. 2010). Intriguingly, NO was also able to activate PPP2 to modulate chromatin folding in HUVE cells (Illi et al. 2008). Thus, the reciprocal interactions between protein phosphatases and NO might play a critical role in mediating placental angiogenesis.
Conclusions and Perspectives
In conclusion, an increasing body of evidence either from our laboratories or other investigators has shown that FGF2 and VEGFA regulate fetoplacental angiogenesis via an extremely complex signaling network involving multiple of protein kinases and phosphatases as well as NO (Fig. 4). While much progress has been made, many difficult challenges still remain for dissecting signaling mechanisms underlying fetoplacental angiogenesis. For example, how can we isolate, establish, and maintain fetoplacental microvascular endothelial cell models with highly homogenous cellular purity? What and how are protein phosphatases activated and how do they modulate activation of protein kinases in fetoplacental angiogenesis? Moreover, to date, almost all endothelial cell models used to study placental angiogenesis have been cultured and expanded under an ambient O2 level (~ 21% or pO2 ~ 160 mmHg), which obviously does not closely reflect in vivo physiological O2 levels under which placental endothelial cells constantly reside (~ 1.5–8.0% O2 or pO2 < 12–60 mmHg) (Meschia 2004, Bertout et al. 2008). These endothelial cells cultured under such a condition might represent a subpopulation of endothelial cells which have adapted to hyperoxia. Therefore, culturing and investigating placental endothelial cells under chronic physiological O2 levels are critical to provide information on fetoplacental angiogenesis and underlying signaling mechanisms, more closely mimicking in vivo states.
Acknowledgments
We thank our collaborators and colleagues (Drs. Dong-bao Chen, Wu-xiang Liao, Eugenia Mata- Greenwood, Ian M. Bird, Ronald R. Magness), current and former graduate students (Drs. Song Yang, Cai-feng Dai, Yi-zhou Jiang, Mr. Yan Li), and laboratory personnel (Ms. Wen YX, Mr. Austin JL, and Mr. Phernetton TM) for their valuable contributions to this work. We thank Dr. Ronald R. Magness (RRM) for his critical reading of the manuscript. We also wish to acknowledge the financial support from the U.S. National Institutes of Health (HL64703 to JZ, HD38843 to RRM/JZ).
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- 1.Aitsebaomo J, Portbury AL, Schisler JC, Patterson C. Brothers and sisters: molecular insights into arterial-venous heterogeneity. Circulation Research. 2008;103:929–939. doi: 10.1161/CIRCRESAHA.108.184937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM, Cohen P. Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Current Biology. 1995;5:283–295. doi: 10.1016/s0960-9822(95)00059-5. [DOI] [PubMed] [Google Scholar]
- 3.Alexander G. Birth weight of lambs. Influences and consequences. In: Elliott K, Knight J, editors. Ciba Foundation Symposium 27: Size at Birth. New York: Elsevier Publisher; 1974. [Google Scholar]
- 4.Aramburu J, Heitman J, Crabtree GR. Calcineurin: a central controller of signaling in eukaryotes. EMBO Reports. 2004;5:343–348. doi: 10.1038/sj.embor.7400133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babaei S, Teichert-Kuliszewska K, Monge JC, Mohamed F, Bendeck MP, Stewart DJ. Role of nitric oxide in the angiogenic response in vitro to basic fibroblast growth factor. Circulation Research. 1998;82:1007–1015. doi: 10.1161/01.res.82.9.1007. [DOI] [PubMed] [Google Scholar]
- 6.Barcroft J, Barron DH. Observations upon the form and relations of the maternal and fetal vessels in the placenta of the sheep. Anatomical Record. 1946;94:569–595. doi: 10.1002/ar.1090940403. [DOI] [PubMed] [Google Scholar]
- 7.Barford D, Das AK, Egloff MP. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annual Review of Biophysics & Biomolecular Structure. 1998;27:133–164. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- 8.Baudin B, Bruneel A, Bosselut N, Vaubourdolle M. A protocol for isolation and culture of human umbilical vein endothelial cells. Nature Protocols. 2007;2:481–485. doi: 10.1038/nprot.2007.54. [DOI] [PubMed] [Google Scholar]
- 9.Benirschke K, Kaufmann P, Baergen R. Architecture of normal villous trees. In: Benirschke K, Kaufmann P, Baergen R, editors. Pathology of the Human Placenta. Vol. 5. New York: Springer Science + Business Media; 2006a. pp. 121–173. [Google Scholar]
- 10.Benirschke K, Kaufmann P, Baergen R. Anatomy and pathology of the umbilical cord. In: Benirschke K, Kaufmann P, Baergen R, editors. Pathology of the Human Placenta. 5. New York: Springer Science + Business Media; 2006b. pp. 380–451. [Google Scholar]
- 11.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nature Review Cancer. 2008;8:967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Black SM, Heidersbach RS, McMullan DM, Bekker JM, Johengen MJ, Fineman JR. Inhaled nitric oxide inhibits NOS activity in lambs: potential mechanism for rebound pulmonary hypertension. American Journal of Physiology. 1999;277:H1849–H1856. doi: 10.1152/ajpheart.1999.277.5.H1849. [DOI] [PubMed] [Google Scholar]
- 13.Blumer KJ, Johnson GL. Diversity in function and regulation of MAP kinase pathways. Trends in Biochemical Sciences. 1994;19:236–240. doi: 10.1016/0968-0004(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 14.Boilly B, Vercoutter-Edouart AS, Hondermarck H, Nurcombe V, Le Bourhis X. FGF signals for cell proliferation and migration through different pathways. Cytokine Growth Factor Reviews. 2000;11:295–302. doi: 10.1016/s1359-6101(00)00014-9. [DOI] [PubMed] [Google Scholar]
- 15.Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. American Journal of Physiology. 2003;285:C499–C508. doi: 10.1152/ajpcell.00122.2003. [DOI] [PubMed] [Google Scholar]
- 16.Brownbill P, McKeeman GC, Brockelsby JC, Crocker IP, Sibley CP. Vasoactive and permeability effects of vascular endothelial growth factor-165 in the term in vitro dually perfused human placental lobule. Endocrinology. 2007;148:4734–4744. doi: 10.1210/en.2007-0180. [DOI] [PubMed] [Google Scholar]
- 17.Bussolati B, Dunk C, Grohman M, Kontos CD, Mason J, Ahmed A. Vascular endothelial growth factor receptor-1 modulates vascular endothelial growth factor-mediated angiogenesis via nitric oxide. American Journal of Pathology. 2001;159:993–1008. doi: 10.1016/S0002-9440(10)61775-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB Journal. 2000;14:6–16. [PubMed] [Google Scholar]
- 19.Challier JC, Kacemi A, Olive G. Mixed culture of pericytes and endothelial cells from fetal microvessels of the human placenta. Cellular and Molecular Biology. 1995;41:233–241. [PubMed] [Google Scholar]
- 20.Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D, Brown PO. Endothelial cell diversity revealed by global expression profiling. Proceedings of the National Academy of Sciences USA. 2003;100:10623–10628. doi: 10.1073/pnas.1434429100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho US, Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature. 2007;445:53–57. doi: 10.1038/nature05351. [DOI] [PubMed] [Google Scholar]
- 22.Cobb MH. MAP kinase pathways. Progress in Biophysics and Molecular Biology. 1999;71:479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 23.Conrad KP, Joffe GM, Kruszyna H, Kruszyna R, Rochelle LG, Smith RP, Chavez JE, Mosher MD. Identification of increased nitric oxide biosynthesis during pregnancy in rats. FASEB Journal. 1993;7:566–571. [PubMed] [Google Scholar]
- 24.Cross MJ, Dixelius J, Matsumoto T, Claesson-Welsh L. VEGF-receptor signal transduction. TRENDS in Biochemical Sciences. 2003;28:488–494. doi: 10.1016/S0968-0004(03)00193-2. [DOI] [PubMed] [Google Scholar]
- 25.Cuevas P, Carceller F, Ortega S, Zazo M, Nieto I, Gimenez-Gallego G. Hypotensive activity of fibroblast growth factor. Science. 1991;254:1808–1810. doi: 10.1126/science.1957172. [DOI] [PubMed] [Google Scholar]
- 26.D’Angelo G, Struman I, Martial J, Weiner RI. Activation of mitogen-activated protein kinases by vascular endothelial growth factor and basic fibroblast growth factor in capillary endothelial cells is inhibited by the antiangiogenic factor 16-kDa N-terminal fragment of prolactin. Proceedings of the National Academy of Sciences USA. 1995;92:6374–6378. doi: 10.1073/pnas.92.14.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis RJ. The mitogen-activated protein kinase signal transduction pathway. Journal of Biological Chemistry. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 28.dela Paz NG, D’Amore PA. Arterial versus venous endothelial cells. Cell and Tissue Research. 2009;335:5–16. doi: 10.1007/s00441-008-0706-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dembinska-Kiec A, Dulak J, Partyka L, Huk I, Mailnski T. VEGF-nitric oxide reciprocal regulation. Nature Medicine. 1997;3:1177. doi: 10.1038/nm1197-1177a. [DOI] [PubMed] [Google Scholar]
- 30.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 31.Du MR, Zhou WH, Yan FT, Zhu XY, He YY, Yang JY, Li DJ. Cyclosporin A induces titin expression via MAPK/ERK signaling and improves proliferative and invasive potential of human trophoblast cells. Human Reproduction. 2007;22:2528–2537. doi: 10.1093/humrep/dem222. [DOI] [PubMed] [Google Scholar]
- 32.Dulak J, Jozkowicz A. Regulation of vascular endothelial growth factor synthesis by nitric oxide: facts and controversies. Antioxidants & Rredox Signaling. 2003;5:123–132. doi: 10.1089/152308603321223612. [DOI] [PubMed] [Google Scholar]
- 33.Dye JF, Vause S, Johnston T, Clark P, Firth JA, D’Souza SW, Sibley CP, Glazier JD. Characterization of cationic amino acid transporters and expression of endothelial nitric oxide synthase in human placental microvascular endothelial cells. FASEB Journal. 2004;18:125–127. doi: 10.1096/fj.02-0916fje. [DOI] [PubMed] [Google Scholar]
- 34.Farivar AS, Mackinnon-Patterson BC, Barnes AD, McCourtie AS, Mulligan MS. Cyclosporine modulates the response to hypoxia-reoxygenation in pulmonary artery endothelial cells. The Annals of Thoracic Surgery. 2005;79:1010–1016. doi: 10.1016/j.athoracsur.2004.08.078. [DOI] [PubMed] [Google Scholar]
- 35.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nature Medicine. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 36.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 37.Frank S, Stallmeyer B, Kampfer H, Schaffner C, Pfeilschifter J. Differential regulation of vascular endothelial growth factor and its receptor fms-like-tyrosine kinase is mediated by nitric oxide in rat renal mesangial cells. Biochemical Journal. 1999;338:367–374. [PMC free article] [PubMed] [Google Scholar]
- 38.Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxidants & Redox Signaling. 2009;11:791–810. doi: 10.1089/ars.2008.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proceedings of the National Academy of Sciences USA. 2001;98:2604–2609. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukuo K, Inoue T, Morimoto S, Nakahashi T, Yasuda O, Kitano S, Sasada R, Ogihara T. Nitric oxide mediates cytotoxicity and basic fibroblast growth factor release in cultured vascular smooth muscle cells. A possible mechanism of neovascularization in atherosclerotic plaques. Journal of Clinic Investigation. 1995;95:669–676. doi: 10.1172/JCI117712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fulton D, Gratton JP, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? Journal of Pharmacology and Experimental Therapeutics. 2001;299:818–824. [PubMed] [Google Scholar]
- 43.Gabel S, Benefield J, Meisinger J, Petruzzelli GJ, Young M. Protein phosphatases 1 and 2A maintain endothelial cells in a resting state, limiting the motility that is needed for the morphogenic process of angiogenesis. Otolaryngol Head Neck Surgery. 1999;121:463–468. doi: 10.1016/S0194-5998(99)70238-X. [DOI] [PubMed] [Google Scholar]
- 44.Gary-Gouy H, Sainz-Perez A, Bismuth G, Ghadiri A, Perrino BA, Dalloul A. Cyclosporin-A inhibits ERK phosphorylation in B cells by modulating the binding of Raf protein to Bcl2. Biochemical and Biophysical Research Communications. 2006;344:134–139. doi: 10.1016/j.bbrc.2006.03.121. [DOI] [PubMed] [Google Scholar]
- 45.Gille H, Kowalski J, Yu L, Chen H, Pisabarro MT, Davis-Smyth T, Ferrara N. A repressor sequence in the juxtamembrane domain of Flt-1 (VEGFR-1) constitutively inhibits vascular endothelial growth factor-dependent phosphatidylinositol 3′-kinase activation and endothelial cell migration. EMBO Journal. 2000;19:4064–4073. doi: 10.1093/emboj/19.15.4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gotz J, Probst A, Ehler E, Hemmings B, Kues W. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit C alpha. Proceedings of the National Academy of Sciences USA. 1998;95:12370–12375. doi: 10.1073/pnas.95.21.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell. 2001;105:863–875. doi: 10.1016/s0092-8674(01)00396-8. [DOI] [PubMed] [Google Scholar]
- 48.Gratton JP, Morales-Ruiz M, Kureishi Y, Fulton D, Walsh K, Sessa WC. Akt down-regulation of p38 signaling provides a novel mechanism of vascular endothelial growth factor-mediated cytoprotection in endothelial cells. Journal of Biological Chemistry. 2001;276:30359–30365. doi: 10.1074/jbc.M009698200. [DOI] [PubMed] [Google Scholar]
- 49.Greif DM, Kou R, Michel T. Site-specific dephosphorylation of endothelial nitric oxide synthase by protein phosphatase 2A: evidence for crosstalk between phosphorylation sites. Biochemistry. 2002;41:15845–15853. doi: 10.1021/bi026732g. [DOI] [PubMed] [Google Scholar]
- 50.Hernandez GL, Volpert OV, Iniguez MA, Lorenzo E, Martinez-MartInez S, Grau S, Fresno M, Redondo JM. Selective inhibition of vascular endothelial growth factor–mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. Journal of Experimental Medicine. 2001;193:607–620. doi: 10.1084/jem.193.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hida A, Kawakami A, Miyashita T, Yamasaki S, Nakashima K, Tanaka F, Izumi Y, Tamai M, Huang M, Ida H, Nakamura H, Origuchi T, Ueki Y, Eguchi K. Nitric oxide acts on the mitochondria and protects human endothelial cells from apoptosis. Journal of Laboratory and Clinical Medicine. 2004;144:148–155. doi: 10.1016/j.lab.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 52.Illi B, Dello Russo C, Colussi C, Rosati J, Pallaoro M, Spallotta F, Rotili D, Valente S, Ragone G, Martelli F, Biglioli P, Steinkuhler C, Gallinari P, Mai A, Capogrossi MC, Gaetano C. Nitric oxide modulates chromatin folding in human endothelial cells via protein phosphatase 2A activation and class II histone deacetylases nuclear shuttling. Circulation Research. 2008;102:51–58. doi: 10.1161/CIRCRESAHA.107.157305. [DOI] [PubMed] [Google Scholar]
- 53.Im SH, Rao A. Activation and deactivation of gene expression by Calcium-calcineurin-NFAT-mediated signaling. Molecular Cells. 2004;18:1–9. [PubMed] [Google Scholar]
- 54.Issbrucker K, Marti HH, Hippenstiel S, Springmann G, Voswinckel R, Gaumann A, Breier G, Drexler HC, Suttorp N, Clauss M. p38 MAP kinase--a molecular switch between VEGF-induced angiogenesis and vascular hyperpermeability. FASEB Journal. 2003;17:262–264. doi: 10.1096/fj.02-0329fje. [DOI] [PubMed] [Google Scholar]
- 55.Itoh H, Zheng J, Bird IM, Nakao K, Magness RR. Basic fibroblast growth factor down-regulates the expression of clearance receptor of natriuretic peptides via mitogen-activated protein kinase cascade in ovine feto-placental artery endothelial cells. American Journal of Physiology. 1999;277:R541–R547. doi: 10.1152/ajpregu.1999.277.2.R541. [DOI] [PubMed] [Google Scholar]
- 56.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochemistry Journal. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kacemi A, Galtier M, Espie MJ, Challier JC. Isolation of villous microvessels from the human placenta. Comptes Rendus des Seances de l Academie des Sciences. Serie III, Sciences de la Vie. 1997;320:171–177. doi: 10.1016/s0764-4469(97)85009-3. [DOI] [PubMed] [Google Scholar]
- 58.Kadota O, Ohta S, Kumon Y, Sakaki S, Matsuda S, Sakanaka M. Role of basic fibroblast growth factor in the regulation of rat basilar artery tone in vivo. Neuroscience Letters. 1995;199:99–102. doi: 10.1016/0304-3940(95)12040-b. [DOI] [PubMed] [Google Scholar]
- 59.Kawasaki K, Smith RS, Jr, Hsieh CM, Sun J, Chao J, Liao JK. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Molecular and Cellular Biology. 2003;23:5726–5737. doi: 10.1128/MCB.23.16.5726-5737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kiely B, Feldman G, Ryan MP. Modulation of renal epithelial barrier function by mitogen-activated protein kinases (MAPKs): mechanism of cyclosporin A-induced increase in transepithelial resistance. Kidney International. 2003;63:908–916. doi: 10.1046/j.1523-1755.2003.00804.x. [DOI] [PubMed] [Google Scholar]
- 61.Kimura H, Esumi H. Reciprocal regulation between nitric oxide and vascular endothelial growth factor in angiogenesis. Acta Biochimica Polonica. 2003;50:49–59. [PubMed] [Google Scholar]
- 62.Klagsbrun M, D’Amore PA. Regulators of angiogenesis. Annual Review of Physiology. 1991;53:217–239. doi: 10.1146/annurev.ph.53.030191.001245. [DOI] [PubMed] [Google Scholar]
- 63.Klein S, Roghani M, Rifkin DB. Fibroblast growth factors as angiogenesis factors: new insights into their mechanism of action. EXS. 1997;79:159–192. doi: 10.1007/978-3-0348-9006-9_7. [DOI] [PubMed] [Google Scholar]
- 64.Kopincova J, Puzserova A, Bernatova I. Biochemical aspects of nitric oxide synthase feedback regulation by nitric oxide. Interdisciplinary Toxicology. 2011;4:63–68. doi: 10.2478/v10102-011-0012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kostyk SK, Kourembanas EL, Medeiros WD, McQuilllan LP, D’Amore PA, Braunhut SJ. Basic fibroblast growth factor increases nitric synthase production in bovine endothelial cells. American Journal of Physiology. 1995;269:H1583–1589. doi: 10.1152/ajpheart.1995.269.5.H1583. [DOI] [PubMed] [Google Scholar]
- 66.Kou R, Greif D, Michel T. Dephosphorylation of endothelial nitric-oxide synthase by vascular endothelial growth factor. Implications for the vascular responses to cyclosporin A. Journal of Biological Chemistry. 2002;277:29669–29673. doi: 10.1074/jbc.M204519200. [DOI] [PubMed] [Google Scholar]
- 67.Kyriakis JM, Avruch J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays. 1996;18:567–577. doi: 10.1002/bies.950180708. [DOI] [PubMed] [Google Scholar]
- 68.Lang I, Pabst MA, Hiden U, Blaschitz A, Dohr G, Hahn T, Desoye G. Heterogeneity of microvascular endothelial cells isolated from human term placenta and macrovascular umbilical vein endothelial cells. European Journal of Cell Biology. 2003;82:163–173. doi: 10.1078/0171-9335-00306. [DOI] [PubMed] [Google Scholar]
- 69.Lang I, Schweizer A, Hiden U, Ghaffari-Tabrizi N, Hagendorfer G, Bilban M, Pabst MA, Korgun ET, Dohr G, Desoye G. Human fetal placental endothelial cells have a mature arterial and a juvenile venous phenotype with adipogenic and osteogenic differentiation potential. Differentiation. 2008;76:1031–1043. doi: 10.1111/j.1432-0436.2008.00302.x. [DOI] [PubMed] [Google Scholar]
- 70.Lechward K, Awotunde OS, Swiatek W, Muszynska G. Protein phosphatase 2A: variety of forms and diversity of functions. Acta Biochimica Polonica. 2001;48:921–33. [PubMed] [Google Scholar]
- 71.Lee T, Kim SJ, Sumpio BE. Role of PP2A in the regulation of p38 MAPK activation in bovine aortic endothelial cells exposed to cyclic strain. Journal of Cellular Physiology. 2003;194:349–355. doi: 10.1002/jcp.10211. [DOI] [PubMed] [Google Scholar]
- 72.Leidi M, Mariotti M, Maier JA. EDF-1 contributes to the regulation of nitric oxide release in VEGF-treated human endothelial cells. European Journal of Cell Biology. 2010;89:654–660. doi: 10.1016/j.ejcb.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 73.Li Y, Zheng J, Bird IM, Magness RR. Effects of pulsatile shear stress on nitric oxide production and endothelial cell nitric oxide synthase expression by ovine fetoplacental artery endothelial cells. Biology of Reproduction. 2003;69:1053–1059. doi: 10.1095/biolreprod.102.013474. [DOI] [PubMed] [Google Scholar]
- 74.Li Y, Zheng J, Bird IM, Magness RR. Mechanisms of shear stress-induced eNOS phosphorylation and expression in ovine feto-placental artery endothelial cells. Biology of Reproduction. 2004;70:785–796. doi: 10.1095/biolreprod.103.022293. [DOI] [PubMed] [Google Scholar]
- 75.Liao Y, Hung MC. A new role of protein phosphatase 2A in adenoviral E1A protein-mediated sensitization to anticancer drug-induced apoptosis in human breast cancer cells. Cancer Research. 2004;64:5938–5942. doi: 10.1158/0008-5472.CAN-04-1533. [DOI] [PubMed] [Google Scholar]
- 76.Liao WX, Feng L, Zhang HH, Zheng J, Moore TR, Chen DB. Compartmentalizing VEGF-induced ERK2/1 signaling in placental artery endothelial cell caveolae: a paradoxical role of caveolin-1 in placental angiogenesis in vitro. Molecular Endocrinology. 2009;23:1428–1444. doi: 10.1210/me.2008-0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liao WX, Feng L, Zheng J, Chen DB. Deciphering mechanisms controlling placental artery endothelial cell migration stimulated by vascular endothelial growth factor. Endocrinology. 2010;151:3432–3444. doi: 10.1210/en.2009-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu Y, Shepherd EG, Nelin LD. MAPK phosphatases–regulating the immune response. Nature Reviews Immunology. 2007;7:202–212. doi: 10.1038/nri2035. [DOI] [PubMed] [Google Scholar]
- 79.Magness RR, Zheng J. Circulatory changes during gestation. In: Gluckman PD, Heymann MA, editors. Scientific Basis of Pediatric and Perinatal Medicine. 2. London: Edward Arnold Publishers; 1996. pp. 762–772. [Google Scholar]
- 80.Magness RR. Maternal cardiovascular and other physiologic responses to the endocrinology of pregnancy. In: Fuller BW, editor. Endocrinology of Pregnancy. Vol. 18. Totowa NJ: Humana Press; 1998. pp. 507–539. [Google Scholar]
- 81.Manjunath N, Wu H, Subramanya S, Shankar P. Lentiviral delivery of short hairpin RNAs. Advanced Drug Delivery Reviews. 2009;61:732–745. doi: 10.1016/j.addr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mata-Greenwood E, Liao WX, Zheng J, Chen DB. Differential activation of multiple signaling pathways dictates eNOS upregulation by FGF2 but not VEGF in placental artery endothelial cells. Placenta. 2008;29:708–717. doi: 10.1016/j.placenta.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mata-Greenwood E, Liao WX, Zheng J, Chen DB. Activation of AP-1 transcription factors differentiates FGF2 and VEGF regulation of endothelial nitric oxide synthase expression in placental artery endothelial cells. Journal of Biological Chemistry. 2010;285:17348–17358. doi: 10.1074/jbc.M109.092791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matsumoto T, Turesson I, Book M, Gerwins P, Claesson-Welsh L. p38 MAP kinase negatively regulates endothelial cell survival, proliferation, and differentiation in FGF-2-stimulated angiogenesis. Journal of Cellular Biology. 2002;156:149–160. doi: 10.1083/jcb.200103096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meschia G. Placental respiratory gas and exchange and fetal oxyenation. In: Creasy RK, Resnik R, Iams JD, editors. Maternal-Fetal Medicine: Principles and Practice. 5. Philadelphia: Else Elsevier Health Sciences; 2004. pp. 199–207. [Google Scholar]
- 86.Michell BJ, Griffiths JE, Mitchelhill KI, Rodriguez-Crespo I, Tiganis T, Bozinovski S, de Montellano PO, Kemp BE, Pearson RB. The Akt kinase signals directly to endothelial nitric oxide synthase. Current Biology. 1999;9:845–848. doi: 10.1016/s0960-9822(99)80371-6. [DOI] [PubMed] [Google Scholar]
- 87.Michell BJ, Chen ZB, Tiganis T, Stapleton D, Katsis F, Power DA, Simi AT, Kemp BE. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. Journal of Biological Chemistry. 2001;276:17625–17628. doi: 10.1074/jbc.C100122200. [DOI] [PubMed] [Google Scholar]
- 88.Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovascular Research. 2004;63:467–475. doi: 10.1016/j.cardiores.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 89.Morales-Ruiz M, Fulton D, Sowa G, Languino LR, Fujio Y, Walsh K, Sessa WC. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circulation Research. 2000;86:892–896. doi: 10.1161/01.res.86.8.892. [DOI] [PubMed] [Google Scholar]
- 90.Morris KV. RNA-directed transcriptional gene silencing and activation in human cells. Oligonucleotides. 2009;19:299–306. doi: 10.1089/oli.2009.0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Murata K, Mills I, Sumpio BE. Protein phosphatase 2A in stretch-induced endothelial cell proliferation. Journal of Cellular Biochemistry. 1996;63:311–319. doi: 10.1002/(SICI)1097-4644(19961201)63:3%3C311::AID-JCB6%3E3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 92.Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. Journal of Clinical Investigation. 1998;101:2567–2578. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Myatt L. Control of vascular resistance in the human placenta. Placenta. 1992;13:329–341. doi: 10.1016/0143-4004(92)90057-z. [DOI] [PubMed] [Google Scholar]
- 94.Myatt L, Brockman DE, Eis AL, Pollock JS. Immunohistochemical localization of nitric oxide synthase in the human placenta. Placenta. 1993;14:487–495. doi: 10.1016/s0143-4004(05)80202-4. [DOI] [PubMed] [Google Scholar]
- 95.Namba T, Koike H, Murakami K, Aoki M, Makino H, Hashiya N, Ogihara T, Kaneda Y, Kohno M, Morishita R. Angiogenesis induced by endothelial nitric oxide synthase gene through vascular endothelial growth factor expression in a rat hindlimb ischemia model. Circulation. 2003;108:2250–2257. doi: 10.1161/01.CIR.0000093190.53478.78. [DOI] [PubMed] [Google Scholar]
- 96.Noiri E, Hu Y, Bahou WF, Keese CR, Giaever I, Goligorsky MS. Permissive role of nitric oxide in endothelin-induced migration of endothelial cells. Journal of Biological Chemistry. 1997;272:1747–1752. doi: 10.1074/jbc.272.3.1747. [DOI] [PubMed] [Google Scholar]
- 97.Osol G, Mandala M. Maternal uterine vascular remodeling during pregnancy. Physiology. 2009;24:58–71. doi: 10.1152/physiol.00033.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Parenti A, Morbidelli L, Cui XL, Douglas JG, Hood JD, Granger HJ, Ledda F, Ziche M. Nitric Oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal-regulated kinase1/2 activation in postcapillary endothelium. Journal of Biological Chemistry. 1998;273:4220–4226. doi: 10.1074/jbc.273.7.4220. [DOI] [PubMed] [Google Scholar]
- 99.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiological Review. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Perrino BA, Wilson AJ, Ellison P, Clapp LH. Substrate selectivity and sensitivity to inhibition by FK506 and cyclosporin A of calcineurin heterodimers composed of the alpha or beta catalytic subunit. European Journal of Biochemistry. 2002;269:3540–3548. doi: 10.1046/j.1432-1033.2002.03040.x. [DOI] [PubMed] [Google Scholar]
- 101.Pipili-Synetos E, Sakkoula E, Maragoudakis ME. Nitric oxide is involved in the regulation of angiogenesis. British Journal of Pharmacology. 1993;108:855–857. doi: 10.1111/j.1476-5381.1993.tb13476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Powers CJSW, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocrine-Related Cancer. 2000;7:165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 103.Rafiee P, Heidemann J, Ogawa H, Johnson NA, Fisher PJ, Li MS, Otterson MF, Johnson CP, Binion DG. Cyclosporin A differentially inhibits multiple steps in VEGF induced angiogenesis in human microvascular endothelial cells through altered intracellular signaling. Cell Communication and Signaling. 2004;2:3. doi: 10.1186/1478-811X-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ravi K, Brennan LA, Levic S, Ross PA, Black SM. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proceedings of the National Academy of Sciences USA. 2004;101:2619–2624. doi: 10.1073/pnas.0300464101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Reynolds LP, Redmer DA. Utero-placental vascular development and placental function. Journal of Animal Sciences. 1995;73:1839–1851. doi: 10.2527/1995.7361839x. [DOI] [PubMed] [Google Scholar]
- 106.Reynolds LP, Borowicz PP, Vonnahme KA, Johnson ML, Grazul-Bilska AT, Redmer DA, Caton JS. Placental angiogenesis in sheep models of compromised pregnancy. Journal of Physiology (London) 2005;565:43–58. doi: 10.1113/jphysiol.2004.081745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rocha SF, Adams RH. Molecular differentiation and specialization of vascular beds. Angiogenesis. 2009;12:139–147. doi: 10.1007/s10456-009-9132-x. [DOI] [PubMed] [Google Scholar]
- 108.Rosenfeld CR, Morriss FH, Makowski El, Meschia G, Battaglia FC. Circulatory changes in the reproductive tissues of ewes during pregnancy. Gynecological Investigation. 1974;5:252–268. doi: 10.1159/000301658. [DOI] [PubMed] [Google Scholar]
- 109.Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 110.Rousseau S, Houle F, Kotanides H, Witte L, Waltenberger J, Landry J, Huot J. Vascular endothelial growth factor (VEGF)-driven actin-based motility is mediated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. Journal of Biological Chemistry. 2000;275:10661–10672. doi: 10.1074/jbc.275.14.10661. [DOI] [PubMed] [Google Scholar]
- 111.Sa G, Murugesan G, Jaye M, Ivashchenko Y, Fox PL. Activation of cytosolic phospholipase A2 by basic fibroblast growth factor via a p42 mitogen-activated protein kinase-dependent phosphorylation pathway in endothelial cells. Journal of Biological Chemistry. 1995;270:2360–2366. doi: 10.1074/jbc.270.5.2360. [DOI] [PubMed] [Google Scholar]
- 112.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 113.Sheehy AM, Burson MA, Black SM. Nitric oxide exposure inhibits endothelial NOS activity but not gene expression: a role for superoxide. American Journal of Physiology. 1998;274:L833–L841. doi: 10.1152/ajplung.1998.274.5.L833. [DOI] [PubMed] [Google Scholar]
- 114.Shi Y. Serine/Threonine Phosphatases: Mechanism through Structure. Cell 2009. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 115.Silverstein AM, Barrow CA, Davis AJ, Mumby MC. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proceedings of the National Academy of Sciences USA. 2002;99:4221–4226. doi: 10.1073/pnas.072071699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sladek SM, Magness RR, Conrad KP. Nitric oxide and pregnancy. American Journal of Physiology. 1997;272:R441–463. doi: 10.1152/ajpregu.1997.272.2.R441. [DOI] [PubMed] [Google Scholar]
- 117.Song Y, Wang K, Chen DB, Magness RR, Zheng J. Suppression of protein phosphatase 2 does not affect VEGF- and FGF2-stimulated ovine fetoplacental artery endothelial cell proliferation. Placenta. 2009;30:907–913. doi: 10.1016/j.placenta.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sontag E. Protein phosphatase 2A: the Trojan Horse of cellular signaling. Cell Signal. 2001;13:7–16. doi: 10.1016/s0898-6568(00)00123-6. [DOI] [PubMed] [Google Scholar]
- 119.Souttou B, Raulais D, Vigny M. Pleiotrophin induces angiogenesis: involvement of the phosphoinositide-3 kinase but not the nitric oxide synthase pathways. Journal of Cellular Physiology. 2001;187:59–64. doi: 10.1002/1097-4652(2001)9999:9999<00::AID-JCP1051>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 120.Sprague BJ, Phernetton TM, Magness RR, Chesler NC. The effects of the ovarian cycle and pregnancy on uterine vascular impedance and uterine artery mechanics. European Journal of Obstetrics & Gynecology and Reproductive Biology. 2009;144:S184–S191. doi: 10.1016/j.ejogrb.2009.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sprague B, Chesler NC, Magness RR. Shear stress regulation of nitric oxide production in uterine and placental artery endothelial cells: experimental studies and hemodynamic models of shear stresses on endothelial cells. International Journal of Developmental Biology. 2010;54:331–339. doi: 10.1387/ijdb.082832bs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Szukiewicz D, Szewczyk G, Watroba M, Kurowska E, Maslinski S. Isolated placental vessel response to vascular endothelial growth factor and placenta growth factor in normal and growth-restricted pregnancy. Gynecologic and Obstetric Investigation. 2005;59:102–107. doi: 10.1159/000082622. [DOI] [PubMed] [Google Scholar]
- 123.Tanaka K, Abe M, Sato Y. Roles of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase in the signal transduction of basic fibroblast growth factor in endothelial cells during angiogenesis. Japanese Journal of Cancer Research. 1999;90:647–654. doi: 10.1111/j.1349-7006.1999.tb00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tsurumi Y, Murohara T, Krasinski K, Chen D, Witzenbichler B, Kearney M, Couffinhal T, Isner JM. Reciprocal relation between VEGF and NO in the regulation of endothelial integrity. Nature Medicine. 1997;3:879–86. doi: 10.1038/nm0897-879. [DOI] [PubMed] [Google Scholar]
- 125.Tuder RM, Flook BE, Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. Journal of Clinical Investigation. 1995;95:1798–1807. doi: 10.1172/JCI117858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ugele B, Lange F. Isolation of endothelial cells from human placental microvessels: effect of different proteolytic enzymes on releasing endothelial cells from villous tissue. In Vitro Cellular & Developmental Biology Animal. 2001;37:408–413. doi: 10.1290/1071-2690(2001)037<0408:IOECFH>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 127.Urbich C, Reissner A, Chavakis E, Dernbach E, Haendeler J, Fleming I, Zeiher AM, Kaszkin M, Dimmeler S. Dephosphorylation of endothelial nitric oxide synthase contributes to the anti-angiogenic effects of endostatin. FASEB Journal. 2002;16:706–708. doi: 10.1096/fj.01-0637fje. [DOI] [PubMed] [Google Scholar]
- 128.Van Kanegan MJ, Adams DG, Wadzinski BE, Strack S. Distinct protein phosphatase 2A heterotrimers modulate growth factor signaling to extracellular signal-regulated kinases and Akt. Journal of Biological Chemistry. 2005;280:36029–36036. doi: 10.1074/jbc.M506986200. [DOI] [PubMed] [Google Scholar]
- 129.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nature Review Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 130.Vonnahme KA, Wilson ME, Li Y, Rupnow HL, Phernetton TM, Ford SP, Magness RR. Circulating levels of nitric oxide and vascular endothelial growth factor throughout ovine pregnancy. Journal of Physiology. 2005;15:565, 101–109. doi: 10.1113/jphysiol.2004.082321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang K, Song Y, Chen DB, Zheng J. Protein phosphatase 3 differentially modulates vascular endothelial growth factor and fibroblast growth factor 2-stimulated cell proliferation and signaling in ovine fetoplacental artery endothelial cells. Biology of Reproduction. 2008;79:704–710. doi: 10.1095/biolreprod.108.068957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang K, Jiang YZ, Chen DB, Zheng J. Hypoxia enhances FGF2- and VEGF-stimulated human placental artery endothelial cell proliferation: Roles of MAP2K1/2/MAPK3/1 and PI3K/AKT1 pathways. Placenta. 2009;30:1045–1051. doi: 10.1016/j.placenta.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang X, Athayde N, Trudinger B. A proinflammatory cytokine response is present in the fetal placental vasculature in placental insufficiency. American Journal of Obstetrics and Gynecology. 2003;189:1445–1451. doi: 10.1067/s0002-9378(03)00652-5. [DOI] [PubMed] [Google Scholar]
- 134.Wang Y, Zhao S. Vascular Biology of the Placenta. In: Granger DN, Granger J, editors. Integrated Systems Physiology: from Molecules to Function to Disease. San Rafael: Morgan & Claypool Life Sciences; 2010. [PubMed] [Google Scholar]
- 135.Wen HC, Huang WC, Ali A, Woodgett JR, Lin WW. Negative regulation of phosphatidylinositol 3-kinase and Akt signaling pathway by PKC. Cell Signal. 2003;15:37–45. doi: 10.1016/s0898-6568(02)00047-5. [DOI] [PubMed] [Google Scholar]
- 136.Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochemical and Biophysical Research Communications. 2004;322:1178–1191. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 137.Williams DJ, Vallance PJ, Neild GH, Spencer JA, Imms FJ. Nitric-oxide mediated vasodilation in human pregnancy. American Journal of Physiology. 1997;272:H748–H752. doi: 10.1152/ajpheart.1997.272.2.H748. [DOI] [PubMed] [Google Scholar]
- 138.Yashima R, Abe M, Tanaka K, Ueno H, Shitara K, Takenoshita S, Sato Y. Heterogeneity of the signal transduction pathways for VEGF-induced MAPKs activation in human vascular endothelial cells. Journal of Cellular Physiology. 2001;188:201–210. doi: 10.1002/jcp.1107. [DOI] [PubMed] [Google Scholar]
- 139.Young MR, Kolesiak K, Meisinger J. Protein phosphatase-2A regulates endothelial cell motility and both the phosphorylation and the stability of focal adhesion complexes. International Journal of Cancer. 2002;100:276–282. doi: 10.1002/ijc.10491. [DOI] [PubMed] [Google Scholar]
- 140.Zarlingo TJ, Eis AL, Brockman DE, Kossenjans W, Myatt L. Comparative localization of endothelial and inducible nitric oxide synthase isoforms in haemochorial and epitheliochorial placentae. Placenta. 1997;18:511–520. doi: 10.1016/0143-4004(77)90004-2. [DOI] [PubMed] [Google Scholar]
- 141.Zhang R, Wang L, Zhang L, Chen J, Zhu Z, Zhang Z, Chopp M. Nitric oxide enhances angiogenesis via the synthesis of vascular endothelial growth factor and cGMP after stroke in the rat. Circulation Research. 2003;92:308–313. doi: 10.1161/01.res.0000056757.93432.8c. [DOI] [PubMed] [Google Scholar]
- 142.Zheng J, Vagnoni KE, Bird IM, Magness RR. Expression of basic fibroblast growth factor, endothelial mitogenic activity, and angiotensin II type-1 receptors in the ovine placenta during the third trimester of pregnancy. Biology of Reproduction. 1997;56:1189–1197. doi: 10.1095/biolreprod56.5.1189. [DOI] [PubMed] [Google Scholar]
- 143.Zheng J, Bird IM, Melsaether AM, Magness RR. Activation of the mitogen-activated protein kinase cascade is necessary but not sufficient for bFGF and EGF stimulated expression of endothelial nitric oxide synthase. Endocrinology. 1999;140:1399–1407. doi: 10.1210/endo.140.3.6542. [DOI] [PubMed] [Google Scholar]
- 144.Zheng J, Yun L, Weiss AR, Bird IM, Magness RR. Expression of endothelial and inducible nitric oxide synthase and nitric oxide production in the ovine placental and uterine tissues during late pregnancy. Placenta. 2000;21:516–524. doi: 10.1053/plac.1999.0504. [DOI] [PubMed] [Google Scholar]
- 145.Zheng J, Bird IM, Chen DB, Magness RR. Angiotensin II regulation of fetoplacental artery endothelial functions. Journal of Physiology (London) 2005;565:59–69. doi: 10.1113/jphysiol.2004.082420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zheng J, Wen YX, Austin JL, Chen DB. Exogenous nitric oxide stimulates cell proliferation via activation of a mitogen activated protein kinase pathway in ovine fetoplacental artery endothelial cells. Biology of Reproduction. 2006;74:375–382. doi: 10.1095/biolreprod.105.043190. [DOI] [PubMed] [Google Scholar]
- 147.Zheng J, Wen YX, Song Y, Wang K, Chen DB, Magness RR. Activation of multiple signaling pathways is critical for fibroblast growth factor 2- and vascular endothelial growth factor-stimulated ovine fetoplacental endothelial cell proliferation. Biology of Reproduction. 2008;78:143–150. doi: 10.1095/biolreprod.107.064477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ziche M, Morbidelli L, Masini E, Granger HJ, Geppetti P, Ledda F. Nitric oxide promotes DNA synthesis and cycle GMP formation in endothelial cells from postcapillary venules. Biochemical and Biophysical Research Communications. 1993;192:1198–1203. doi: 10.1006/bbrc.1993.1543. [DOI] [PubMed] [Google Scholar]
- 149.Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, Geppetti P, Ledda F. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. Journal of Clinical Investigation. 1994;94:2036–2044. doi: 10.1172/JCI117557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ziche M, Morbidelli L, Mchoudhuri R, Zhang HT, Donnini S, Granger HJ, aggi CA, Bicknell R. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. Journal of Clinical Investigation. 1997a;99:2625–2634. doi: 10.1172/JCI119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ziche M, Parenti A, Ledda F, Dell’Era P, Granger HJ, Maggi CA, Presta M. Nitric oxide promotes proliferation and plasminogen activator production by coronary venular endothelium through endogenous bFGF. Circulation Research. 1997b;80:845–852. doi: 10.1161/01.res.80.6.845. [DOI] [PubMed] [Google Scholar]
- 152.Zubilewicz A, Hecquet C, Jeanny JC, Soubrane G, Courtois Y, Mascarelli F. Two distinct signalling pathways are involved in FGF2-stimulated proliferation of choriocapillary endothelial cells: a comparative study with VEGF. Oncogene. 2001;20:1403–1413. doi: 10.1038/sj.onc.1204231. [DOI] [PubMed] [Google Scholar]