

Abstract
Protein Ser/Thr kinase CK2 (casein kinase II) is involved in a myriad of cellular processes including cell growth and proliferation by phosphorylating hundreds of substrates, yet the regulation process of CK2 function is poorly understood. Here we report that the CK2 catalytic subunit CK2α is modified by O-GlcNAc on Ser347, proximal to a cyclin-dependent kinase phosphorylation site (Thr344) on the same protein. We use protein semisynthesis to show that Thr344 phosphorylation increases CK2α cellular stability via Pin1 interaction whereas Ser347 glycosylation appears to be antagonistic to Thr344 phosphorylation and permissive to proteasomal degradation. By performing kinase assays with the site-specifically modified phospho- and glyco-modified CK2α in combination with CK2β and Pin1 binding partners on human protein microarrays, we show that CK2 kinase substrate selectivity is modulated by these specific posttranslational modifications. This study suggests how a promiscuous protein kinase can be regulated at multiple levels to achieve particular biological outputs.
Introduction
Protein Kinase CK2 (also known as casein kinase II) is a Ser/Thr kinase implicated in cell proliferation and many disease processes1. CK2 is ubiquitously expressed and is proposed to phosphorylate hundreds, if not thousands, of distinct cellular protein substrates, but its mechanisms of regulation are poorly understood2,3. The 45 kDa catalytic CK2 polypeptide (CK2α) can exist as an active monomer in cells, but it can also phosphorylate substrates when part of a tetrameric complex containing two CK2α and two 25 kDa CK2β subunits4. The substrate specificity and catalytic activity of CK2α is reported to be modulated through its association with the CK2β subunit, but this has only been studied for a handful of substrates5–7. CK2α is modified by C-terminal phosphorylation on four sites (T344, T360, S362, S370) by Cdk1/cyclin B (Fig.1a), but the role of such phosphorylation on CK2 function is not clear8,9. Mutation of these positions to Glu, a rather crude phosphoSer/phosphoThr mimic, has not yielded an obvious change in activity.
Figure 1. Preparation of CK2α semisynthetic proteins.
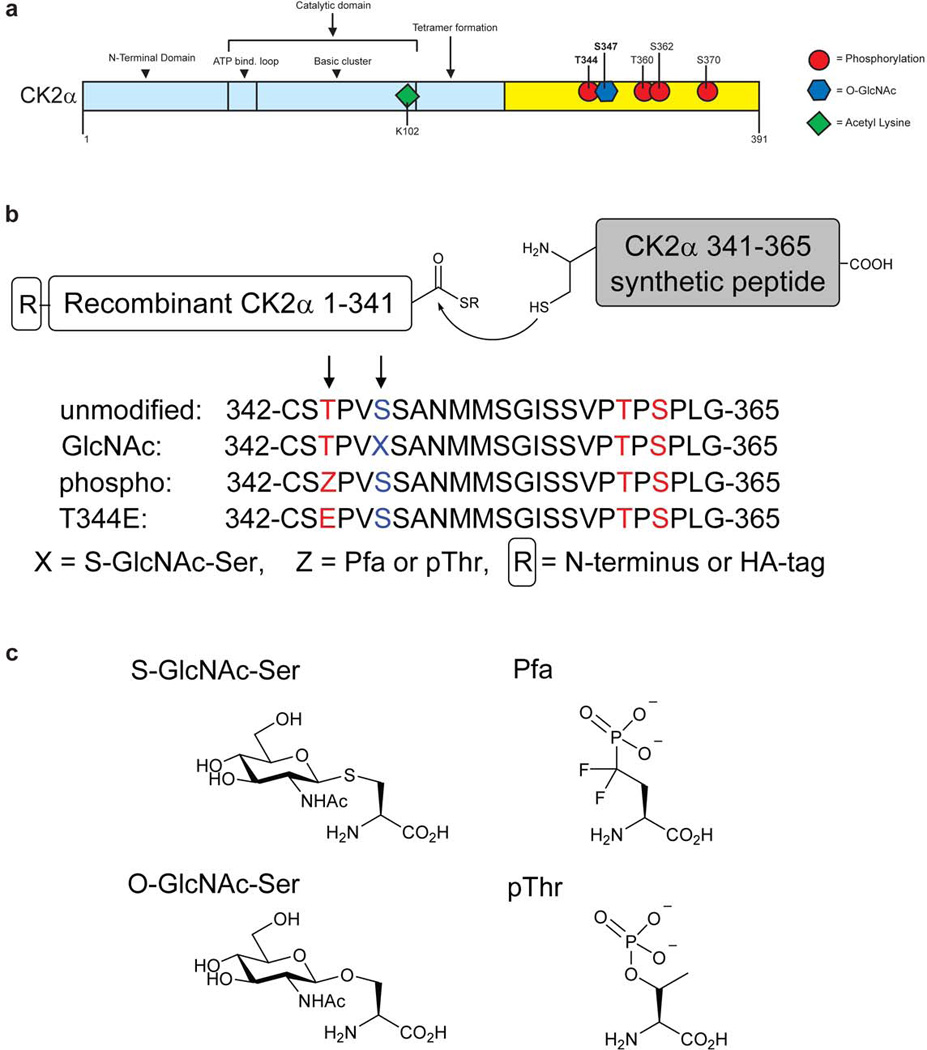
(a) The posttranslational modifications on CK2. The known posttranslational modifications on CK2α include: phosphorylation at positions Thr344, Thr360, S362, S370 and lysine acetylation at K10250. Here we demonstrate CK2α is O-GlcNAc modified at Ser347. (b) Scheme for expressed protein ligation and list of the peptides used for semisynthesis. (c) Posttranslational modifications and the cleavage-resistant mimics used.
In many instances, nuclear, cytoplasmic, and mitochondrial proteins can be modified dynamically by O-linked β-N-acetyl-glucosamine (O-GlcNAc) at or near sites of phosphorylation10,11. The addition of O-GlcNAc to protein Ser/Thr residues is receiving increasing attention in cell signaling studies as more of these sites are mapped11–14, but it has generally been difficult to elucidate the detailed functions of these modifications at specific sites. Classical mutagenesis of the specific sites modified and/or inhibition of O-GlcNAc transfer enzymes by drugs or RNAi have been standard techniques to analyze O-GlcNAcylation, but they lack the precision needed to pinpoint biochemical effects of particular PTMs (posttranslational modifications). In fact, the same challenges pertain to sorting out phosphorylation-site specific effects.
As discussed below, we reveal here that CK2α is O-GlcNAc-modified on Ser347, near the Cdk1/cyclin B-mediated Thr344 phosphorylation site. Protein semisynthesis15 is used in this study to install metabolically stable O-GlcNAc equivalent, S-GlcNAc-, and phosphonate, Pfa, mimics site-specifically into CK2α. Furthermore, the effects of these modifications on kinase activity, substrate selectivity, as well as cellular stability have been analyzed. We show that phosphorylation at Thr344 appears to stabilize CK2α by enhancing Pin1 interaction. In contrast, O-GlcNAcylation at Ser347 inhibits Thr344 phosphorylation and reciprocal CK2α phosphorylation and O-GlcNAcylation modulate protein kinase substrate selectivity.
Results
CK2α is O-GlcNAc modified at Ser347
Although it has been shown that O-GlcNAc transferase (OGT) can modify CK2 in vitro11,14, it has not been previously reported that CK2α is O-GlcNAc modified in cells. We purified CK2α from bovine brain, fractionated it on a WGA column, and tested fractions for CK2 activity (Supplementary Results, Supplementary Fig. 1a). We found that fractions that bound to the column and eluted with 0.5M GlcNAc contained the highest CK2 activity, suggesting that a majority of the active CK2 protein is modified by terminal GlcNAc or sialic acid residues. We used reaction with UDP-[3H]galactose and galactosyltransferase to probe for terminal GlcNAc residues and demonstrated that CK2α but not CK2β, contains terminal GlcNAc residues (Supplementary Fig. 1b). The radiolabel was lost when we subjected the samples to alkali-induced β-elimination, consistent with an O-glycosidic bond to Ser/Thr. We found that sizing of the released saccharide by chromatography matched standard Galβ1,4GlcNAcitol disaccharide, which is the expected product for a single GlcNAc residue labeled by galactosyltransferase and galactose. Using cyanogen bromide cleavage followed by HPLC purification of peptides and Edman sequencing, we identified Ser347 as the CK2α O-GlcNAc modification site, a position that is close to one of the phosphorylation sites (Thr344) in the C-terminal tail of CK2α (Supplementary Fig. 1c,d).
CK2α semisynthetic protein preparation
To investigate the potential functions of Thr344 phosphorylation and Ser347 O-GlcNAcylation, we generated modified and unmodified CK2α semisynthetic proteins using expressed protein ligation16–18. For this strategy, we expressed in E. coli an N-terminal recombinant CK2α protein fragment (residues 1–341, with or without an N-terminal hemagglutinin (HA) epitope tag) bearing a C-terminal thioester using an in-frame intein. In addition, we synthesized N-Cys containing peptides (aa 342–365) carrying substitutions of interest at positions 344 (Thr, pThr, or phosphonodifluoromethylene alanine (Pfa)) or 347 (SGlcNAc-Ser or Ser) using solid phase peptide synthesis (Fig. 1b,c and Supplementary Fig. 2)16–19. Ligation of the recombinant protein and synthetic peptide fragments proceeded smoothly, and we obtained the desired semisynthetic CK2α proteins in milligram quantities in >95% purity judged by Coomassie-stained SDS-PAGE and MALDI-MS (Supplementary Figs. 3,4). In control experiments, we showed that the necessary introduction of a Cys at position 342 (in place of Gly) and a modest (26 residue) C-terminal truncation of the CK2α relative to the full-length wild-type protein were well tolerated and did not alter the kinase activity with synthetic peptide substrate. Specific kinase activities for semisynthetic unmodified CK2α agreed within 20% of those for the recombinant full-length wild-type and G342C CK2α proteins (see Supplementary Table 1). There were some experiments where either the naturally occurring pThr or Pfa containing proteins were used, but in experiments where both proteins were used, it was demonstrated that Pfa effectively mimics the pThr residue.
Determination of kinetic parameters of CK2α proteins
We analyzed the kinase activities of the Pfa344, S-GlcNAc-Ser347, and unmodified semisynthetic CK2α proteins using three different peptide substrates: an optimized peptide20, a peptide based on casein21, and a peptide based on the XRCC1 protein22. We determined the apparent kcat and Km values for each of the semisynthetic CK2αs with each peptide substrate and ATP. Turnover numbers for CK2 activity with the optimized substrate were within 2-fold of those previously described23,24. As shown in Table 1, there were little or no (<2-fold) changes in the catalytic parameters among the semisynthetic CK2αs bearing different modifications. The role of CK2β on CK2α activity has been investigated previously6,7,25,26, and we confirm here that it can modulate peptide phosphorylation efficiency (Table 1). For the optimized and casein peptide substrates, there was an increase in Km values in the presence of the CK2β subunit; however, this increase in Km was not impacted by 344 or 347 PTM. These data suggest that the 344 and 347 PTMs in CK2α did not have a significant effect in modulating CK2 catalytic efficiencies (kcat/Kms) with peptide substrates regardless of the presence of CK2β.
Table 1. Kinetic Parameters for CK2 semisynthetic enzymes.
| Substrate | Enzyme | Apparent kcat (min−1) |
Apparent Km (µM) |
kcat / Km (min−1 µM−1) |
|---|---|---|---|---|
| Optimized Peptide | unmodified α | 260 ± 10 | 37 ± 7 | 7.1 |
| Pfa α | 260 ± 10 | 33 ± 5 | 7.8 | |
| S-GlcNAc α | 190 ± 10 | 33 ± 5 | 5.7 | |
| unmodified α+β | 180 ± 20 | 170 ± 50 | 1.0 | |
| Pfa α+β | 200 ± 20 | 160 ± 50 | 1.2 | |
| S-GlcNAc α+β | 160 ± 10 | 160 ± 50 | 0.90 | |
| Casein Peptide | unmodified α | 22 ± 1 | 400 ± 50 | 0.054 |
| Pfa α | 22 ± 1 | 340 ± 60 | 0.065 | |
| S-GlcNAc α | 15 ± 1 | 340 ± 30 | 0.044 | |
| unmodified α+β | NS | >1600 | 0.014 | |
| Pfa α+β | NS | >1600 | 0.015 | |
| S-GlcNAc α+β | NS | >1600 | 0.013 | |
| XRCC1 Peptide | unmodified α | 180 ± 10 | 250 ± 10 | 0.74 |
| Pfa α | 240 ± 30 | 350 ± 90 | 0.69 | |
| S-GlcNAc α | 190 ± 10 | 250 ± 30 | 0.75 | |
| unmodified α+β | 75 ± 4 | 260 ± 40 | 0.29 | |
| Pfa α+β | 130 ± 20 | 460 ± 120 | 0.28 | |
| S-GlcNAc α+β | 100 ± 10 | 440 ± 120 | 0.24 | |
| ATP | unmodified α | 320 ± 10a | 20 ± 2 a | 16 |
| Pfa α | 370 ± 10 a | 21 ± 2 a | 18 | |
| S-GlcNAc α | 270 ± 10 a | 16 ± 2 a | 17 | |
| unmodified α+β | 250 ±20b | 39 ± 6b | 6.3 | |
| Pfa α+β | 250 ± 20b | 30 ± 4b | 8.3 | |
| S-GlcNAc α+β | 190 ± 10b | 31 ± 4b | 6.2 | |
± values indicate standard error obtained from non-linear curve-fitting parameters. NS = non-saturated with peptide substrate, Km value was not determined. Kmapp peptide and kcat values were obtained with 100 µM ATP.
Kmapp ATP and kcat values were obtained with 360 µM optimized peptide.
Kmapp ATP and kcat values were obtained with 800 µM optimized peptide.
Localization and stability of semisynthetic CK2αs
Prior experiments suggest that mitotic and phosphorylated CK2α might be driven to the cell nucleus27. To examine the potential effect of 344-phosphorylation and 347-glycosylation of CK2α on cellular localization, we microinjected N-terminally HA-tagged semisynthetic CK2α proteins containing Pfa344, S-GlcNAc-Ser347, and no modifications into the cytoplasm of rat embryonic fibroblasts (REF52 cells) and performed immunocytochemistry using an anti-HA Ab (Supplementary Figs. 5,6). It should be noted that the Pfa18 and S-GlcNAc19 phospho- and glyco-mimics are resistant to enzymatic cleavage28 which makes them useful for cellular studies. Unexpectedly, there did not appear to be any significant differences in cellular localization (nuclear vs. cytoplasmic) of CK2α due to the different modifications on its C-terminal tail (Supplementary Fig. 5). At 2 h following microinjection in the cytoplasm, much of the CK2α had translocated into the nucleus although there was residual cytoplasmic protein (Supplementary Fig. 5). Interestingly, when CK2β was co-injected with the semisynthetic CK2αs, we found that the CK2α subunit was apparently retarded in moving into the cell nucleus (Supplementary Fig. 5). However, the Pfa- and S-GlcNAc-containing CK2αs behaved the same as unmodified CK2α in this regard (Supplementary Fig. 5). Taken together, our data suggest that phosphorylation at Thr344 site or O-GlcNAcylation at Ser347 does not have a major impact on cellular localization regardless of the presence of the CK2β subunit.
We also carried out microinjection studies to examine the cellular stability of CK2α in response to the different modifications on its tail (Fig. 2a–c and Supplementary Fig. 6). Unexpectedly, the Pfa344-CK2α was significantly more stable than the S-GlcNAc-Ser347 or unmodified CK2αs, most clearly evident 4 and 8 h after microinjection (Fig. 2a and Supplementary Fig. 6). We also observed this enhanced stabilization of CK2α mediated by the phospho-mimetic after co-injection with stoichiometric CK2β, although CK2β slowed the breakdown of S-GlcNAc-Ser347 and unmodified CK2α (Fig. 2b). The idea that degradation of unmodified CK2α is mediated by the proteasome was bolstered by our finding that proteasome inhibitor MG132 led to stabilization of the microinjected kinase (Fig. 2c).
Figure 2. Cellular stability of CK2α.

(a) Relative CK2α protein levels over time following cytoplasmic microinjection of REF52 cells with the HA-tagged semisynthetic proteins bearing different modifications using immunocytochemistry with anti-HA Ab. Data represent mean values ± s.d. (b) Relative CK2α protein levels following co-injection of β subunit with CK2α proteins. Data represent mean values ± s.d. (c) Relative CK2α protein levels following co-injection of proteasome inhibitor (1 µM MG132) with unmodified CK2α protein. Data represent mean values ± s.d. (d) Western blot for total CK2α protein in HeLa cells following treatment with Cdk inhibitors. Numbers indicate relative quantification normalized for loading control, mean values (n = 3) shown ± s.d. (e) Western blot for total CK2α protein in HeLa cells following treatment with nocodazole to arrest cells in mitosis. See Supplementary Fig. 8 for full gels corresponding to those in panels d, e.
CK2α cellular stabilization, Cdks, and Pin1 interaction
As shown above, the Pfa phospho-mimic at Thr344 of CK2α protects the kinase from degradation in REF52 cells. To further examine how phosphorylation of CK2α enhances its cellular stability, we analyzed endogenous CK2α protein levels in HeLa cells using Western blots following pharmacologic manipulation predicted to perturb the phosphorylation state of CK2α (Fig. 2d and 2e). Since Cdk1/cyclin B has been reported to be responsible for cellular phosphorylation of CK2α8, we investigated the effects of two small molecule Cdk kinase inhibitors flavopiridol29 and 9-cyanopaullone30 (Fig. 2d and Supplementary Fig. 7). We treated HeLa cells with either flavopiridol or 9-cyanopaullone vs. vehicle (control DMSO) which caused cell cycle changes (Supplementary Fig. 7) consistent with their known broad spectrum Cdk inhibition31,32 and led to considerable decrease in total CK2α protein levels (Fig. 2e). Prior studies report that the mitotic spindle poison nocodazole can induce a state where CK2α is highly phosphorylated8,33. Here we show that HeLa cells treated with nocodazole, which caused G2/M phase arrest (Supplementary Fig. 9), had higher levels of both phosphorylated and total CK2α compared with vehicle-treated cells (Fig. 2e). Taken together, these pharmacologic and microinjection experiments support the concept that phosphorylation by Cdks can stabilize CK2α in the cell.
It has been shown that phosphorylation of CK2α can confer interaction with the phosphoSer/Thr adaptor protein Pin134, a peptidyl-prolyl cis/trans isomerase (PPIase) active on only phospho-Ser/Thr-Pro motifs35. We thus performed pull-down assays with HA-tagged semisynthetic CK2αs, which demonstrated that only pThr344 CK2α and Pfa344 could efficiently bind Pin1, whereas Thr344, Glu344 (a crude phospho-mimic), and S-GlcNAc-Ser347 showed minimal evidence of Pin1 interaction (Fig. 3a). The concentrations of Pin1 used in our pull-down assays were selected based on estimated Pin1 concentrations in the cell36. Co-immunoprecipitation of endogenous Pin1 from REF52 cell lysates with Pfa344 CK2α but not unmodified CK2α provides further evidence for the phosphorylation dependence of this interaction (Fig. 3b). To determine whether this pThr-dependent interaction with Pin1 plays a role in CK2α cellular stability, we evaluated for the impact of polyclonal anti-Pin1 antibody IgY, (which competed for pThr-CK2 binding to Pin1 in vitro, Fig. 3c), on HA-tagged Pfa344 CK2α level after cytoplasmic microinjection in REF52 cells. Indeed, co-injection of anti-Pin1 IgY with Pfa344 CK2α into REF52 cells resulted in degradation of CK2α relative to cells co-injected with Pfa344 CK2α and vehicle or control non-specific IgY Ab (Fig. 3d and Supplementary Fig. 12). These results suggest that disruption of the Pin1/phospho-CK2α interaction was sufficient to eliminate the enhanced cellular stability of CK2α imparted by the presence of Pfa at Thr344.
Figure 3. CK2 interactions with Pin1.

(a) Pull-down experiments with semisynthetic HA-CK2α protein and recombinant Pin1. CK2α was immobilized using anti-HA antibodies bound to protein G beads. Pin1 was incubated with immobilized CK2α for 20 min at 4°C. (b) Co-immunoprecipitation of endogenous Pin1 with CK2α. Pfa containing and unmodified HA-CK2α semisynthetic proteins were spiked into REF52 cell lysates and immobilized using anti-HA antibodies. Input samples are shown for loading control. (c) Pull-down experiments with Pfa344 HA-CK2α protein and recombinant Pin1 in the presence of anti-Pin1 or non-specific IgY. (d) Relative CK2α protein levels over time following microinjection of Pfa344 CK2α with anti-Pin1 IgY or non-specific IgY (control). Data represent mean values ± s.d. See Supplementary Figs 10, 11 for full gels corresponding to those in panels a–c.
O-GlcNAc influences phosphorylation and stability of CK2α
The CK2α O-GlcNAc modification (Ser347) lies proximal to its Thr344 phosphorylation site. We thus examined whether the presence of S-GlcNAc at Ser347 can influence the extent to which Thr344 can be phosphorylated by recombinant Cdk1/cyclin B in the context of a synthetic peptide substrate derived from the CK2α C-terminal tail (residues 337–352). Using a 32P-ATP kinase assay, we showed that the peptide containing S-GlcNAc at Ser347 was phosphorylated by Cdk1/cyclin B very weakly compared with the unmodified peptide substrate (Fig. 4a and Supplementary Fig. 13), indicating that the S-GlcNAc group interfered with enzyme-substrate binding, catalysis, or both. To test whether GlcNAc modification could influence phosphorylation-induced stabilization of endogenous CK2α in cells, we examined the pharmacologic influence of selective O-GlcNAcase enzyme inhibitor ThiametG (TMG)37 in cell culture. First, we showed that addition of TMG (20 µM) for 12 h could enhance the level of O-GlcNAc modification on CK2α from HeLa cells as judged by immunoprecipitation-western blot analysis (Fig. 4b). However, 12–16 h after TMG exposure, there was a modest but reproducible (n=4) reduction in overall CK2α protein amounts as judged by western blot (Fig. 4c). We interpret the results of these experiments as evidence that enhancing CK2α O-GlcNAcylation reduced Cdk-mediated phosphorylation and may have subsequently conferred reduced cellular stability of CK2α.
Figure 4. Role of O-GlcNAc modification in CK2α.

(a) Phosphorylation of C-terminal tail CK2α peptides (residues 337 to 352) by Cdk1/cyclin B. In vitro Cdk1/cyclin B kinase assays were performed using the following peptide substrates: SSMPGGSTPVXSANMMK(εBiotin) where X = serine or S-GlcNAc-serine. After reactions were quenched, peptides were separated from reaction mixture on Tris-tricine gels. Data represent mean values ± s.d. (b) IP-Western blot for O-GlcNAc CK2α in HeLa cells following treatment with O-GlcNAcase inhibitor, TMG, for 12 h. Numbers indicate relative quantification normalized for total CK2α levels. (c) Western blot for total CK2α protein in HeLa cells following treatment with TMG for 12 or 16 h. Numbers indicate relative quantification normalized for loading control, mean values (n = 4) shown ± s.d. See Supplementary Figs. 14, 15 for full gels corresponding to those in panel b,c.
CK2 kinase activity with protein substrates
While the work described above establishes that 344 phosphorylation and 347 O-GlcNAcylation of CK2α do not influence CK2’s activity in simple peptide substrate phosphorylation, we considered the possibility that protein substrate selectivity might be more sensitive to CK2α PTMs. Initial support for this idea came from our observation that calmodulin, known to be more efficiently phosphorylated by CK2α monomer versus CK2α:CK2β tetramer5, showed modestly greater inhibition of phosphorylation by pThr344-CK2α relative to S-GlcNAc-Ser347-CK2α and unmodified CK2α in the presence of sub-saturating CK2β (Supplementary Fig. 16).
To more comprehensively determine the interplay between CK2α PTMs and CK2β effects on catalytic activity, we performed kinase assays on a home-made microarray of 17,000 human proteins fabricated using the same method as described previously38. Using the standard deviation (SD) from the normal distribution of the signal intensities of all of the proteins, we used a stringent cutoff of three SDs to identify positive hits. The initial set of CK2 kinase assays on the microarray identified approximately 200 substrates that were phosphorylated on duplicate microarrays by at least one of the CK2 enzyme forms tested (unmodified CK2α ±β, S-GlcNAc CK2α ±β, Pfa CK2α ±β) (Supplementary Fig. 17). Proteins identified as “positive hits” in both duplicate experiments were qualified as a “substrate” for a given experiment. The results of these kinase assays on the microarray were computationally validated by statistical analysis that demonstrated enrichment for known CK2 substrates (see Supplementary Tables 2 and 3)3,39.
Of the substrates identified in the microarrays, 23 proteins could be classified as substrates that were selectively phosphorylated by specific PTM forms of CK2. These are listed in Table 2. To be included in Table 2, the “positive hit” could not have shown to be positive in either of the duplicate experiments for the other CK2 enzyme forms. For example, AHCYL2 was phosphorylated in duplicate on both microarrays treated with S-GlcNAc CK2α+β, but not on any of the microarrays treated with the other CK2 enzyme combinations. There were also examples of substrates whose phosphorylation by CK2 was inhibited by the presence of a PTM. PCDL2 was phosphorylated in duplicate on both microarrays treated with unmodified CK2α as well as both microarrays treated with Pfa344 CK2α, but not phosphorylated on either microarray treated with S-GlcNAc-Ser347 CK2α. To confirm the findings with microarrays, we selected several of these substrates, expressed and purified them on a larger scale (Supplementary Fig. 18), and tested them in solution phase kinase assays. Consistent with the protein microarray results, the S-adenosylhomocysteine hydrolase homolog AHCYL2 was most efficiently phosphorylated by S-GlcNAc-Ser347-CK2α in the presence of CK2β compared to all five other combinations (Fig. 5a). In contrast, the histone chaperone NAP1L3 was most effectively phosphorylated by pThr344 (and Pfa344) CK2α in the presence of CK2β relative to the other five possibilities, as observed in the microarray experiments (Fig. 5b). Interestingly, the T344E mutant could not substitute adequately for the pThr344 or Pfa344 protein, indicating a role for the authentic PTM or its close phosphonate mimic (Pfa) for achieving NAP1L3 substrate specificity (Supplementary Fig. 20). Other recombinant protein solution phase kinase assays that recapitulated protein microarray results included eEF-2 kinase and WASL (best phosphorylated by O-GlcNAc- and unmodified CK2α in the presence of CK2β), and UBE2O and PCDL2 (best phosphorylated by phospho- and unmodified CK2α in the presence of CK2β) (Supplementary Figs. 21, 22). Taken together, these results demonstrate that single phosphorylation and O-GlcNAc modifications at sites remote from the active site of a kinase can significantly influence protein substrate selectivity without necessarily modulating kinase activity on short peptides.
Table 2. Protein microarray results for substrate selectivity from CK2α PTM’s.
Gene names listed with common protein name in parentheses. See Supplementary Methods for criteria for positive hits.
| Form of CK2 | α only | α + β |
|---|---|---|
| S-GlcNAc | DAPK2 (death-associated protein kinase 2) | AHCYL2 (S-adenosylhomocysteine hydrolase) |
| Phospho (Pfa) | CPNE1 (copine I isoform a), PGRMC1(progesterone receptor membrane component 1), GOLGA4 (GOLGA4 protein) |
NAP1L3 (nucleosome assembly protein1-like 3 or MB20 protein), COL4A3BP (collagen binding protein isoform 2) |
| Unmodified | MAPK9 (stress activated kinase JNK2), VPS25 (Vacuolar sorting protein 25) |
CLGN (calmegin precursor), MLPH (melanophilin), RADP51AP1 (RAD51 associated protein 1) |
| Both S-GlcNAc & phospho (Pfa) | None | APLP2 (APLP2 protein) |
| Both S-GlcNAc & unmodified | None | EEF2K (eEF-2 kinase), WASL (Wiskitt-Aldrich syndrome-like protein) SLC7A6OS (Solute carrier protein family7 member 6) |
| Both Phospho (Pfa) & unmodified | PDCD2L (programmed cell death protein 2-like), EAF2 (ELL associated factor 2), DNAJB2 (DnaJ (Hsp40) homolog B2), PFKM (Phosphofructokinase from muscle) |
UBE2O (ubiquitin-conjugating enzyme E2O), HNRNPC (hnRNP C 1/2), MTF2 (Metal-response element-binding transcription factor 2) |
Figure 5. Substrate selectivity for CK2.

Phosphorimage analysis of in vitro CK2 kinase assays with protein substrates. All reactions were performed at 30 °C. Numbers indicate nM product for each kinase reaction. (a) AHCYL2 substrate (25 ng/µL) with 100 µM ATP and 10 nM CK2α ± 11 nM CK2β for 6 min. (b) NAP1L3 substrate (15 ng/µL) with 100 µM ATP and 10 nM CK2α ± 11 nM CK2β for 6 min. (c) NIPBL substrate (50 ng/µL) with 100 µM ATP and 5 nM CK2α ± 11 nM Pin1 for 5 min. See Supplementary Figs. 19, 25 for full gels corresponding to those in panel a–c.
We performed related protein microarray kinase assays to determine if Pin1 modulates phospho-CK2α activity toward protein substrates. We found no significant differences in kcat or Km values for Pfa344-CK2α with a synthetic peptide substrate in the presence versus the absence of Pin1 (Supplementary Fig. 23), suggesting that this protein-protein interaction does not have a general activity effect on kinase action. However, there are subsets of substrate proteins whose phosphorylation was positively or negatively influenced by Pin1-pThr344-CK2α interaction (Supplementary Table 4, Supplementary Fig. 24). In a solution phase assay with recombinant NIPBL protein substrate, we confirmed that Pin1 dramatically inhibits NIPBL phosphorylation by pThr344-CK2α but has no effect on unmodified CK2α phosphorylation of NIPBL (Fig. 5c and Supplementary Figs. 18b, 24). These results further highlight the combinatorial matrix of PTM-induced effects on CK2α patterns of protein phosphorylation in the proteome.
Discussion
The finding that protein kinase CK2α was posttranslationally modified by O-GlcNAc on a site near a known Cdk-phosphorylation site led us to explore how these modifications may impact CK2 regulation. Expressed protein ligation is a strategy that was well suited for this task, since the modification sites are near the C-terminus of the recombinant protein. We provide evidence that Thr344 phosphorylation and Ser347 O-GlcNAcylation of CK2α transmit reciprocal information important for cell signaling networks. Thr344 phosphorylation enhances the stability of CK2α by Pin1 interaction and can also shape the pattern of protein substrates targeted by CK2. In contrast, it appears that Ser347 O-GlcNAcylation of CK2α may limit its cellular stability by altering Thr344 phosphorylation, and this sugar residue can further induce a distinct pattern of protein substrates phosphorylated by the kinase. Even though we did not see differences in stability between the unmodified and S-GlcNAc-Ser347 CK2α proteins in the microinjection experiments at the time points analyzed, we did observe a modest but reproducible reduction in endogenous CK2α levels when treating with TMG for a longer period. These results may differ since it is presumed that only a minute fraction of the unmodified microinjected protein is phosphorylated in cells during the course of the experiment.
Phosphorylation-dependent interactions with Pin1 have been shown to be stabilizing for other proteins including Nanog40 and p27Kip141. How Pin1 binding can stabilize CK2 from proteasome-mediated destruction is not yet settled, although one possible mechanism is that the Pin1 complex prevents ubiquitination by sequestration from a requisite E3 ligase42. A consequence of our findings is that CK2α levels can spike in cells during the period of mitosis, which might lead to a special set of phosphorylation events. In addition, the function of the different patterns of substrate phosphorylation conferred by Cdk-mediated modification of CK2α and Pin1 interaction might have important roles in mitotic regulation.
The ability of pThr344 and S-GlcNAc347 to influence changes in phosphorylation by CK2 via direct contacts with the purified protein substrates summarized in Table 2 is a noteworthy finding. PTM regulation of the function of kinases is typically associated with general activation or inhibition of a kinase, or to serve as a specific recruiting site for phosphoprotein binding adaptor molecules like SH2 domain or 14-3-3 domain containing proteins. Such specific PTM binding domains are not readily apparent in the CK2 substrates in Table 2, so the mechanism for specificity will be an interesting structural biology challenge for future studies. The implications of the particular CK2 substrate interactions that we identified in Table 2 and Supplementary Table 4 suggest a wide range of biologically processes being affected. For example, CK2 catalyzed phosphorylation of eEF2 kinase and WASL (N-WASP), inhibited by CK2 phosphorylation on Thr344, could influence translation43 and cytoskeletal dynamics44,45, respectively. In contrast, pThr344-CK2 is more active as a kinase toward histone chaperone NAP1L3, which could modulate chromatin dynamics46. The reversible kinetics of CK2 PTMs in vivo, not captured here using fixed forms of CK2s in the microarray studies, could indeed create a mosaic of complex cellular effects on the proteome.
The marrying of expressed protein ligation to generate site-specific and stoichiometrically modified forms of a protein kinase with protein microarray phosphorylation assays, performed here for the first time, can help illuminate distinct connections in signaling cascades that would be very difficult to discern using other biochemical approaches. While mass-spectrometric based phosphoproteomics47 using inhibitors or chemical complementation48 in cellular assays is powerful, it cannot readily provide insights into the substrate-altering behavior of specific PTMs on a kinase. As shown in our study, the common mutagenesis phospho mimic approach, Thr to Glu, fails to replicate the properties of pThr344 in CK2α in Pin1 interaction or in selective phosphorylation of NAP1L3. Moreover, there is no natural amino acid residue that can even be considered a plausible mimic of an O-GlcNAc moiety.
In summary, the complex regulation of CK2 by competing phosphorylation and O-GlcNAcylation provides a new framework for understanding this key signaling enzyme. This study also provides a comprehensive approach to investigating how protein PTMs can mediate multiple actions in a cellular network.
Methods
As described in more detail below and in Supplementary Methods, we used the phospho-mimic Pfa containing protein in all cell microinjection studies. In the Pin1 pull-down assays, we used both the Pfa and pThr containing proteins to demonstrate the ability of Pfa to mimic pThr. To examine and validate protein substrate selectivity influenced by phosphorylation at Thr344, we initially used Pfa, but subsequently validated the results using the natural pThr residue.
Microinjection and immunocytochemistry
For individual microinjections (performed at rt), semisynthetic HA-CK2α proteins, FLAG-CK2β, and other samples were diluted in microinjection buffer and injected into REF52 cells. Following microinjection, cells were incubated (37°C in 5% CO2 for 1–8 h) or fixed immediately (t=0 h). At least 85 cells were injected for each sample and condition in at least three repeated independent experiments. For the experiments, final sample concentrations in the microinjection capillary were as follows: 0.25 µg/µL CK2α; 0.25 µg/µL CK2α + 0.138 µg/µL CK2β (molar equivalent); 0.25 µg/µL CK2α + 1 µM MG132 (LC Biolabs); or 0.25 µg/µL CK2α + 2 µg/µL IgY. Cells were fixed and permeabilized prior to immunostaining with anti-HA mouse antibody (Covance HA.11 Clone 16B12, 1:4000) and DyLight488-conjugated goat anti-mouse IgG (KPL, 1:950). Imaging was performed using an Olympus BX61 upright fluorescence microscope and FITC filter cube. By using SlideBook5 software, fluorescence intensities of all positive cells were quantified, and the sum was multiplied by the number of positive cells divided by the number of injected cells, giving a relative fluorescence intensity value as the indication of the stability of the microinjected proteins inside the cells. Data are presented as the mean ± SD of at least three separate experiments. These values were then normalized to the sample’s t = 0 time point.
Western blots and IPs
For Cdk inhibitor studies, HeLa cells were treated with 0.1% DMSO (control), 2 µM flavopiridol (SantaCruz), or 3 µM 9-cyanopaullone (SantaCruz) for 18 h and membranes were probed with 1:1000 anti-CK2α (Cell Signaling). For nocodazole studies, HeLa cells were treated with 0.1% DMSO (control) or 100 ng/mL nocodazole (Sigma) for 24 h and membranes were probed with 1:800 anti-CK2α (SC-9030 SantaCruz). For O-GlcNAcase inhibitor studies, HeLa cells were treated with 20 µM TMG (synthesized by custom order by SD ChemMolecules) or 50 µM Tris (control) for 12 h or 16 h and membranes were probed with 1:1000 anti-CK2α (Cell Signaling). For IP-Western, HeLa cells were treated with 20 µM TMG or 50 µM Tris (control) for 12 h. CK2α was immunoprecipitated with 2 µg mouse monoclonal anti-CK2α (Abcam 8E5) and membranes were probed with 1:3000 anti-O-GlcNAc mouse IgM (antigen purified CTD110.6 as previously described49) or 1:1000 rabbit anti-CK2α (Cell Signaling).
Pull-downs for Pin1
Pin1 was expressed and purified from E. coli as previously described. Pull-down assays were performed with HA-CK2α proteins (700 nM final) immobilized on Protein G Dynabeads (Invitrogen) using anti-HA mouse monoclonal antibody (Covance). Beads were incubated with anti-CK2 antibody for 15 min at rt and then with various CK2α proteins for 40 min at 4 °C before being washed and incubated with recombinant human Pin1 (0.05 µM) at 4 °C for 20 min. To demonstrate that the presence of anti-Pin1 IgY, but not non-specific IgY, can block the binding interactions between pCK2α and Pin1, pull-down assays were performed after recombinant Pin1 (0.1 µM) was allowed to pre-incubate with IgY (0.03 µg/µL) for 10 min at rt.
For co-immunoprecipitation of Pin1, cell lysates were prepared from REF52 cells using Cell Lytic-M (Sigma) reagent diluted in PBS plus protease inhibitors and spiked with either 2.3 µM Pfa344 CK2α or 2.3 µM unmodified CK2α. Lysates were allowed to incubate with semisynthetic HA-CK2α proteins for 30 min at 4 °C prior to the addition of protein G beads bound to anti-HA antibody (for 2 h at 4°C).
Kinase assays on chips
Kinase assays were performed on human protein microarrays provided by the Protein Microarray Core at the High Throughput Biology Center at Johns Hopkins. Each kinase group was run in duplicate. For the first set of kinase assays on the chips, we examined differences in substrate selectivity for the following kinases: unmodified CK2α alone, unmodified CK2α+β, S-GlcNAc-CK2α alone, S-GlcNAc-CK2α+β, Pfa-CK2α alone, Pfa-CK2α+β. Kinase reaction conditions for this set of assays were 50 mM Hepes pH 7.5, 80 mM NaCl, 10 mM MgCl2, 1 mM DTT, 0.1% NP-40, 100 µM ATP, 6 µCi [33P]γ-ATP, with 20 nM CK2α ± 24 nM CK2β (Supplementary Fig. 17). For the second set of kinase assays on the chips, we examined differences in substrate selectivity for the following kinases: unmodified CK2α alone, unmodified CK2α + Pin1, pThr-CK2α alone, pThr-CK2α + Pin1. Kinase reaction conditions for this set of assays were the same as above with the addition of 1 mM MnCl2 and with 50 nM CK2α ± 100 nM Pin1 (Supplementary Fig. 24). Since this set of kinase assays was performed subsequent to the first set reported, we added MnCl2 to enhance the signal of the controls used to align the protein chip results. Chips were exposed to film for 4 weeks. The signal intensity of each spot on the microarray chips was quantified and since the distribution of signal intensities of all of the proteins followed a normal distribution, standard deviation of the signal intensity for each protein was used to qualify a protein as a hit or not (see Supplementary Dataset 1 and Supplementary Results for description of file). We used a stringent cutoff of three standard deviation (SD) values and only proteins identified as “positive hits” in both duplicate experiments were qualified as a “substrate” for a given experiment. To be included in Table 2 rows 1–3, the “positive hit” must have been unique for a CK2 enzyme form, that is it could not be positive in either of the duplicate experiments for any of the other CK2 enzyme forms. To be included in Table 2 rows 4–6, the “positive hit” was positive for two CK2 enzyme forms and it could not have been positive in either duplicate experiments for the alternative CK2 enzyme form.
Supplementary Material
Acknowledgments
We thank D. Schwarzer, L. Szewczuk, S. Taverna, and Y. Zhang as well as the JHU SOM Microscope Facility for advice and assistance and the National Institutes of Health (CA42486, GM62437, RR020839) for support.
Footnotes
Accession codes. Microarray data was deposited in the GEO database under GSE33151.
Author Contributions:
P.A.C., M.K.T., G.W.H., H.Z., Y.I., C.G., Y.J., and J.Q. conceived of the research and planned the experiments. M.K.T., H.R., Z. X., C.G., Y.J., N.Z., G.Y., and T.M. performed the experiments. All authors contributed to data analysis and interpretation. J.C., F.E., J.J., and S.B. prepared key reagents. M.K.T. and P.A.C. wrote the manuscript with the support of all of the authors.
Competing financial interest statement: G.W.H. receives royalties for the sale of anti-GlcNAc Ab.
References
- 1.Pagano MA, Cesaro L, Meggio F, Pinna LA. Protein kinase CK2: a newcomer in the 'druggable kinome'. Biochem Soc Trans. 2006;34:1303–1306. doi: 10.1042/BST0341303. [DOI] [PubMed] [Google Scholar]
- 2.Pinna LA. Protein kinase CK2: a challenge to canons. J Cell Sci. 2002;115:3873–3878. doi: 10.1242/jcs.00074. [DOI] [PubMed] [Google Scholar]
- 3.Salvi M, Sarno S, Cesaro L, Nakamura H, Pinna LA. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim Biophys Acta. 2009;1793:847–859. doi: 10.1016/j.bbamcr.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 4.Niefind K, Guerra B, Ermakowa I, Issinger OG. Crystal structure of human protein kinase CK2: insights into basic properties of the CK2 holoenzyme. Embo J. 2001;20:5320–5331. doi: 10.1093/emboj/20.19.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarno S, et al. Cooperative modulation of protein kinase CK2 by separate domains of its regulatory beta-subunit. Biochemistry. 2000;39:12324–12329. doi: 10.1021/bi0011431. [DOI] [PubMed] [Google Scholar]
- 6.Meggio F, Boldyreff B, Marin O, Pinna LA, Issinger OG. Role of the beta subunit of casein kinase-2 on the stability and specificity of the recombinant reconstituted holoenzyme. Eur J Biochem. 1992;204:293–297. doi: 10.1111/j.1432-1033.1992.tb16636.x. [DOI] [PubMed] [Google Scholar]
- 7.Marin O, Meggio F, Boldyreff B, Issinger OG, Pinna LA. Dissection of the dual function of the beta-subunit of protein kinase CK2 ('casein kinase-2'): a synthetic peptide reproducing the carboxyl-terminal domain mimicks the positive but not the negative effects of the whole protein. FEBS Lett. 1995;363:111–114. doi: 10.1016/0014-5793(95)00295-k. [DOI] [PubMed] [Google Scholar]
- 8.Litchfield DW, Luscher B, Lozeman FJ, Eisenman RN, Krebs EG. Phosphorylation of casein kinase II by p34cdc2 in vitro and at mitosis. J Biol Chem. 1992;267:13943–13951. [PubMed] [Google Scholar]
- 9.Bosc DG, Slominski E, Sichler C, Litchfield DW. Phosphorylation of casein kinase II by p34cdc2. Identification of phosphorylation sites using phosphorylation site mutants in vitro. J Biol Chem. 1995;270:25872–25878. doi: 10.1074/jbc.270.43.25872. [DOI] [PubMed] [Google Scholar]
- 10.Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 11.Lazarus MB, Nam Y, Jiang J, Sliz P, Walker S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature. 2011;469:564–567. doi: 10.1038/nature09638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo B, et al. Chronic hexosamine flux stimulates fatty acid oxidation by activating AMP-activated protein kinase in adipocytes. J Biol Chem. 2007;282:7172–7180. doi: 10.1074/jbc.M607362200. [DOI] [PubMed] [Google Scholar]
- 13.Zeidan Q, Hart GW. The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci. 2010;123:13–22. doi: 10.1242/jcs.053678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lubas WA, Hanover JA. Functional expression of O-linked GlcNAc transferase. Domain structure and substrate specificity. J Biol Chem. 2000;275:10983–10988. doi: 10.1074/jbc.275.15.10983. [DOI] [PubMed] [Google Scholar]
- 15.Vila-Perello M, Muir TW. Biological applications of protein splicing. Cell. 2010;143:191–200. doi: 10.1016/j.cell.2010.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muir TW, Sondhi D, Cole PA. Expressed protein ligation: a general method for protein engineering. Proc Natl Acad Sci U S A. 1998;95:6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piontek C, et al. Semisynthesis of a homogeneous glycoprotein enzyme: ribonuclease C: part 1. Angew Chem Int Ed Engl. 2009;48:1936–1940. doi: 10.1002/anie.200804734. [DOI] [PubMed] [Google Scholar]
- 18.Zheng W, et al. Cellular stability of serotonin N-acetyltransferase conferred by phosphonodifluoromethylene alanine (Pfa) substitution for Ser-205. J Biol Chem. 2005;280:10462–10467. doi: 10.1074/jbc.M412283200. [DOI] [PubMed] [Google Scholar]
- 19.Ohnishi Y, Ichikawa M, Ichikawa Y. Facile synthesis of N-Fmoc-serine-S-GlcNAc: a potential molecular probe for the functional study of O-GlcNAc. Bioorg Med Chem Lett. 2000;10:1289–1291. doi: 10.1016/s0960-894x(00)00223-7. [DOI] [PubMed] [Google Scholar]
- 20.Marin O, Meggio F, Pinna LA. Design and synthesis of two new peptide substrates for the specific and sensitive monitoring of casein kinases-1 and -2. Biochemical and biophysical research communications. 1994;198:898–905. doi: 10.1006/bbrc.1994.1128. [DOI] [PubMed] [Google Scholar]
- 21.Liu Q, Huang SS, Huang JS. Kinase activity of the type V transforming growth factor beta receptor. J Biol Chem. 1994;269:9221–9226. [PubMed] [Google Scholar]
- 22.Loizou JI, et al. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004;117:17–28. doi: 10.1016/s0092-8674(04)00206-5. [DOI] [PubMed] [Google Scholar]
- 23.Tiganis T, House CM, Kemp BE. Protein kinase CK2: biphasic kinetics with peptide substrates. Arch Biochem Biophys. 1996;325:289–294. doi: 10.1006/abbi.1996.0036. [DOI] [PubMed] [Google Scholar]
- 24.Sarno S, et al. Mutational analysis of residues implicated in the interaction between protein kinase CK2 and peptide substrates. Biochemistry. 1997;36:11717–11724. doi: 10.1021/bi9705772. [DOI] [PubMed] [Google Scholar]
- 25.Salvi M, et al. Discrimination between the activity of protein kinase CK2 holoenzyme and its catalytic subunits. FEBS Lett. 2006;580:3948–3952. doi: 10.1016/j.febslet.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 26.Sarno S, Marin O, Ghisellini P, Meggio F, Pinna LA. Biochemical evidence that the N-terminal segments of the alpha subunit and the beta subunit play interchangeable roles in the activation of protein kinase CK2. FEBS Lett. 1998;441:29–33. doi: 10.1016/s0014-5793(98)01516-6. [DOI] [PubMed] [Google Scholar]
- 27.Homma MK, Homma Y. Cell cycle and activation of CK2. Mol Cell Biochem. 2008;316:49–55. doi: 10.1007/s11010-008-9823-4. [DOI] [PubMed] [Google Scholar]
- 28.Macauley MS, Stubbs KA, Vocadlo DJ. O-GlcNAcase catalyzes cleavage of thioglycosides without general acid catalysis. J Am Chem Soc. 2005;127:17202–17203. doi: 10.1021/ja0567687. [DOI] [PubMed] [Google Scholar]
- 29.Senderowicz AM. Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Invest New Drugs. 1999;17:313–320. doi: 10.1023/a:1006353008903. [DOI] [PubMed] [Google Scholar]
- 30.Gussio R, et al. Structure-based design modifications of the paullone molecular scaffold for cyclin-dependent kinase inhibition. Anticancer Drug Des. 2000;15:53–66. [PubMed] [Google Scholar]
- 31.Shapiro GI, Koestner DA, Matranga CB, Rollins BJ. Flavopiridol induces cell cycle arrest and p53-independent apoptosis in non-small cell lung cancer cell lines. Clin Cancer Res. 1999;5:2925–2938. [PubMed] [Google Scholar]
- 32.Fischer PM, Lane DP. Inhibitors of cyclin-dependent kinases as anti-cancer therapeutics. Curr Med Chem. 2000;7:1213–1245. doi: 10.2174/0929867003374048. [DOI] [PubMed] [Google Scholar]
- 33.Bosc DG, Luscher B, Litchfield DW. Expression and regulation of protein kinase CK2 during the cell cycle. Mol Cell Biochem. 1999;191:213–222. [PubMed] [Google Scholar]
- 34.Messenger MM, et al. Interactions between protein kinase CK2 and Pin1. Evidence for phosphorylation-dependent interactions. J Biol Chem. 2002;277:23054–23064. doi: 10.1074/jbc.M200111200. [DOI] [PubMed] [Google Scholar]
- 35.Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 36.Shen M, Stukenberg PT, Kirschner MW, Lu KP. The essential mitotic peptidyl-prolyl isomerase Pin1 binds and regulates mitosis-specific phosphoproteins. Genes Dev. 1998;12:706–720. doi: 10.1101/gad.12.5.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuzwa SA, et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol. 2008;4:483–490. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- 38.Hu S, et al. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell. 2009;139:610–622. doi: 10.1016/j.cell.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? Faseb J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 40.Moretto-Zita M, et al. Phosphorylation stabilizes Nanog by promoting its interaction with Pin1. Proc Natl Acad Sci U S A. 2010;107:13312–13317. doi: 10.1073/pnas.1005847107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou W, et al. Pin1 catalyzes conformational changes of Thr-187 in p27Kip1 and mediates its stability through a polyubiquitination process. J Biol Chem. 2009;284:23980–23988. doi: 10.1074/jbc.M109.022814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siepe D, Jentsch S. Prolyl isomerase Pin1 acts as a switch to control the degree of substrate ubiquitylation. Nat Cell Biol. 2009;11:967–972. doi: 10.1038/ncb1908. [DOI] [PubMed] [Google Scholar]
- 43.Mahoney SJ, Dempsey JM, Blenis J. Cell signaling in protein synthesis ribosome biogenesis and translation initiation and elongation. Prog Mol Biol Transl Sci. 2009;90:53–107. doi: 10.1016/S1877-1173(09)90002-3. [DOI] [PubMed] [Google Scholar]
- 44.Canton DA, Litchfield DW. The shape of things to come: an emerging role for protein kinase CK2 in the regulation of cell morphology and the cytoskeleton. Cell Signal. 2006;18:267–275. doi: 10.1016/j.cellsig.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 45.Cory GO, Cramer R, Blanchoin L, Ridley AJ. Phosphorylation of the WASP-VCA domain increases its affinity for the Arp2/3 complex and enhances actin polymerization by WASP. Mol Cell. 2003;11:1229–1239. doi: 10.1016/s1097-2765(03)00172-2. [DOI] [PubMed] [Google Scholar]
- 46.Shen HH, Huang AM, Hoheisel J, Tsai SF. Identification and characterization of a SET/NAP protein encoded by a brain-specific gene, MB20. Genomics. 2001;71:21–33. doi: 10.1006/geno.2000.6397. [DOI] [PubMed] [Google Scholar]
- 47.Choudhary C, Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nat Rev Mol Cell Biol. 2010;11:427–439. doi: 10.1038/nrm2900. [DOI] [PubMed] [Google Scholar]
- 48.Qiao Y, Molina H, Pandey A, Zhang J, Cole PA. Chemical rescue of a mutant enzyme in living cells. Science. 2006;311:1293–1297. doi: 10.1126/science.1122224. [DOI] [PubMed] [Google Scholar]
- 49.Comer FI, Vosseller K, Wells L, Accavitti MA, Hart GW. Characterization of a mouse monoclonal antibody specific for O-linked N-acetylglucosamine. Anal Biochem. 2001;293:169–177. doi: 10.1006/abio.2001.5132. [DOI] [PubMed] [Google Scholar]
- 50.Huang R, et al. Site-Specific Introduction of an Acetyl-Lysine Mimic into Peptides and Proteins by Cysteine Alkylation. J Am Chem Soc. 2010;132:9986–9987. doi: 10.1021/ja103954u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.