

Abstract
Cellular senescence is an irreversible arrest of cell growth. Biochemical and morphological changes occur during cellular senescence, including the formation of a unique cellular morphology such as flattened cytoplasm. Function of mitochondria, endoplasmic reticulum and lysosomes are affected resulting in the inhibition of lysosomal and proteosomal pathways. Cellular senescence can be triggered by a number of factors including, aging, DNA damage, oncogene activation and oxidative stress. While the molecular mechanism of senescence involves p16 and p53 tumor suppressor genes and telomere shortening, this review is focused on the mechanism of p16 control. The p16 mediated senescence acts through the retinoblastoma (Rb) pathway inhibiting the action of the cyclin dependant kinases leading to G1 cell cycle arrest. Rb is maintained in a hypophosphorylated state resulting in the inhibition of transcription factor E2F1. Regulation of p16 expression is complex and involves epigenetic control and multiple transcription factors. PRC1 (Pombe repressor complex 1) and PRC2 (Pombe repressor complex 2) proteins and histone deacetylases play an important role in the promoter hypermethylation for suppressing p16 expression. While transcription factors YY1 and Id1 suppress p16 expression, transcription factors CTCF, Sp1, and Ets family members activate p16 transcription. Senescence occurs with the inactivation of suppressor elements leading to the enhanced expression of p16.
Keywords: Senescence, Tumor suppressor gene p16
Introduction
Cellular senescence is a state of irreversible cellular arrest involving several cellular changes. These changes include protein aggregation in endoplasmic reticulum, enlarged and functionally defective mitochondria and non functional lysosomes (Figure 1) [1, 2]. Levels of BiP chaperone which is critical for protein folding declines in endoplasmic reticulum [3]. Calnexin, a protein that is responsible for glycosylation in endoplasmic reticulum, has also been shown to decline significantly during senescence [4, 5]. The SERCA pump is impaired resulting in increased cytoplsmic concentrations of calcium and therefore the calcium storage role of the endoplasmic reticulum is impaired [5, 6]. During senescence, free radical levels increase and these reactive species damage mitochondria [6]. Structural deterioration in mitochondria includes loss of cristae, and swelling and destruction of the inner membrane [7-9]. Aging related senescence also results in increased frequency of mutations in mtDNA and defective mitochondria leading to various pathological conditions including myopathies and cardiomyopathies [10, 11]. Autophagy which is a mechanism used by cells to degrade dysfunctional organelles also decreases in post mitotic cells resulting in the accumulation of defective mitochondria [12]. Lysosomes accumulate lipofuscin, a brown pigment composed of insoluble membrane free protein aggregates [13, 14]. Lipofuscin inhibits lysosomal and proteosomal protein degradation pathways thereby preventing recycling of amino acids. Additionally, aging negatively impacts the integrity of cell membrane and the free radicals generated by incomplete oxidation breaks the double bond of cholesterol and unsaturated fatty acids in the membrane. These molecules are highly toxic to the cell and can lead to apoptosis and cell death [15, 16]. The level of beta galactosidase in lysosomes increases significantly in senescent cells and is thus used as an experimental marker. While senescence is reflected in the phenotypic changes of different organelles, the molecular mechanism of cellular senescence involves p16 and p53 mediated pathways and telomere shortening. In the present review, we provide details on the relationship between negative and positive regulators of p16 driving cell growth or senescence.
Figure 1.
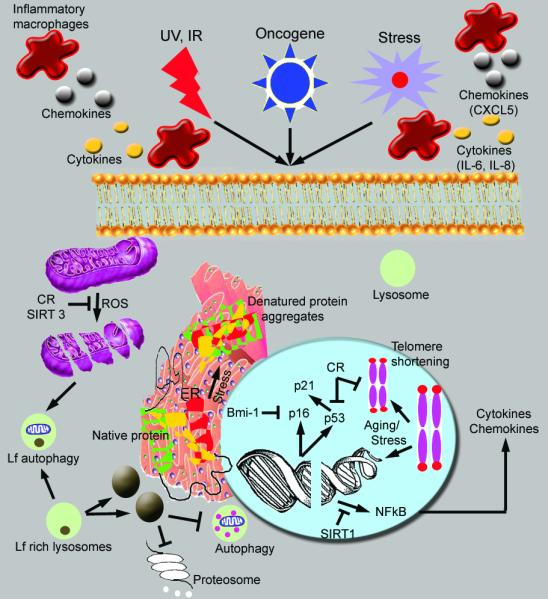
Molecular mechanism of senescence. Induction of reactive oxygen species (ROS) induces mitochondrial breakdown as well as double stranded breaks. Activation of p16 and p53 induce growth arrest at G1/S and activation of NFkB leads to increased expression of cytokines and chemokines. Telomere shortening involved in aging or under stress conditions could result in double stranded breaks. ROS production as well as aging induced stress responses could be blocked by sirtuins, NAD dependent protein deacetylases [125]. Aggregates and crystals of denatured proteins (as in β amyloid crystals in Alzeimer’s disease) are visualized in the endoplasmic reticulum. Lipofuscin (Lf), a brown pigment consisting of insoluble membrane free protein aggregates, formed due to ROS and stress inhibits normal autophagy as well as proteosomal pathway of protein recycling machinery. Calorie restriction (CR) and sirtuins (SIRT) inhibit the effects of ROS and senescence.  Activation
Activation  Inhibition
Inhibition
P16 and cell cycle control
Progression through cell cycle is dependent on the cell passing through four cell cycle checkpoints, the G1-S transition, S phase checkpoint, G2 to M transition and the Mitotic spindle checkpoint. The G1 to S transition is controlled by the p16-Rb pathway. P16 binds to CDK4/6 inhibiting its kinase activity thereby preventing Rb phosphorylation (Figure 2). Rb remains associated with transcription factor E2F1 localizing it to the cytoplasm and thus preventing transcription of E2F1 target genes which are crucial for the G1/S transition. It has also been shown that premature senescence could be induced by Rb-p16-19Arf proteins in the presence of E2F activation in a mouse pituitary tumor model [17]. Although the sustained activation of E2F results in hyper-cell proliferation, cells still undergo senescence. Here the cell cycle arrest seems to occur through the formation of repressor complexes between Rb family (pRb, p107, and p130) of pocket proteins and E2F in the nucleus [17, 18]. Rb is therefore an important tumor suppressor gene, and its inactivation in human tumors through the inhibition of p16 has a positive impact on nuclear reprogramming; increasing both the number of reprogrammed cells and the kinetics of de-differentiation in proliferating cells [19]. P16 expression is also regulated by Rb: phosphorylation of Rb results in increased p16 expression which inhibits CDK4/6 resulting in increased levels of hypophosphorylated Rb which in turn leads to decreased p16 expression [20]. Although there is a feedback loop between p16 and Rb, it has been shown that the level of p16 expression does not change appreciably during cell cycle to correlate with the activation status of Rb [21]. Further, p16 expression is elevated in Rb negative cells indicating other mechanisms of p16 regulation. In aging, there is increased expression of p16 that is independent of telomere shortening. In addition, exposure to ionizing radiation or DNA damaging agents have also been shown to increase expression of p16 resulting in senescence [22, 23]. Finally, it has been shown that p16/Rb pathway collaborates with the mitogenic signaling cascade for the induction of ROS (reactive oxygen species) which in turn activates the protein kinase C delta (PKC delta) [24]. This in turn enhances ROS production resulting in a positive feedback loop between ROS and PKC delta leading to an irreversible cell cycle arrest. Thus p16 seems to play an important role in the initiation as well as in the maintenance of cellular senescence. Control of p16 expression is therefore an important mechanism for the suppression of senescence.
Figure 2.

Regulation of p16 expression and senescence. Bmi-1 binds to PRC2 (Pombe Repressor Complex) proteins, which then bind to p16 promoter resulting in methylation and repression of transcription. Bmi-1 expression is up-regulated by C-MYC which in turn is positively regulated by the E2F-1 transcription factor. P16 levels are up-regulated by the Ets-1 and HBP-1 transcription factors. Binding of p16 to Cyclin D1 and cdk4/6 inhibits the cyclin dependent kinase activity leading to reduced levels of E2F1 released from the Rb. However, HDAC is involved in the release of E2F1 which in turn could up-regulate p14ARF. Mdm2 activity is then inhibited leading to the activation of p53 and induction of senescence through p21. RAS mediated senescence is inhibited by phosphorylation at Ser 62 of C-MYC by cyclin E dependent cdk2. The cyclin E/Cdk2 function however can be abrogated by p53 induced p21 protein. Cisplatin induced senescence is mediated by the activation of nuclear p16 and p53 proteins.  Activation
Activation  Inhibition
Inhibition
PRC (polycomb repressor complex) mediated inhibition of p16 expression
The p16 gene is contained within the locus encompassing the Ink4b/ARF/Ink4a genes on chromosome 9p21 [25]. Two distinct reading frames are used for the transcription of p14ARF (alternate reading frame) and p16 tumor suppressor genes (Figure 3) [26, 27]. While p14ARF is transcribed from exon 1β and exon 2, p16 is transcribed from exon 1α localized 20 kb downstream of 1β and exons 2 and 3 [28].
Figure 3.

Transcriptional regulation of p16/p14ARF by PRC1 and PRC2 complexes. A) Binding of PRC1 and PRC2 complex proteins to the p16/p14ARF promoter results in formation of heterochromatin leading to suppression of transcription. Release of these factors results in the formation of euchromatin and transcription of p16/p14ARF genes. B) Locus of p16/p14ARF genes showing exons involved in alternate splicing. Promoter regions are shown in red. The map is not drawn to scale.
Expression of p16 and p14ARF are regulated by promoter hypermethylation through proteins of the PRC1 and PRC2 complexes. The polycomb group (PcG) of transcriptional repressor proteins was originally characterized in drosophila as maintenance proteins of pluripotency [29] [30]. PRC2 functions as an initiator of transcription repression and PRC1 functions as a repressor maintenance complex. Thus the methylation mediated by PRC2 is a pre-requisite for the binding of PRC1 to the chromatin [31].
The PRC1 complex is comprised of Polycomb (Pc), Posterior sex comb (Psc), Polyhomeotic (Ph) and Sex combs extra(Sce) [32, 33]. In mammals each category of proteins has numerous subtypes leading to multiple potential combinations. There are 5 Pc proteins (CXB2, CBX4, CBX6, CBX7 and CBX8), 6 Psc proteins, one of them being Bmi-1, 3 Ph proteins (HPH1, HPH2 and HPH3) and 2 Sce proteins (RING1 and RING 2) [34, 35]. The primary mechanism of transcriptional repression by the PRC1 involves monoubiquination of histone H2A K119 by histone H2A ubiquitin ligase [36].
The PRC2 complex is composed of 3 core proteins, Ezh2 which mediates trimethylation of histone H3K27, Eed and Suz12 [37-39]. The SET domain of Ezh2 possesses methyltransferase activity specific for histone H3K27 [40, 41]. This results in long term reversible suppression of Ink4a genes p14ARF and p16. In cells that are senescing, Ezh2 levels are decreased leading to decreased levels of H3K27 trimethylation, disassociation of Bmi-1 from the chromatin and increased transcription of Ink4a genes [42]. Trimethylation of histone H3 at lysine 9 by PRC2 complex is an irreversible epigenetic modification of the genome [43] which mediates the binding of the adaptor molecule HP-1 allowing irreversible binding of PRC1 complex proteins [44].
Bmi-1 and other transcription factors of PRC1 Complex
Bmi-1 is a polycomb transcription factor and a member of the PRC1 complex that inhibits senescence by inhibiting transcription of p16. Thus Bmi-1 behaves as an oncogene and is a marker of tumor stem cells [28, 45]. The ability of Bmi-1 to decrease transcription from the p16 locus depends on the presence of Ezh2 and other components of the PRC2 complex. Bmi-1 also mediates the interaction of the Zinc finger protein 277(Zfp 277) with the PRC1 complex. Loss of Zfp277 in mouse embryonic cells results in disassociation of the PcG proteins from the Ink4a locus leading to an increase in expression of p16 and senescence [46]. Similarly, deletion of Bmi-1 is associated with increased p16 expression and retardation of growth. However, deletion of both Bmi-1 and p16 results only in partial cell growth indicating a requirement for both Bmi-1 and p16 in normal cell growth [47, 48]. Kotake et al [49] showed that the removal of pRb from cells resulted in the loss of histone H3K27 trimethylation leading to the loss of Bmi-1 recruitment to the p16 locus. Thus pRb is also shown to be necessary for Bmi-1 function in the transcription repression of p16.
The PRC1 complex containing the Pc protein CXB8, the Psc protein Bmi-1 and 2 Sce proteins RING 1 and RING2 is the most widely known PRC1 complex to regulate p16 expression [50]. However, studies have shown that PRC1 complex comprised of CXB7 and Bmi-1 could also regulate p16 expression [51]. Maartens et al [52] suggested that Pc proteins CXB7 and CXB8 form at least 4 different PRC1 complexes including Bmi-1 and Mel18. Knockdown of anyone of these proteins results in derepression of p16 locus and premature senescence. Bmi-1 and Mel18 are also regulated by USP7 and USPII, ubiquitin proteases that co-localize with PRC1 complex through interactions with Bmi-1 and Mel18. Removal of USP7 or USPII results in increased turnover of Bmi-1 and Mel18 proteins, loss of PRC1 binding to the p16 locus, and increased senescence [52].
Maartens et al also [52] demonstrated that Mel18, CXB7 and CXB8 could bind to the p16 locus indicating repression of p16 by multiple PRC1 complexes. Both in vitro and in vivo studies have shown that CXB7 interacts with additional partners including a non coding antisense RNA, ANRIL, for regulation of p16 [39, 41]. CXB7 has distinct binding sites for PRC1 proteins and ANRIL RNA and the disruption of the interaction between CXB7 and ANRIL leads to disruption of the repressor complex [53]. Furthermore, CXB7 interacts with SUV39H1, SUV39H2, and G9a, histone methyl transferases for the trimethylation of histone H3K9 which also results in irreversible transcriptional repression of p16 [31, 44, 54-57].
Studies have further shown interdependence between these proteins for binding to the p16 locus. Knockdown of CXB7 results in decreased binding of CXB8 without changes in the expression level of CXB8. Similarly, reduced expression of Bmi-1 leads to decreased binding of Mel18 without changes in the level of Mel18 expression. Knockdown of SUV39H1 reverses the repressor activity of CBX7 indicating a role for H3K9 trimethylation in the regulation of p16. Similarly, over expression of PRC1 complex proteins decreases p16 expression and delays the onset of senescence [58, 59].
Epigenetic regulators of PRC2 Complex
Polycomblike protein 2 (Pcl2), an associated protein of the PRC2 complex, exhibits considerable binding to the p16 locus along with other PRC2 core proteins Eed and Suz 12 [56, 57]. Studies have shown that Pcl2 is an important mediator of cellular senescence in mouse embryonic fibroblasts and that cells that had a deleted Pcl2 gene divide exponentially compared to the wild type cells. Moreover, ectopic expression of Pcl2 in immortalized cells results in senescence, confirming the role of Pcl2 as a mediator of cellular senescence. It has also been speculated that Pcl2 decreases the catalytic activity of the PRC2 complex protein Ezh2 resulting in decreased histone H3K27 trimethylation [60, 61].
The PRC2 complex is also known to interact with class 1 histone deactylases (HDAC) such as HDAC 1 [62-64]. Yamaguchi et al [65] showed that knockdown of Ezh2, the H3K27 methyl transferase gene, along with HDAC1 results in a synergistic increase in cell death indicating a role for both of these gene complexes in the epigenetic regulation of p16 [47, 65] .
Recent studies have shown that SWI/SNF protein complex could counteract PcG mediated transcription repression [66, 67]. It has been shown that the loss of function of the hSNF5 component of the SWI/SNF chromatin remodeling complex can lead to malignant tumor formation [68, 69]. On the other hand, overexpression of hSNF5 protein leads to the disassociation of PRC1 and PRC2 complexes from the p16 locus without affecting the expression level of these proteins. The increased p16 expression resulting from these changes is related to the removal of BRG1 ATPase, a central protein of the chromatin remodeling complex [70].
Jung et al [71] have described two different mechanisms whereby histone deactylases play a role in transcription repression. They demonstrate that histone deacetylases promote the disassociation of E2F1 from Rb. E2F1 then binds to the promoter of C-MYC and increases the levels of this protein which is a positive transcriptional regulator of Bmi-1 (Figure 2). The other mechanism involves binding of E2F1 directly to the promoter sequences of the PRC2 proteins Ezh2 and Suz12 leading to increased transcription of these genes and binding of Bmi-1 to the p16 locus [71].
Histone deacetylases have also been shown to function directly as inhibitors of transcription, including that of p16. Feng et al [72] have shown that ZBP-89, a four zink finger transcription factor plays a central role in the recruitment of HDACs to the p16 promoter. This results in decreased expression of p16. Furthermore, this repression seems to require binding of both HDAC1 and ZBP-89 proteins to the p16 locus as knocking down ZBP-89 expression leads to reduced binding of HDAC1 to the p16 promoter. Additionally, administration of histone acetyalse inhibitors results in increased HDAC activity and decreased p16 levels confirming the role of HDACs in p16 repression. The inhibitory role of HDACs is also influenced by transcription factor YY1. It has been suggested that the increased expression of YY1 results in the increased binding of HDACs 3 and 4 to the p16 promoter for the repression of transcription [73]. In addition to transcription factors, it has been shown that the replication factor CDC6 recruits polycomb proteins to the origin of replication for the control of p16 expression [67, 68]. Gonzalez and Serrano [74] have proposed that an origin of replication is situated close to the regulatory domain of p16 and during replication CDC6 is recruited to the origin leading to unwinding of the DNA and recruitment of PRC1 and PRC2 proteins. Song et al [75] have further demonstrated that CDC6 levels are regulated by a feed-back loop between transcription factor MipP130/LIN-9 and p14ARF. MipP130/LIN-9 binds to the CDC6 promoter and increases its transcription for the repression of p16 locus. P14ARF reverses this effect by translocating Mip130/LIN-9 to the nucleolus for post translational modification and ubiquitination [75]. Sugimoto et al [76] have also demonstrated that PIWI proteins that have RNAse activity and involved in RNA maturation play role in regulating p16 expression. Their results show that overexpression of PIWI like molecule 4, the most ubiquitous of the PIWI proteins, results in increased methylation of lysine 9 on H3 at the p16 locus. This decreases p16 expression levels in the cell, inhibiting senescence [76, 77]. Thus various studies demonstrate that the epigenetic regulation of p16 is a complex process involving a number of transcription factors and histone deacetylases. Similarly activation of p16 as described below is also a complex process during cell cycle.
AP1 and other transcription factor mediated activation of p16
AP-1 proteins are transcription factors that are homo or hetero dimers consisting of Jun proteins c-Jun and Jun B, Jun dimerization partners (JDP1 and JDP2) or ATF proteins that interact with each other through a leucine zipper motif [78]. There are three sites for AP-1 proteins to bind to the p16 promoter [79]. JDP-2 inhibits recruitment of the PRC-1 and PRC-2 complexes to the p16 thereby preventing promoter methylation [80]. Ectopic expression of JDP2 promotes cell differentiation in p16 deficient mouse embryonic cells confirming the role of JDP2 in p16 transcription activation [81, 82]. In addition, Jun B activates p16 expression through its inhibitory role on the expression of cyclin D1. Finally, there is a feed-back loop between c-Jun and p16 where p16 controls c-Jun mediated repression by inhibiting c-Jun kinase, which is necessary for c-Jun activation [83]. Besides the promoter region, factors have also been identified that activate p16 expression through binding to upstream sequences. A chromosomal boundary 2kb upstream of the transcriptional start site divides the p16 locus into domains characterized by the presence or absence of epigenetic marks and by the presence of H2A.Z [84]. This functionally diverse protein, in association with the transcription factor CTCF is shown to inhibit epigenetic silencing through chromatin remodeling of the p16 locus [85-87]. Deletion of CTCF through siRNA resulted in hypermethylation of the p16 promoter and down regulation of p16 expression. Hence CTCF interaction with the chromosomal boundary is important for the maintenance of p16 expression [84]. Another factor involved in p16 activation is p300 that contains histone acetylase activity and an interaction motif for Sp1 [88, 89]. Wang et al [89] showed that the interaction between p300 and Sp1 at the p16 locus leads to hypermethylation of histone H4 and activation p16.
Gan et al [90] demonstrated that the PPAR gamma transcription factor binds to the p16 promoter and that treatment of cells with troglitazone a PPAR gamma agonist, resulted in increased p16 expression contributing to senescence. In addition, siRNA knockdown of PPAR gamma decreased the p16 levels in these cells. Moreover, these authors also demonstrated that dephosphorylation of PPAR gamma results in increased p16 transcription from the promoter indicating dephosphorylation as a molecular mechanism of control of p16 transcription [90].
Oncogene and stress mediated activation of p16 transcription
Oncogene mediated premature senescence is a tumor suppressor mechanism that has been well described in the RAS activated MAP kinase pathway [19, 91, 92]. The Ets family of transcription factors is often activated by the RAS-MAP kinase pathway [93]. Through reporter assays Ohtani et al [94] have shown that Ets transcription factors, Ets 1 and Ets 2 increase transcription of p16 and addition of activated RAS further enhances transcription of the p16 gene (Figure 2). Id1 is a helix loop helix (HLH) transcription factor which inhibits the activity of Ets transcription factors decreased the transcription of p16. These authors further show that mutating the binding site on Ets1 for Id1 prevents transcriptional repression of p16. These results clearly demonstrated an Ets1 dependent pathway for Id1 inhibition of p16 transcription. Another mechanism of RAS mediated senescence occurs through the expression of HBP-1 a High mobility group box containing protein 1 that is homologous to the HMG family of transcription factors [95]. Ectopic HBP-1 expression raises the level of p16 expression and the degradation of HBP-1 with siRNA results in decreased expression of p16. Finally, RAS could also promote senescence through the activation of the H3K27 histone demethylase JMJD3 and down regulation of the methyl transferase Ezh2 [96].
Oncogene induced senescence seems to go through a two step process. In the initial phase, as in the early papilloma stage of skin cancer, cells undergo hyper-cell proliferation through the MAPK/MEK growth signaling pathway [97]. Activation of E2F transcription factors takes place resulting in increased transcription of DMNT1, the histone H3 lysine 9 dimethyl transferase (H3K9 me2). DMNT1 is then involved in enhanced methylation of H3K9 at the p16 promoter site. Thus, histone modification and not DNA methlyation seems to be the cause of repression of the p16 gene by oncogene activation [97]. However with increased oncogene signaling, as in the late papilloma stage, hyper-cell proliferation leads to DNA damage and induction of ROS which in turn inhibits DNMT1 expression through the inhibition of E2F. This results in reduced methylation of H3K9 and derepression of the p16 gene.
In the presence of wild type p53, DNA damage response activates ATM/ATR kinase pathway for the activation of p53 to induce senescence through the expression of p21 and apoptosis through the expression of anti-apoptotic proteins [97-99]. Activation of p53 is therefore an important event in blocking proliferation of the damaged cells that could cause further DNA damage. When p53 is inactivated as in cancer cells, accumulation of DNA damage due to hyper-cell proliferation leads to induction of senescence through ROS mediated p16 expression. These authors therefore suggest that p16 serves as a backup tumor suppressor gene for p53 and hence is inactivated by homozygous deletion or promoter hypermethlyation in cancer cells. ROS signaling as described earlier establishes a feedback loop with DNA damage response and thus is a mediator of oncogene induced senescence. It is therefore suggested that ROS plays an important role in the initiation as well as maintenance of cellular senescence [24]. P38, a mitogen activated protein kinase (MAPK), plays a role in inducing senescence in response to stress due to reactive oxygen species, UV light, heat, osmotic stress and growth factors [100, 101]. Activated p38 phosphorylates transcription factors including ESE-3, a transcription factor that belongs to the Ets family, leading to an increase in p16 expression [102]. ESE-3 activates transcription of p16 through a direct binding to the GGAA motif of the p16 promoter and the level of transcription is similar to that of Ets transcription factors [102].
P16 and aging
Aging is associated with replicative senescence and p16 levels increase with aging in most mammalian tissues [25, 103]. The levels of p16 in an individual can be predicted by stochastic model that takes into consideration the subjects’ age [104]. According to this model, p16 levels exponentially increase with age in a p16 dependent manner and reach a plateau. As proof of this model, early fibroblasts have low levels of p16 and aging senescing fibroblasts have significantly increased levels of p16 expression. This model also demonstrates that increases in p16 levels due to environmental factors such as smoking are associated with a decrease in p16 degradation and not due to an increase in synthesis [104].
Increased p16 levels in the pancreas during aging inhibit the proliferation of beta cells and decreases their ability to respond to injury. While the beta cells of the p16 knock-out mice were able to proliferate in response to injury, beta cells with ectopic expression of p16 showed reduced proliferative response confirming the association between p16 and beta cell senescence [25, 103, 105, 106]. During aging, the regenerative capacity of the stem cells also decreases in different tissues including CNS, and hematopoietic cells [107, 108]. Studies in mice have shown that the decreased proliferation of stem cells is due to enhanced expression of p16 confirming the relationship between p16 expression and stem cell regeneration [109]. In human studies, investigation into the proliferation of skin and kidney tissues have confirmed the inverse relationship between p16 and Bmi-1 during aging indicating the role played by p16 in cellular senescence [48, 109, 110].
P16 and cellular processes in senescence
P16 mediated senescence results in chromatin reorganization which is related to the repression of genes regulated by transcription factor E2F1 [92, 111]. Chromatin reorganization in oncogene induced premature senescence is visualized as SAHF (senescence associated heterochromatin foci), represented by densely stained nuclear DNA and enriched H3K9 trimethylation [112, 113]. According to these recent publications, SAHF are rarely seen in fibroblasts undergoing replicative senescence and are not observed in premalignant lesions containing enhanced p16 expression and other features of senescence. These studies indicate that SAHF are markers of oncogene activation, are involved in the restriction of DDR mediated apoptotic response, and are dispensable for cellular senescence [112, 113]. A major characteristic of senescence and aging is also increased inflammation and oxidative damage (Figure 1). Experiments show that aging in rats is associated with a significant increase in reactive oxygen species produced by COX1 and COX2 enzymes which are involved in prostaglandin synthesis [114]. NFkB is the most important transcription factor that responds to inflammatory molecular signals [115]. In aging, there is an increase in the duration of inflammation resulting in increased activation of NFkB which in turn increases transcription of inflammatory molecules [116]. In addition, aging results in down regulation of PPARα (Peroxisome proliferator activated receptor α) signaling which normally functions to negatively modulate NFkB [117]. In senescence, molecules that possess anti-inflammatory properties such as macrophage-derived chemotactic factor-II are down regulated while pro-inflammatory and apoptotic signaling pathways such as caspase 3 signaling are up-regulated [118]. Moreover, senescent cells express an increased amount of secreted proteins such as metalloproteinases, inflammatory cytokines and growth factors related to NFkB activation [119]. Thus senescence can lead to cellular damages that are irreversible and prevent growth and differentiation of aging cells.
Relationship between p16 and p53 in senescence
Senescence is also mediated by the p53 pathway leading to G1 arrest of the cell cycle. This p53 activity is independent of p16 mediated senescence (Figure 2). Here again, senescence can be induced by DNA damage as in UV irradiation and by enhanced expression of oncogenes [120, 121]. P14ARF of the Ink4a/p16 locus plays a major role in senescence through its affect on p53. Mdm2 is an ubiquitin ligase that recognizes a region in the N terminus of p53 and hence targets p53 for degradation. P14ARF traps Mdm2 ubiquitin ligase to the nucleolus preventing p53 ubiquitination. This allows p53 to form a functional tetramer and activate transcription of p21. Activities of CDK4 and cyclin E dependant kinase CDK2 are suppressed by p21 resulting in cell cycle arrest at G1/S. There is also a negative feedback for progression through G1/S by the up-regulated expression of Mdm2. Here, p14ARF expression is suppressed by E2F1 released from the Rb complex by CDK4 dependent phosphorylation of Rb. Thus p16 and p53 pathways are connected by a feedback mechanism involving p14ARF and E2F1 [122]. As described earlier, there is evidence in the literature that p16 might be playing a backup tumor suppressor role for p53 [97, 123]. While up-regulation of p16 is observed in p53 deficient mouse fibroblasts, re-expression of p53 does not abolish p16 expression but brings it back to the normal level [123]. Further, under stress induced p53 activation, expression of p16 is not abolished. However in the absence of p53, p16 level goes up possibly through the up-regulation of Ets1, the positive regulator of p16 [102]. Finally, in a mouse skin development model, it has been shown that the expression of p63, a family member of p53, is required for normal skin development through its control of p16 and p194ARF gene expression [124]. Absence of p63 is accompanied by abnormal keratinocyte differentiation that is correlated with the enhanced expression of p16 and p19ARF genes. These studies therefore indicate that p16, p53 and p63 gene activities are interconnected and evolution has developed p16 and p53 mediated senescence as complementary pathways for the control of human tumor development.
Conclusion
Senescence is an important event in cancer prevention. However, senescence also plays a major role in aging. It is also suggested that senescent cells release cytokines and chemokines which could have non-cell autonomous growth activation of neighboring cells (a paracrine effect). Thus senescence has a positive and a negative role in human growth and differentiation. Increased expression of p16 tumor suppressor gene leads to senescence and the cancer cells overcome this effect by inactivating this gene by homozygous deletion or hypermethylation. Normal cell homeostasis needs to balance the expression of p16 between growth and differentiation. It has therefore built a complex mechanism for the activation as well as for the inactivation of p16. While p53 is a gate keeper gene of cell growth playing a role in growth arrest and DNA repair, p16 plays an important role in preventing tumorigenesis. In fact inactivation of p16 is the second most mechanism besides the inactivation of p53 in human tumors. Chemotherapeutic drugs such as cisplatin induce senescence by enhancing the activities of p16 both in cancer and normal cells resulting in increased toxicity to normal cells. Therefore, induction of senescence with enhanced expression of p16 in cancer cells alone will be a valuable therapeutic strategy in cancer treatment.
Acknowledgements
We thank Eugene Han for his help with the figures. The study was supported by funds from VAGLAHS, West Los Angeles Surgical Education Research Center, NIH (R21 CA116826-01 to Marilene B. Wang) and Merit grant from the Veterans Administration, Washington, DC (Eri S. Srivatsan).
Footnotes
Conflict of Interest None of the authors have financial interest in this review article
Authors’ contributions HR carried out literature survey and in association with ESS contributed to the design and draft of the manuscript and the figure. MBW participated in the design and coordination of the manuscript. ESS conceived of the study, and participated in the design and drafting of the manuscript. All authors read and approved the final manuscript.
References
- 1.Nuss JE, Choksi KB, DeFord JH, Papaconstantinou J. Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem Biophys Res Commun. 2008;365:355–61. doi: 10.1016/j.bbrc.2007.10.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clinical Inves. 2002;110:1389–98. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rabek JP, Boylston H., III Carbonylation of ER chaperone proteins in aged mouse liver. Biochem Biophys Res Commun. 2003;305:566–72. doi: 10.1016/s0006-291x(03)00826-x. [DOI] [PubMed] [Google Scholar]
- 4.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–64. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 5.Erickson RR, Dunning LM, Holtzman JL. The effect of aging on the chaperone concentrations in the hepatic, endoplasmic reticulum of male rats: the possible role of protein misfolding due to the loss of chaperones in the decline in physiological function seen with age. J Gerontol A Biol Sci Med Sci. 2006;61:435–43. doi: 10.1093/gerona/61.5.435. [DOI] [PubMed] [Google Scholar]
- 6.Yin D, Kuczera K, Squier TC. The sensitivity of carboxyl-terminal methionines in calmodulin isoforms to oxidation by H2O2 modulates the ability to activate the plasma membrane Ca-ATPase. Chem Res Toxicol. 2000;13:103–10. doi: 10.1021/tx990142a. [DOI] [PubMed] [Google Scholar]
- 7.De Grey AD. A proposed refinement of the mitochondrial free radical theory of aging. Bioessays. 1997;19:161–66. doi: 10.1002/bies.950190211. [DOI] [PubMed] [Google Scholar]
- 8.Harman D. The biologic clock: the mitochondria? J Am Geriatric Soc. 1972;20:145–47. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 9.Terman A, Gustafsson B, Brunk UT. Mitochondrial damage and intralysosomal degradation in cellular aging. Mol Aspects Med. 2007;27:471–82. doi: 10.1016/j.mam.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Khrapko K, Bodyak N, Thilly WG, van Orsouw NJ, Zhang X, Coller HA, Perls TT, Upton M, Vijg J, Wei JY. Cell by- cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999;27:2434–41. doi: 10.1093/nar/27.11.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao ZWJ, McKiernan SH, Aiken JM. Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: analysis by laser capture microdissection. Nucleic Acids Res. 2001;29:4502–08. doi: 10.1093/nar/29.21.4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miwa SLC, von Zglinicki T. Mitochondrial turnover in liver is fast in vivo and is accelerated by dietary restriction: application of a simple dynamic model. Aging Cell. 2008;7:920–23. doi: 10.1111/j.1474-9726.2008.00426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brunk UT, Terman A. Lipofuscin: mechanisms of agerelated accumulation and influence on cell functions. Free Radic Biol Med. 2002;33:611–19. doi: 10.1016/s0891-5849(02)00959-0. [DOI] [PubMed] [Google Scholar]
- 14.Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269:1996–02. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 15.Spiteller G. Linoleic acid peroxidation—the dominant lipid peroxidation process in low density lipoprotein—and its relationship to chronic diseases. Chem Phys Lipids. 1998;95:105–62. doi: 10.1016/s0009-3084(98)00091-7. [DOI] [PubMed] [Google Scholar]
- 16.Dudda A, Spiteller G, Kobalt F. Lipid oxidation products in ischemic porcine heart tissue. Chem Phys Lipids. 1996;82:39–51. doi: 10.1016/0009-3084(96)02557-1. [DOI] [PubMed] [Google Scholar]
- 17.Denchi E Lazzerini, Attwooll C, Pasini D, Helin K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol Cell Biol. 2005;25:2660–72. doi: 10.1128/MCB.25.7.2660-2672.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rowland BD, Bernards R. Re-evaluating cell-cycle regulation by E2Fs. Cell. 2006;127:871–74. doi: 10.1016/j.cell.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 19.Li CM, Villasante A, Strati K, Ortega S, Cañamero M, Blasco MA, Serrano M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136–39. doi: 10.1038/nature08290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Nichols MA, Shay JW, Xiong Y. Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb. Cancer Res. 1994;54:6078–72. [PubMed] [Google Scholar]
- 21.Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996;16:859–67. doi: 10.1128/mcb.16.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng A, Wang Y, Van Zant G, Zhou D. Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res. 2003;63:5414–19. [PubMed] [Google Scholar]
- 23.Wang Y, Schulte BA, Larue AC, Ogawa M, Zhou D. Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood. 2006;107:359–66. doi: 10.1182/blood-2005-04-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006;8:1291–97. doi: 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- 25.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clinical Invest. 2004;114:1299–07. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes and Dev. 1995;9:1831–45. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- 27.Kim WY, Sharpless NE. The regulation of INK4/ ARF in cancer and aging. Cell. 2006;127:265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cyle arrest. Cell. 1995;8:993–00. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 29.Sparmann A, Van Louhizen LM. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–56. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genetics. 2007;8:9–22. doi: 10.1038/nrg1981. [DOI] [PubMed] [Google Scholar]
- 31.Hernandez-Munoz I, Taghavi P, Kuijl C, Neefjes J, Van Lohuizen M. Association of BMI1 with polycomb bodies is dynamic and requires PRC2/ EZH2 and the maintenance DNA methyltransferase DNMT1. Mol Cell Biol. 2005;25:11047–58. doi: 10.1128/MCB.25.24.11047-11058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saurin AJ, Shao Z, Erdjument-Bromage H, Tempst P, Kingston RE. A Drosophila Polycomb group complex includes Zeste and dTAFII proteins. Nature. 2001;412:655–60. doi: 10.1038/35088096. [DOI] [PubMed] [Google Scholar]
- 33.Shao Z, Raible F, Mollaaghababa R, Guyon JR, Wu CT, Bender W, Kingston RE. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell Biology. 1999;98:37–46. doi: 10.1016/S0092-8674(00)80604-2. [DOI] [PubMed] [Google Scholar]
- 34.Gil J, Bernard D, Martinez D, Beach D. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–77. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- 35.Whitcomb SJ, Basu A, Allis CD, Bernstein E. Polycomb Group proteins: an evolutionary perspective. Trends Genet. 2007;23:494–02. doi: 10.1016/j.tig.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 36.Maertens G, Messaoudi-Aubert S. Ubiquitin-specific proteases 7 and 11 modulate Polycomb regulation of the INK4a tumour suppressor. The EMBO Journal. 2010;29:2553–65. doi: 10.1038/emboj.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones R, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 38.Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyl-transferase activity that marks chromosomal Polycomb sites. Cell Biology. 2002;111:185–96. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 39.Müller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O’Connor MB, Kingston RE, Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell Biology. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 40.Cao RZY. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opinion in Gene and Dev. 2004;14:155–64. doi: 10.1016/j.gde.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Kuzmichev A, Tempst P, Reinberg D. Different EZH2- containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell. 2004;14:183–93. doi: 10.1016/s1097-2765(04)00185-6. [DOI] [PubMed] [Google Scholar]
- 42.Bracken AP, Dietrich N, Pasini D, Gargiulo G. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes and Dev. 2007;21:525–30. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bannister AJ, Zegerman P. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 44.Ball LJ, Murzina NV, Broadhurst RW, Raine AR, Archer SJ, Stott FJ, Murzin AG, Singh PB, Domaille PJ, Laue ED. Structure of the chromatin binding (chromo) domain from mouse modifier protein 1. EMBO J. 1997;16:273–81. doi: 10.1093/emboj/16.9.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes and Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 46.Negishi M, Saraya A, Mochizuki S, Helin K, Koseki H, Iwama A. A Novel Zinc Finger Protein Zfp277 Mediates Transcriptional Repression of the Ink4a/Arf Locus through Polycomb Repressive Complex 1. PLos One. 2010;5:e12373. doi: 10.1371/journal.pone.0012373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruggeman SW, Valk-Lingbeek ME, van der Stoop PP, Jacobs JJ, Kieboom K, Tanger E, Hulsman D, Leung C, Arsenijevic Y, Marino S, Van Lohuizen M. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes and Dev. 2005;19:1438–43. doi: 10.1101/gad.1299305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Molofsky AV, He S, Bydon M, Morrison SJ, Pardal R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes and Dev. 2005;19:1432–37. doi: 10.1101/gad.1299505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kotake YRC, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4a tumor suppressor gene. Genes and Dev. 2006;21:49–54. doi: 10.1101/gad.1499407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dietrich N, Bracken AP, Trinh E, Schjerling CK, Koseki H, Rappsilber J, Helin K, Hansen KH. Bypass of senescence by the polycomb group protein CBX8 through direct binding to the INK4A-ARF locus. EMBO J. 2007;26:1637–48. doi: 10.1038/sj.emboj.7601632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernard D, Martinez-Leal JF, Rizzo S, Martinez D, Hudson D, Visakorpi T, Peters G, Carnero A, Beach D, Gil J. CBX7 controls the growth of normal and tumor-derived prostate cells by repressing the Ink4a/Arf locus. Oncogene. 2005;24:5543–51. doi: 10.1038/sj.onc.1208735. [DOI] [PubMed] [Google Scholar]
- 52.Maertens GN, El Messaoudi-Aubert S, Racek T, Stock JK, Nicholls J, Rodriguez-Niedenführ M, Gil J, Peters G. Several Distinct Polycomb Complexes Regulate and Co-Localize on the INK4a Tumor Suppressor Locus. PloS One. 2009;4:e6380. doi: 10.1371/journal.pone.0006380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yap KL, Li S, Muñoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou MM. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol Cell. 2010;38:662–74. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lachner M, O’Caroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–20. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 55.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–24. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 56.Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–19. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 57.Terranova R, Agherbi H, Boned A, Meresse S, Djabali M. Histone and DNA methylation defects at Hox genes in mice expressing a SET domaintruncated form of Mll. Proc Natl Acad Sci. 2006;103:6629–34. doi: 10.1073/pnas.0507425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dietrich N, Bracken AP, Trinh E, Schjerling CK, Koseki H, Rappsilber J, Helin K, Hansen KH. Bypass of senescence by the polycomb group protein CBX8 through direct binding to the INK4A-ARF locus. Embo J. 2007;26:1637–48. doi: 10.1038/sj.emboj.7601632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gil J, Bernard D, Martínez D, Beach D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat Cell Biol. 2004;6:67–72. doi: 10.1038/ncb1077. [DOI] [PubMed] [Google Scholar]
- 60.Nekrasov M, Klymenko T, Fraterman S, Papp B, Oktaba K, Köcher T, Cohen A, Stunnenberg HG, Wilm M, Müller J. Pcl-PRC2 isneeded to generate high levels of H3-K27 trimethylation at Polycomb target genes. EMBO J. 2007;26:4078–88. doi: 10.1038/sj.emboj.7601837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, Isono K, Yamada D, Endo TA, Endoh M, Shinga J, Mizutani-Koseki Y, Otte AP, Casanova M, Kitamura H, Kamijo T, Sharif J, et al. Mammalian Polycomb-Like Pcl2/Mtf2 Is a Novel Regulatory Component of PRC2 That Can Differentially Modulate Polycomb Activity both at the Hox Gene Cluster and at Cdkn2a Genes. Mol Cell. 2011;31:351–64. doi: 10.1128/MCB.00259-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes and Dev. 2002;16:2893–05. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van der Vlag J, Otte AP. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat Genet. 1999;23:474–78. doi: 10.1038/70602. [DOI] [PubMed] [Google Scholar]
- 64.Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. Clinical Oncology. 2005;23:3971–93. doi: 10.1200/JCO.2005.16.600. (2005) [DOI] [PubMed] [Google Scholar]
- 65.Yamaguchi J, Sasaki M, Sato Y, Itatsu K, Harada K, Zen Y, Ikeda H, Nimura Y, Nagino M, Nakanuma Y. Histone deacetylase inhibitor (SAHA) and repression of EZH2 synergistically inhibit proliferation of gallbladder carcinoma. Japanese Cancer Asso. 2009;10:355–62. doi: 10.1111/j.1349-7006.2009.01387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ringrose L, Paro R. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development. 2007;134:223–32. doi: 10.1242/dev.02723. [DOI] [PubMed] [Google Scholar]
- 67.Schuettengruber B, Chourrout B, Vervoort M, Leblanc B, Cavalli G. Genome regulation by Polycomb and Trithorax proteins. Cell Biology. 2007;128:735–45. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 68.Klochendler-Yeivin A, Muchardt M, Yaniv M. SWI/SNF chromatin remodeling and cancer. Curr Opinion in Gene Development. 2002;12:73–79. doi: 10.1016/s0959-437x(01)00267-2. [DOI] [PubMed] [Google Scholar]
- 69.Sevenet N, Sheridan E, Amram D, Schneider D, Handgretinger R, Delattre O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet. 1999;65:1342–48. doi: 10.1086/302639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kia SK, Gorski MM, Giannakopoulos S, Verrijzer CP. SWI/SNF Mediates Polycomb Eviction and Epigenetic Reprogramming of the INK4b-ARF-INK4a Locus. Mol Cell Biol. 2008;28:3457–64. doi: 10.1128/MCB.02019-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jung JW, Lee S, Seo MS, Park SB, Kurtz A, Kang KS. Histone deacetylase controls adult stem cell aging by balancing the expression of polycomb genes and jumonji domain containing 3. Cell Mol Life Sci. 2010;7:1165–76. doi: 10.1007/s00018-009-0242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feng Y, Wang X, Xu L, Pan H, Zhu S, Liang Q, Huang B, Lu J. The transcription factor ZBP-89 suppresses p16 expression through a histone modification mechanism to affect cell senescence. FEBS J. 2009;15:4197–06. doi: 10.1111/j.1742-4658.2009.07128.x. [DOI] [PubMed] [Google Scholar]
- 73.Wang X, Feng Y, Xu L, Chen Y, Zhang Y, Su D, Ren G, Lu J, Huang B. YY1 restrained cell senescence through repressing the transcription of p16. Biochim Biophys Acta. 2008;1783:1876–73. doi: 10.1016/j.bbamcr.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 74.Gonzalez S, Serrano M. A new mechanism of inactivation of the INK4/ ARF locus. Cell Cycle. 2006;5:1382–84. doi: 10.4161/cc.5.13.2901. [DOI] [PubMed] [Google Scholar]
- 75.Song J, Sandoval R, Pilkinton MA, Tian X, Raychaudhuri P, Colamonici OR. ARF-induced downregulation of Mip130/LIN-9 protein levels mediates a positive feedback that leads to increased expression of p16Ink4a and p19Arf. Oncogene. 2010;29:1976–86. doi: 10.1038/onc.2009.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sugimoto K, Kage H, Aki N, Sano A, Kitagawa H, Nagase T, Yatomi Y, Ohishi N, Takai D. The induction of H3K9 methylation by PIWIL4 at the p16Ink4a locus. Biochem Biophys Res Comm. 2007;359:497–02. doi: 10.1016/j.bbrc.2007.05.136. [DOI] [PubMed] [Google Scholar]
- 77.Tahbaz N, Kolb FA, Zhang H, Jaronczyk K, Filipowicz W, Hobman TC. Characterization of the interactions between mammalian PAZ PIWI domain proteins and Dicer. EMBO Reports. 2004;5:189–94. doi: 10.1038/sj.embor.7400070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1079:129–57. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 79.Gil J, Peters G. Regulation of the INK4b-ARFINK4a tumor suppressor locus: All for one or one for all. Nat Rev. 2006;7:667–77. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- 80.Huang YC, Saito S, Yokoyama KK. Histone chaperone Jun dimerization protein 2 (JDP2): role in cellular senescence and aging. Kaohsiung. J Med Sci. 2010;26:515–31. doi: 10.1016/S1607-551X(10)70081-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ostrovsky O, Bengal E, Aronheim A. Induction of terminal differentiation by the c-Jun dimerization protein JDP2 in C2 myoblasts and rhabdomyosarcoma cells. J Biol Chem. 2002;277:40043–54. doi: 10.1074/jbc.M205494200. (2002) [DOI] [PubMed] [Google Scholar]
- 82.Nakade K, Pan J, Yamasaki T, Murata T, Wasylyk B, Yokoyama KK. JDP2 (Jun Dimerization Protein 2)-deficient mouse embryonic fibroblasts are resistant to replicative senescence. J Biol Chem. 2009;284:24415–24. doi: 10.1074/jbc.M808333200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Choi BY, Choi HS, Ko K, Cho Y-Y, Zhu F, Kang BS, Ermakova SP, Ma W-Y, Bode AM, Dong Z. The tumor suppressor p16INK4a prevents cell transformation through inhibition of c-Jun phosphorylation and AP-1 activity. Nat Struct Mol Biol. 2005;12:699–07. doi: 10.1038/nsmb960. [DOI] [PubMed] [Google Scholar]
- 84.Witcher M, Emerson BM. Epigenetic Silencing of the p16INK4a Tumor Suppressor is Associated with Loss of CTCF Binding and a Chromatin Boundary. Mol Cell. 2009;34:271–84. doi: 10.1016/j.molcel.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raisner RM, Madhani HD. Patterning chromatin: form and function for H2A.Z variant nucleosomes. Curr Opin Genet Dev. 2006;16:119–24. doi: 10.1016/j.gde.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 86.Filippova GN. Genetics and epigenetics of the multifunctional protein CTCF. Curr Top Dev Biol. 2008;80:337–60. doi: 10.1016/S0070-2153(07)80009-3. [DOI] [PubMed] [Google Scholar]
- 87.Wallace JA, Felsenfeld G. We gather together: insulators and genome organization. Curr Opin Genet Dev. 2007;17:400–07. doi: 10.1016/j.gde.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffold. J Cell Sci. 2001;114:2363–73. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- 89.Wang X, Pan L, Feng Y, Wang Y, Han Q, Han L, Han S, Guo J, Huang B, Lu J. p300 plays a role in p16INK4a expression and cell cycle arrest. Oncogene. 2008;27:1894–04. doi: 10.1038/sj.onc.1210821. [DOI] [PubMed] [Google Scholar]
- 90.Gan Q, Huang J, Zhou R, Niu J, Zhu X, Wang J, Zhang Z, Tong T. PPAR accelerates cellular senescence by inducing p16INK4 expression in human diploid fibroblasts. J Cell science. 2008;131:2235–45. doi: 10.1242/jcs.026633. [DOI] [PubMed] [Google Scholar]
- 91.Lin AW, Barradas M, Stone JC, Van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes and Dev. 1998;12:3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, Pehrson JR, Berger JM, et al. Formation of macroH2A-containing senescence- associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005;8:19–31. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 93.Foos G, García-Ramírez JJ, Galang CK, Hauser CA. Elevated expression of Ets2 or distinct portions of Ets 2 can reverse ras mediated celullar transformation. J Biol Chem. 1998;273:18871–80. doi: 10.1074/jbc.273.30.18871. [DOI] [PubMed] [Google Scholar]
- 94.Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, Sharrocks AD, Peters G, Hara E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–70. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- 95.Xiu M, Kim J, Sampson E, Huang CY, Davis RJ, Paulson KE, Yee AS. The transcriptional repressor HBP1 is a target of the p38 mitogen-activated protein kinase pathway in cell cycle regulation. Mol Cell. 2003;23:8890–901. doi: 10.1128/MCB.23.23.8890-8901.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Barradas M, Anderton E, Acosta JC, Li S, Banito A, Rodriguez-Niedenführ M, Maertens G, Banck M, Zhou MM, Walsh MJ, Peters G, Gil J. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes and Dev. 2009;23:1177–82. doi: 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yamakoshi K, Takahashi A, Hirota F, Nakayama R, Ishimaru N, Kubo Y, Mann DJ, Ohmura M, Hirao A, Saya H, Arase S, Hayashi Y, et al. Real-time in vivo imaging of p16Ink4a reveals cross talk with p53. J Cell Biol. 2009;186:393–407. doi: 10.1083/jcb.200904105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Cell Biol. 2007;8:275–83. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 99.Zhang D, Shimizu T, Araki N, Hirota T, Yoshie M, Ogawa K, Nakagata N, Takeya M, Saya H. Aurora A overexpression induces cellular senescence in mammary gland hyperplastic tumors developed in p53-deficient mice. Oncogene. 2008;27:4305–14. doi: 10.1038/onc.2008.76. [DOI] [PubMed] [Google Scholar]
- 100.Jung MS, Jin DH, Chae HD, Kang S, Kim SC, Bang YJ, Choi TS, Choi KS, Shin DY. Bcl-xL and E1B-19K proteins inhibit p53- induced irreversible growth arrest and senescence by preventing reactive oxygen species-dependent p38 activation. J Biol Chem. 2004;279:17765–71. doi: 10.1074/jbc.M305015200. [DOI] [PubMed] [Google Scholar]
- 101.Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells. 2003;8:131–34. doi: 10.1046/j.1365-2443.2003.00620.x. [DOI] [PubMed] [Google Scholar]
- 102.Fujikawa M, Katagiri T, Tugores A, Nakamura Y, Ishikawa F. ESE-3, an Ets family transcription factor, is up-regulated in cellular senescence. Cancer Sci. 2007;98:1468–75. doi: 10.1111/j.1349-7006.2007.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–11. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]
- 104.Tsygankov D, Liu Y, Sanoff HK, Sharpless NE, Elston TC. A quantitative model for age-dependent expression of the p16INK4a tumor suppressor. Proc Natl Acad Sci USA. 2009;106:16562–67. doi: 10.1073/pnas.0904405106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nielsen GP, Stemmer-Rachamimov AO, Shaw J, Roy JE, Koh J, Louis DN. Immunohistochemical survey of p16INK4A expression in normal human adult and infant tissues. Lab Invest. 1999;79:1137–43. [PubMed] [Google Scholar]
- 106.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–57. doi: 10.1038/nature05092. (2006) [DOI] [PubMed] [Google Scholar]
- 107.Chen J, Astle CM, Harrison DE. Genetic regulation of primitive hematopoietic stem cell senescence. Exp Hematology. 2000;28:442–50. doi: 10.1016/s0301-472x(99)00157-5. [DOI] [PubMed] [Google Scholar]
- 108.Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–77. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- 109.Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell selfrenewal from progenitor proliferation. Nature. 2003;425:962–67. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, Jansen-Dürr P, Wlaschek M. p16INK4Ais a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006;5:379–89. doi: 10.1111/j.1474-9726.2006.00231.x. [DOI] [PubMed] [Google Scholar]
- 111.Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during Cellular Senescence. Cell. 2003;113:703–16. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 112.Di Micco R, Sulli G, Dobreva M, Liontos M, Botrugno OA, Gargiulo G, dal Zuffo R, Matti V, d’Ario G, Montani E, Mercurio C, Hahn WC, et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol. 2011;13:292–02. doi: 10.1038/ncb2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a) Cell Cycle. 2011;10:457–68. doi: 10.4161/cc.10.3.14707. [DOI] [PubMed] [Google Scholar]
- 114.Kim HJ, Kim K, Yu BP, Chung HY. The effect of age on cyclooxygenases-2 gene expression: NF-KB activation and IKK degradation. Free Radic Biol Med. 2000;28:683–92. doi: 10.1016/s0891-5849(99)00274-9. [DOI] [PubMed] [Google Scholar]
- 115.Haddad JJ. Science review: Redox and oxygen-sensitive transcription factors in the regulation of oxidant-mediated lung injury: role for nuclear factor-kappa B. Crit Care. 2002;6:481–90. doi: 10.1186/cc1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Handel ML, McMorrow LB, Gravallese EM. Nuclear factor-kappa B in rheumatoid synovium. Localization of p50 and p65. Arthritis Rheum. 1995;38:1762–70. doi: 10.1002/art.1780381209. [DOI] [PubMed] [Google Scholar]
- 117.Poynter ME, Daynes RA. Peroxisome proliferatoractivated receptor activation modulates cellular redox status, represses nuclear factor-kB signaling, and reduces inflammatory cytokine production in aging. J Biol Chem. 1998;273:32833–41. doi: 10.1074/jbc.273.49.32833. [DOI] [PubMed] [Google Scholar]
- 118.Lee MY, Wang Y, Vanhoutte PM. Senescence of Cultured Porcine Coronary Arterial Endothelial Cells Is Associated with Accelerated Oxidative Stress and Activation of NFK B. J Vas Res. 2009;47:287–98. doi: 10.1159/000265563. [DOI] [PubMed] [Google Scholar]
- 119.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc Natl Acad Sci USA. 2001;11:12072–7. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibrobalsts. Science. 1997;275:1649–52. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 121.Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, Yu Ferrans VJ, Howard BH, Finkel T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274:7936–40. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- 122.Rowland BD, Denissov SG, Douma S, Stunnenberg HG, Bernards R, Peeper DS. E2F transcriptional repressor complexes are critical downstream targets of p19(ARF)/p53-induced proliferative arrest. Cancer Cell. 2002;2:55–65. doi: 10.1016/s1535-6108(02)00085-5. [DOI] [PubMed] [Google Scholar]
- 123.Leong WF, Chau JF, Li B. p53 Deficiency leads to compensatory up-regulation of p16INK4a. Mol Cancer Res. 2009;7:354–60. doi: 10.1158/1541-7786.MCR-08-0373. [DOI] [PubMed] [Google Scholar]
- 124.Su X, Cho MS, Gi YJ, Ayanga BA, Sherr CJ, Flores ER. Rescue of key features of the p63-null epithelial phenotype by inactivation of Ink4a and Arf. EMBO J. 2009;28:1904–15. doi: 10.1038/emboj.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guarente L. Sirtuins, aging, and Medicine. N Engl J Med. 2011;364:2235–44. doi: 10.1056/NEJMra1100831. [DOI] [PubMed] [Google Scholar]