

Abstract
This paper describes a method for the selective precipitation and purification of a monovalent protein (carbonic anhydrase is used as a demonstration) from cellular lysate using ammonium sulfate and oligovalent ligands. The oligovalent ligands induce the formation of protein-ligand aggregates, and at an appropriate concentration of dissolved ammonium sulfate, these complexes precipitate. The purification involves three steps: i) the removal of high-molecular weight impurities through the addition of ammonium sulfate to the crude cell lysate; ii) the introduction of an oligovalent ligand and the selective precipitation of the target protein-ligand aggregates from solution; and iii) the removal of the oligovalent ligand from the precipitate by dialysis to release the target protein. The increase of mass and volume of the proteins upon aggregate formation reduces their solubility, and results in the selective precipitation of these aggregates. We recovered human carbonic anhydrase, from crude cellular lysate, in 82% yield and 95% purity with a trivalent benzene sulfonamide ligand. This method provides a chromatography-free strategy of purifying monovalent proteins—for which appropriate oligovalent ligands can be synthesized—and combines the selectivity of affinity-based purification with the convenience of salt-induced precipitation.
Introduction
The solubility of a protein in an aqueous solution depends on the complex interaction of four parameters: i) its physical properties (shape, flexibility, molecular weight, and isoelectric point), ii) the distribution of the hydrophobic, hydrophilic, and charged groups on its surface, iii) the temperature and pH of the solution, and iv) the composition and concentration of various co-solutes.1 Current theory suggests the relative importance of each of these parameters in determining the solubility of a protein. While the theory is unable to predict protein solubility in an experimental context,2 the purification of proteins by precipitation is often a very convenient procedure, despite the fact that it remains a largely empirical process.3,4
This paper describes a method for the precipitation of (and thus, the purification of) monomeric proteins, selectively, with a combination of oligovalent ligands and ammonium sulfate. The interaction of multiple ligands—where a ligand is defined as a small molecule that specifically binds to a protein or receptor of interest—attached to a single entity with multiple receptors on another entity is common in biology and, especially, immunology: a multivalent interaction. We define an oligovalent ligand, in the context of this work, as a single organic molecular scaffold containing less than ten ligands, of the same chemical structure, that target a single protein receptor.
The formation of a protein-ligand aggregate (i.e., multiple proteins interacting with a single oligovalent ligand) increases the molecular mass and volume of the protein of interest and decreases the solubility of the aggregate; this increase allows the aggregate to be removed from solution as a precipitate (Scheme 1). Modulating the solubility of a given protein—by introducing oligovalent ligands that form protein-ligand aggregates of known stoichiometries—provides a strategy for purifying proteins, for which appropriate ligands are available or can be synthesized without the need of chromatographic methods.
Scheme 1.
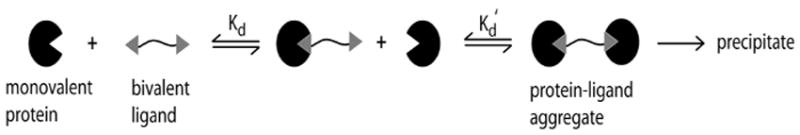
The selective precipitation proteins with oligovalent ligands. The dissociation constant (Kd) dictates the formation and stability of a bivalent protein-ligand aggregate, in which a bivalent ligand of known binding affinity is introduced into a solution containing the protein. The Kd of the monovalent and bivalent complex is assumed to be the same in this system. The increase in mass (and volume) of the aggregates, in solution, results in the decreased solubility of the protein(s), and promote precipitation.
The salt-induced precipitation of proteins is a simple, rapid, and inexpensive method of purification.3-5 Many proteins have similar solubility in aqueous solutions of a salt, however, and precipitate simultaneously; salt-induced precipitation of proteins is, for this reason, generally the first step in a series of steps of purification. The lack of selective and simple protein purification methods has led to the development of chromatographic techniques (e.g., ion exchange, size-exclusion, and affinity).3-5 Affinity chromatography is a selective method for purifying proteins when a tight-binding (Kd < 100 μM) ligand for the protein is known; affinity chromatography, however, i) is time-consuming, ii) requires large volumes of eluent to remove the protein from the solid support (thus diluting the sample), iii) requires pumps and fraction collectors (as well as supporting auxiliary equipment), and iv) is limited in throughput. An ideal method of purification would have the selectivity of affinity chromatography, but the speed, low cost, and throughput of salt-induced precipitation.
We and others have developed several methods that aim to combine the convenience of salt-induced precipitation with the selectivity of affinity chromatography. Bilgiçer et al. used a combination of bivalent ligands and ammonium sulfate (AMS) precipitation to purify antibodies from ascites fluid with yields of > 80% and with > 95 % purity.6
Another method of purification—based on altering the solubility of proteins—is “affinity precipitation”,5,7-9 which relies on multivalent ligands to modify the solubility of proteins, but does not involve the addition of AMS. Affinity precipitation was developed initially for the selective precipitation of multimeric proteins with bivalent ligands; the ligands cause precipitation by inducing the formation of an insoluble, cross-linked, macromolecular network of the protein complex.7 This strategy was utilized to purify monomeric proteins with polymeric scaffolds that contain both an affinity ligand and another functional group that controls the solubility of the polymer.8,9 The solubility of the polymeric ligand can be regulated, typically, by changes in pH, temperature, or ionic strength of the medium.10 This method has three limitations: i) the ligands immobilized on a polymeric scaffold tend to bind less strongly than their monovalent analogs; ii) the resolubilization of the protein in the presence of the polymer is slow; and iii) the cross-linked aggregates of proteins and polymers can trap impurities during precipitation.
Selective Isolation of Protein by Combining Oligovalent Aggregation with Ammonium Sulfate (AMS) Precipitation
This paper describes a method that combines the selectivity of affinity-based methods with the convenience of AMS precipitation. We use non-polymeric, oligovalent ligands (with molecular weights < 5000 Da) to induce the aggregation of monomeric proteins—containing a single ligand binding site—by selectively binding the protein to the oligovalent small molecules. The formation of a higher molecular-weight aggregate, at a concentration of ammonium sulfate that is just below that required to precipitate the monomeric protein, selectively converts the targeted protein from a soluble monomer to an insoluble aggregate.
The procedure has three steps (Figure 1): i) impurities that are higher in molecular weight than the target protein are removed by precipitation with AMS (the concentration of AMS is such that the protein of interest does not precipitate); ii) the target protein is caused to aggregate and precipitate by adding an oligovalent ligand; the precipitate is isolated and redissolved in solution by the addition of buffer that does not contain AMS; iii) the ligand is removed by dialysis.
Figure 1.
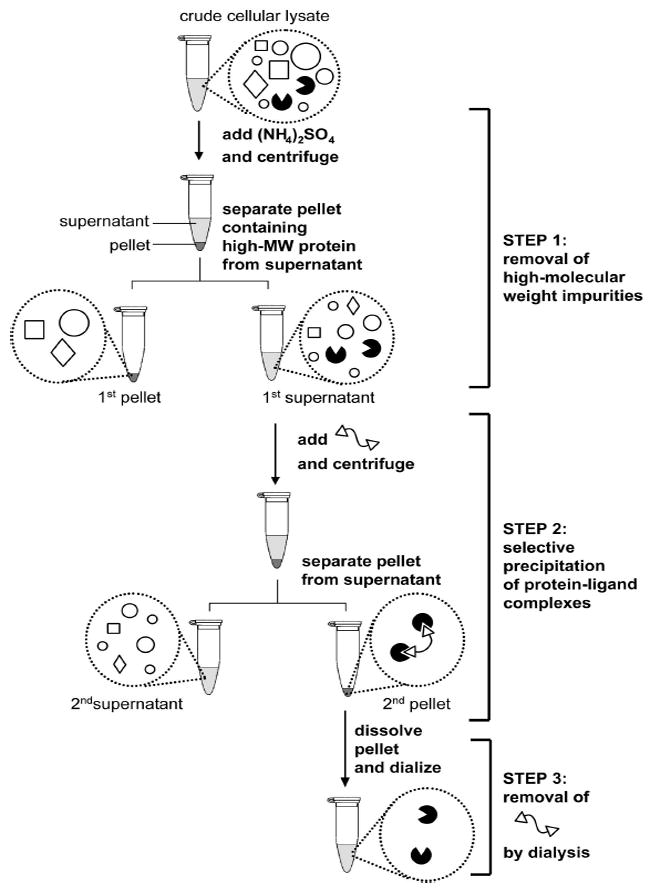
General strategy for purifying proteins using oligovalent ligands. The ligands induce aggregation of monovalent proteins and reduce their solubility in aqueous solutions containing dissolved ammonium sulfate.
This technique of purifying proteins has five useful characteristics: i) it is selective; ii) it can be adapted easily to improve the efficiency of existing processes that rely on salt-induced precipitation; iii) it is rapid; iv) it should scale easily to large volumes (> 1L); and v) it both purifies and concentrates the target protein. This method also has limitations: i) it requires both a small-molecule ligand that binds tightly (Kd < 100 μM) to the target protein, and a practical synthetic route to prepare the oligovalent ligand; ii) it requires separating the protein from the ligand after precipitation. In general, i) is the most constraining of these limitations.
Experimental Design and Rationale
Choice of Protein-Ligand System
We chose carbonic anhydrase (CA, E.C. 4.2.1.1) as our model system as the binding of benzene sulfonamides to CA is conserved, has been extensively studied, and is well characterized with X-ray crystallography. There are numerous benzene sulfonamides that are available commercially, with a range of binding affinities, that are attached easily to oligovalent scaffolds.11,12 We have reviewed the relevant characteristics of CA as a model system for biophysical studies in great depth,11 and have extensive experience studying the interaction of benzene sulfonamide ligands with CA.
In this study, we precipitated bovine carbonic anhydrase II (BCA), selectively, from buffered solutions with oligovalent benzene sulfonamide ligands (monovalent to trivalent). We used pure BCA as a model system to examine the relationships between the relevant experimental parameters (e.g., dissociation (Kd) constants of the ligand, the stoichiometry of the aggregate, and the concentration of AMS). We also purified human carbonic anhydrase II (HCA), selectively, from crude E. coli lysate with a trivalent benzene sulfonamide ligand.
Choice of Salt
We precipitate aggregates of CA and an oligovalent benzene sulfonamide ligand in the presence of AMS. This salt is commonly used in preparative protein purification3-5 as it is commercially available in high purity (> 99.9 %) and low cost (< $0.01 per gram), is soluble in water at concentrations up to 4 M, possesses a low enthalpy of dissolution, and is non-buffering and non-denaturing.
Oligovalent Ligand Scaffolds
We linked the benzene sulfonamide molecules with units of oligoethylene glycol (OEG) because of their flexibility and relative inertness. We synthesized the oligovalent ligands (Figure 2) from commercially available starting materials, by linking the nucleophilic amine of the OEG scaffold with the N-hydroxysuccinimide (NHS)-activated 4-carboxybenzenesulfonamide (unless otherwise noted) using standard condensation chemistries.13 We also synthesized a monovalent control (Figure 2) to show that the presence of the oligoethylene glycol tail does not inhibit protein-ligand binding and, in turn, interfere with protein precipitation. The Supporting Information outlines the details of the synthetic procedures used to make each benzene sulfonamide ligand, as well as their corresponding 1H NMR and mass spectra.
Figure 2.
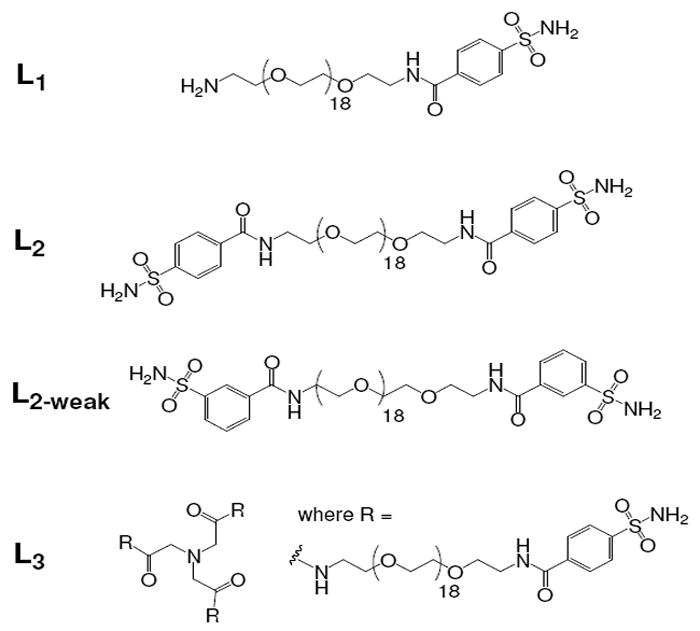
Structures of ligands used for selective precipitation of CA from aqueous solutions of ammonium sulfate.
Results and Discussion
Characterization of the Binding of the Oligovalent Ligands to CA
We measured the monovalent dissociation constant (Kd) of each of the benzene sulfonamide scaffolds from BCA with a fluorescence-based competition assay.14-16 To determine Kd, we used a model that assumed independent and equivalent binding of each benzene sulfonamide to BCA. This model yielded the following monovalent dissociation constants for the benzene sulfonamides within these ligands, from BCA: Kd(monovalent, L1) = 0.5 ± 0.1 μM, Kd(bivalent, L2) = 0.4 ± 0.1 μM, Kd(trivalent, L3) = 0.6 ± 0.6 μM.
Effect of Oligovalent Ligands on the Solubility of Proteins in Aqueous Solutions of AMS
We precipitated BCA—in a buffered solution containing ammonium sulfate, ranging from 1.5 – 3.6 M—in the presence of no ligand, L1, L2, and L3 (Figure 3). In these experiments, we combined BCA, AMS, and the ligand in 20 mM NaH2PO4 buffer, pH = 7.5 (see Supporting Information for details of this experiment), vortexed this mixture for ~10 s to promote rapid mixing, gently agitated the mixture using a rocker-type shaker for 4 hours at 4 °C to allow sufficient time for precipitation to occur,17 and then centrifuged the mixture to separate the precipitate from the supernatant. We resuspended the pellet in 20 mM NaH2PO4 buffer (pH = 7.5) and determined the concentration of protein in the pellet with UV-VIS absorption measurements. Figure 3 reports the fraction of protein recovered in the pellet in relation to the total amount of protein.
Figure 3.
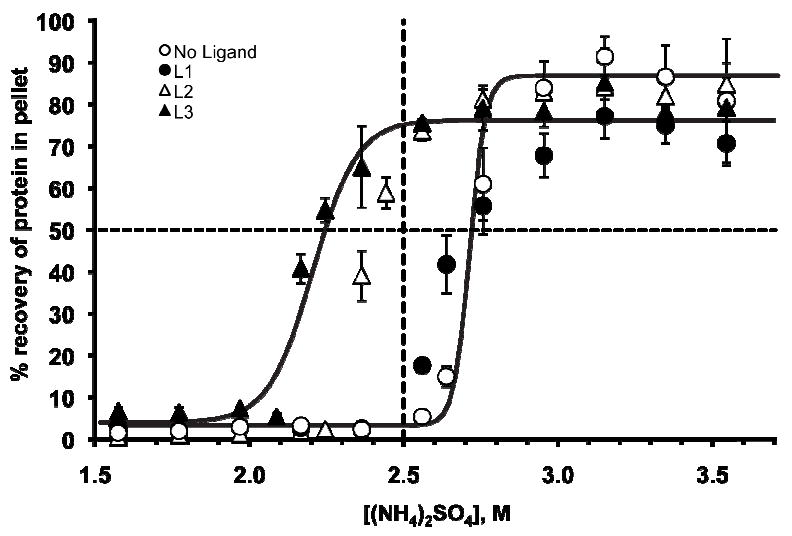
The precipitation of BCA from AMS in Tris-Gly buffer (pH = 7.5, 4 °C) in the presence of L1, L2, L3 and no ligand. The concentration of each component is: [BCA]0 = 15 μM, [L1]0 = 90 μM, [L2]0 = 45 μM, and [L3]0 = 30 μM. The ratio of ligand to protein was optimized empirically to maximize the yield of precipitate. The error bars represent the deviation from three independent measurements; for clarity, we have only included the best fits (solid lines) for the experiments with no added ligand, and with L3. The horizontal dashed line represents the point at which fifty percent of the BCA was precipitated from solution and the vertical dashed line corresponds to 2.5 M AMS.
Figure 3 illustrates the ligand-dependent solubility for BCA in AMS; the solubility of BCA decreases as the size of the putative oligovalent aggregate increases for a given concentration of AMS. The horizontal dashed line in Figure 3 corresponds to the concentration of AMS required to precipitate 50% of the BCA present in solution. The vertical dashed line shows that no BCA precipitates in the absence of ligand at a concentration of 2.5 M, whereas over 70% of the BCA precipitates in the presence of the bi- and tri-valent ligand. Together, these results serve as a basis for the expected selectivity, and thus selective precipitation, for a protein in a complex mixture.
Experimental Parameters for Protein Precipitation
The choice of experimental parameters—the type of buffer, the pH of the AMS solution, the length of OEG linker, the Kd of the ligand, and the concentration of protein in solution—may all affect the efficiency of protein precipitation with oligovalent ligands from a solution of AMS.
Linker length
The distance between binding sites, in an oligovalent ligand, must sufficiently separate the proteins of interest to enable the oligovalent aggregate to form. We estimated—based on X-ray crystallographic structural information of CA—that a bivalent ligand must separate the two binding sites of CA by a total distance of 3 nm, and hypothesized that combinations of ligands and linkers that fall short of this 3 nm requirement would fail to promote the precipitation of CA from an AMS solution. We synthesized several analogs of the L2 ligand (Figure 2) with linkers of various oligoethylene glycol (OEG) units: OEG 1, OEG 2, OEG 10, and OEG 19. We found that L2 connected by OEG 19 (~5.5 nm between each ligand) was the most efficient at reducing the solubility of BCA in AMS, and that analogs having shorter linkers of OEG were less effective, and less reproducible.18
Protein concentration and Kd
We examined the relationship between the Kd of the ligand and the concentration of protein. We expected that the ligand would determine the solubility of the protein if the protein-ligand aggregate were thermodynamically stable (e.g., had low values of Kd and Kd′), as the formation of a precipitate drives the equilibrium toward the formation of the bivalent aggregate (Scheme 1). We previously proposed an analytical model to predict the concentration of bivalent ligand needed to form the maximum amount of the protein-ligand complex in a solution containing the monomeric, monovalent protein.19 Eq. 1 describes the optimum concentration of bivalent ligand needed to maximize the formation of a protein-ligand complex19 between L2 (Kd = 0.4 ± 0.1 μM) and 20 μM of BCA:
| (Eq. 1) |
We designed two experiments to compare the concentration of L2 needed (as determined experimentally) with the values predicted by Eq. 1: (i) we examined aggregate precipitation at several concentrations of BCA (2 – 20 μM) with a fixed concentration (40 μM) of L2; (ii) we synthesized a chemical analog of L2 (L2-weak, Figure 2) with a reduced Kd: Kd(L2) = 0.4 ± 0.1 μM vs. Kd(L2-weak) = 10.8 ± 0.7 μM.
The precipitation of BCA with L2 was most effective for the highest concentration of BCA (20 μM): 4 equivalents of L2 per mole of BCA. This concentration of ligand exceeds the value predicted by Eq. 1 by a factor of 4. We attribute the disparity in the observed and predicted optimum concentrations of L2 to differences in the experimental conditions and the conditions assumed in the model.19 Eq. 1 applies to binding processes under equilibrium conditions where the individual binding partners and their binding complexes are dispersed uniformly in solution. The system we present here is not under equilibrium conditions, and thus we do not expect the equilibrium model described by Mack et al.19 to predict the exact concentration of ligand needed, as the: i) the binding occurs in a highly concentrated AMS solution that promotes the aggregation and precipitation of the protein-ligand complexes from solution; ii) the kinetic process of precipitation alters the concentration of protein, ligand, and their complex in solution during the course of the experiment. We see this model, however, as a means of predicting a starting point for semi-empirical experimental optimization.
L2-weak was less effective than L2 (Figure 2) at promoting the precipitation of 20 μM BCA from a solution of AMS. Less than 20% of the BCA (20 μM) precipitated in the presence of a large excess of L2-weak ligand (e.g., 200 μM, 20 equivalents of ligands per mole of BCA), a result that agrees, qualitatively, with Eq. 1: the lower the value of Kd for a ligand, the lower the concentration of ligand is needed to aggregate and precipitate the protein.
Solution pH and buffer conditions
We examined the effect of the buffer composition and pH on protein precipitation. We anticipated that neither would influence precipitation; the excess of AMS dominates the ionic strength of the aqueous solutions used for precipitation. We confirmed this hypothesis experimentally, and conclude that both the type of salt used to buffer the AMS solution, and the pH of solution (6.0 – 7.5) has negligible effects on the efficiency of this precipitation process.20
Selective Precipitation of CA from Cellular Lysate
We selectively precipitated human CA (HCA) from a complex mixture of other proteins (i.e., crude lysate) with a trivalent ligand. First, we added a known amount of HCA to E. coli lysate (1 mg / 10 mL, 3.4 μM); this experiment allowed us to quantify the efficiency of the precipitation process. In a separate experiment, we purified HCA from crude E. coli lysate in which the protein was over-expressed.11, 21 The selective precipitation of over-expressed protein from cellular lysate provides a means of purifying protein produced on a small scale without the need of chromatography.
To determine the optimal salt concentration required to precipitate the majority of the proteins in the lysate while retaining HCA in the supernatant, we incubated E. coli lysate—containing a known amount of HCA—in different concentrations of AMS (1.6 – 3.2 M, final concentration in 1.0 mL samples). We resuspended the precipitate in an equal volume of phosphate buffered saline (1X PBS), determined the amount of protein contained in the supernatant and in the precipitate with SDS-PAGE, and quantified the total protein concentration for each supernatant and precipitate sample with a standard Bradford assay.22 The majority of the proteins from the cellular lysate samples were precipitated (62% of the total proteins, as determined with a standard Bradford assay) in 2.4 M AMS, while greater than 70% of the HCA was retained in solution at that concentration of AMS, as estimated by SDS PAGE.
To determine the optimum ligand concentration needed to precipitate a known amount of HCA in cellular lysate (100 μg of HCA total; 3.4 μM), we added known amounts of trivalent ligand (0, 5, 12.5 and 25 μM) to a 1.0 mL suspension of E. coli supernatant in 2.4 M AMS, and rocked the mixture at 4 °C for 4 hours.17 The precipitate contained a small amount of unwanted proteins, which we removed by washing and recentrifuging the pellet: the pellet was resuspended in 100 μL of 2.4 M AMS, centrifuged, and the supernatant removed. The pellet, precipitated with the trivalent ligand, contained 82 ± 2 % of the HCA (n = 7 measurements) in the cellular lysate, and was >95% pure, as judged by gel electrophoresis. A fraction of the total HCA, 11 ± 4 % (n = 7 measurements) was precipitated in the absence of ligand.
We also precipitated a sample of over-expressed HCA from crude E. coli lysate.11, 21 Figure 4 summarizes the results of a representative precipitation experiment, as analyzed with SDS PAGE, in which 50 μM of the trivalent ligand was added to 1.0 mL of E. coli crude lysate, the sample was incubated at 4 °C for four hours, and the protein-ligand aggregate precipitated. In this case, addition of trivalent ligand precipitated HCA that was ~90% pure, and a single wash of the precipitated protein provided HCA that was >95% pure as determined via Bradford assay.
Figure 4.
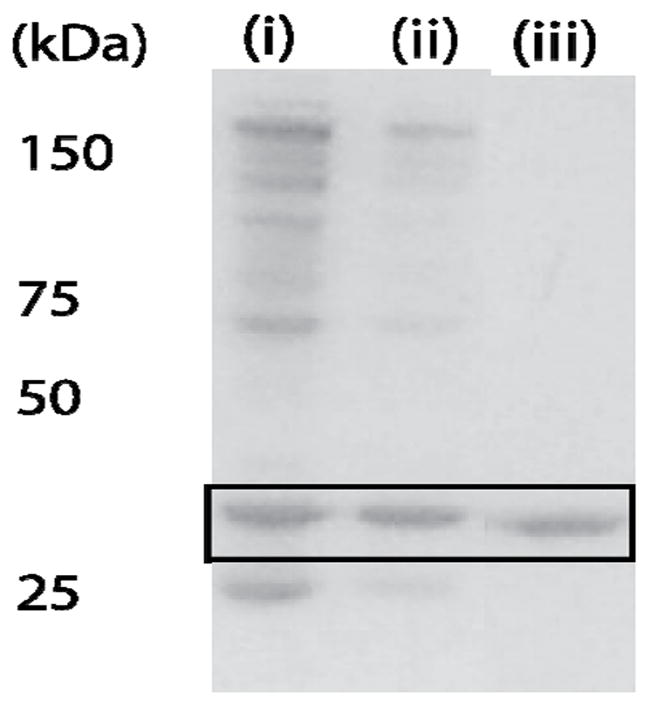
Precipitation of HCA with AMS and L3 from a cellular lysate. Images of SDS PAGE lanes showing (i) the total number of proteins remaining in 2.4 M AMS after centrifugation, (ii) the total number of proteins precipitated after the addition of 50 μM L3 ligand, (iii) the remaining HCA after the pellet of precipitated protein obtained from (ii) was washed with 1X PBS solution. The black box marks the bands corresponding to HCA.
Formation of an oligomeric aggregate between HCA and an oligovalent ligand is necessary for selective precipitation
We performed an experiment analogous to that presented in Figure 3 and precipitated a known amount of HCA (100 μg; 3.4 μM)—in the presence of L1, L2, and L3—from 2.5 mL of crude E. coli lysate. Each reaction contained an equivalent amount of benzene sulfonamide groups available for binding to HCA (i.e., 75 μM L1, 37.5 μM L2, and 25 μM L3). A control experiment was also performed in which HCA was precipitated in the absence of ligand. We washed the precipitated pellets containing aggregates of protein and ligand to remove non-specifically bound proteins and then analyzed the precipitates qualitatively with SDS PAGE, and quantitatively with a Bradford assay: L1 ~ 15 ± 6 %, L2 = 55 ± 3 %, L3 = 82 ± 2 % of the total HCA contained in each lysate sample. These data are consistent with those obtained for the BCA samples (Figure 3); the amount of protein recovered increased with the size of the protein-ligand aggregate.
A small number of unwanted proteins were present in both the bi- and trivalent ligand precipitates, but were successfully removed by a single washing step. The total protein content of each pellet was determined directly after precipitation, after a single washing step, and after four washing steps. The trivalent pellet was ~90 % pure HCA after precipitation, before any wash steps. The amount of HCA present in the pellet was not statistically different after one and four washes; only a single band was present in the SDS PAGE.
The monovalent ligand precipitated 25 ± 4 % of the total protein contained in the E. coli lysate, while 11 ± 4 % of the protein was precipitated in the absence of ligand. L1 should not induce the precipitation of HCA from solution, as the mass and volume of the protein-ligand aggregate has not changed significantly. The majority of the precipitated proteins for L1, and for experiments containing no added ligand were not HCA, as determined with SDS PAGE. We repeatedly washed the L1 precipitate several times with 1X PBS to remove the non-specifically adsorbed proteins; the pellet contained ~ 15 ± 6 % of the HCA after four washes. This value is a relative estimate, as bands from protein impurities were still evident in the SDS PAGE.
Considerations for the Design of an Oligovalent Ligand
The design of the oligovalent ligand determines its compatibility with the strategy—the selective precipitation and purification with an oligovalent ligand, via the formation of an aggregate of the protein and the ligand—presented here. The ligand also determines the efficiency of the formation of the stable protein-ligand aggregate that precipitates in the presence of AMS. There are three important parameters that must be considered before designing and synthesizing the oligovalent ligand: i) the Kd of the ligand, ii) the number of ligands attached to a central scaffold (i.e., the valency of the ligand), and iii) the length of the linker that connects the individual ligands.
The value of Kd of the ligand—the strength the protein-ligand association—determines the concentration of ligand needed to precipitate an aggregate composed of the monovalent protein and the ligand from the AMS solution. An increase in the Kd of the bivalent ligand resulted in a reduced amount of BCA precipitating from a 2.36 M AMS solution: 60% of the BCA in solution precipitated in the presence of 40 μM of the L2 ligand (Kd = 0.4 μM) while 20% of the BCA in solution precipitated in the presence of 200 μM of L2-weak (Kd = 10 μM).
The amount of precipitated protein increases with the valency of the oligovalent ligand; the number of ligands present corresponds to an increased mass and volume of the protein-ligand aggregate. The trend observed for mono-, bi-, and trivalent ligands is a decrease in the solubility of the aggregate with an increase in valency of the ligand. The synthesis of oligovalent ligands with higher valency should further decrease the solubility of the protein-ligand aggregate, thereby, in principle, increasing the amount of precipitated protein.
Lengthening the linker that connects the individual benzene sulfonamide molecules to the oligovalent ligand, increased the efficiency of BCA precipitation. For our purification strategy to be successful, the length of the linker must be long enough to span the distance between the binding sites of two protein molecules without enthalpic penalty from steric interactions between atoms of the proteins. This distance, of course, will depend on the structure and size of the protein of interest.
Conclusions
This paper describes the combination of oligovalent ligands and AMS for the selective precipitation and purification of a model protein, carbonic anhydrase, from cellular lysate. An optimized procedure produced HCA in 82 % yield, and approximately 95% purity.
This method combines the ease and speed of salt-induced precipitation with the selectivity of affinity chromatography and offers a rapid and selective approach to purifying monomeric proteins that have known ligands. This method has five useful characteristics for protein purification: i) it is rapid (formation of precipitate occurs within minutes); ii) it is selective for target protein, and ensures that the purified protein contains intact active site; iii) it is mild and non-denaturing (AMS promotes folding of proteins and protects proteins from degradation by proteases); iv) it concentrates the protein during purification; v) it should scale easily to large volumes. This method has also two limitations: i) it requires a ligand (with a sufficiently low Kd, < 100 μM) that binds to the protein; ii) it involves the separation of the ligand from the protein after purification.
We believe this method may be particularly attractive for: i) improving the efficiency of existing purification processes that rely on salt-induced precipitation; ii) streamlining purification schemes that involve a series of chromatographic and non-chromatographic steps; iii) replacing affinity chromatography to reduce the cost, time, and volume of eluent in purification. This method provides a simple means that can be applied to the routine purification of proteins of interest, at natively- and over-expressed concentrations, from cellular lysate.
Supplementary Material
Acknowledgments
This work was partially supported by NIH award GM030367 and the Wyss Institute of Biologically Inspired Engineering. NDS thanks the NIH for a postdoctoral fellowship. K.A.M. thanks Başar Bilgiçer for helpful discussions and assistance with synthesizing L3, Scott T. Phillips for helpful discussions and help synthesizing L2, and Demetri T. Moustakas for his help with Figure S5.
Footnotes
Supporting Information Available. General methods; detailed synthetic procedures and 1H NMR and mass spectra; additional figures (Figures S1 – S8). This material is available free of charge via Internet at http://pubs.acs.org.
References
- 1.Arakawa T, Timasheff SN. Theory of Protein Solubility. Methods Enzymol. 1985;114:49–77. doi: 10.1016/0076-6879(85)14005-x. [DOI] [PubMed] [Google Scholar]
- 2.Chiew YC, Kuehner D, Blanch HW, Prausnitz JM. Molecular Thermodynamics for Salt-Induced Protein Precipitation. AIChE J. 1995;41:2150–2159. [Google Scholar]
- 3.Hatti-Kaul R, Mattiasson B. Isolation and purification of proteins. Marcel Dekker; New York: 2003. [Google Scholar]
- 4.Wheelwright SM. Protein purification: design and scale up of downstream processing. Hanser Publishers; Distributed in the U.S.A. and in Canada by Oxford University Press; New York: 1991. [Google Scholar]
- 5.Scopes RK. Protein purification: principles and practice. Springer-Verlag; New York: 1994. [Google Scholar]
- 6.Bilgicer B, Thomas SW, Shaw BF, Kaufman GK, Krishnamurthy VM, Estroff LA, Yang J, Whitesides GM. A Non-Chromatographic Method for the Purification of a Bivalently Active Monoclonal IgG Antibody from Biological Fluids. J Am Chem Soc. 2009;131:9361–9367. doi: 10.1021/ja9023836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flygare S, Griffin T, Larsson PO, Mosbach K. Affinity Precipitation of Dehydrogenases. Anal Biochem. 1983;133:409–416. doi: 10.1016/0003-2697(83)90102-1. [DOI] [PubMed] [Google Scholar]
- 8.Gupta MN, Kaul R, Guoqiang D, Dissing U, Mattiasson B. Affinity Precipitation of Proteins. J Mol Recognit. 1996;9:356–359. doi: 10.1002/(sici)1099-1352(199634/12)9:5/6<356::aid-jmr328>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 9.Hilbrig F, Freitag R. Protein Purification by Affinity Precipitation. J Chromatogr, B: Anal Technol Biomed Life Sci. 2003;790:79–90. doi: 10.1016/s1570-0232(03)00081-3. [DOI] [PubMed] [Google Scholar]
- 10.Mammen M, Choi SK, Whitesides GM. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew Chem Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 11.Krishnamurthy VM, Kaufman GK, Urbach AR, Gitlin I, Gudiksen KL, Weibel DB, Whitesides GM. Caronbic Anhydrase as a Model for Biophysical and Physical-Organic Studies of Proteins and Protein-Ligand Binding. Chem Rev. 2008;108:946–1051. doi: 10.1021/cr050262p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mack ET, Snyder PW, Perez-Castillejos R, Whitesides GM. Using Covalent Dimers of Human Carbonic Anhydrase II to Model Bivalency in Immunoglobulins. J Am Chem Soc. 2011;133:11701–11715. doi: 10.1021/ja2038084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain A, Huang SG, Whitesides GM. Lack of Effect of the Length of Oligoglycine-Derived and OligoEthylene-Glycol-Derived Para-Substituents on the Affinity of Benzenesulfonamides for Carbonic-Anhydrase-II in Solution. J Am Chem Soc. 1994;116:5057–5062. [Google Scholar]
- 14.Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, Whitesides GM. Dependence of Effective Molarity on Linker Length for an Intramolecular Protein-Ligand System. J Am Chem Soc. 2007;129:1312–1320. doi: 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z-X. An Exact Mathematical Expression for Describing Competitive Binding of Two Different Ligands to a Protein Molecule. FEBS Lett. 1995;360:111–114. doi: 10.1016/0014-5793(95)00062-e. [DOI] [PubMed] [Google Scholar]
- 16.The experimental details of the fluorescence-based competition assay can be found in the Supporting Information section.
- 17.We found, experimentally, that the yield of the precipitation reaction did not change significantly after a 4 h incubation period, at 4 °C.
- 18.Figure S4 of the Supporting Information contains the experimental results comparing the number of the oligoethylene units in linker and its effectiveness at precipitating BCA from solution.
- 19.Mack ET, Perez-Castillejos R, Suo Z, Whitesides GM. Exact Analysis of Ligand-Induced Dimerization of Monomeric Receptors. Analytical Chemistry. 2008;80:5550–5555. doi: 10.1021/ac800578w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Figures S2 and S3 of the Supporting Information contain the experimental results comparing the effectiveness of different buffer (S2) and pH (S3) conditions at precipitating BCA from solution.
- 21.Nair SK, Calderone TL, Christianson DW, Fierke CA. Altering the Mouth of the Hydrophobic Pocket. Structure and Kinetics of Human Carbonic Anhydrase II Mutants at Residue Val-121. J Biol Chem. 1991;266:17320–17325. [PubMed] [Google Scholar]
- 22.Figure S7 contains the total concentration of protein contained in each precipitate and supernatant sample, as determined with a standard Bradford assay. The values reported in S7 are from n = 10 samples. Figure S8 contains a representative SDS PAGE image of the total protein content for each precipitate and supernatant sample.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.