

Abstract
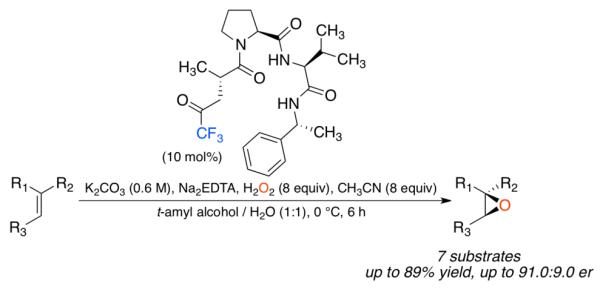
The development of peptide-based oxidation catalysts that use a transiently generated dioxirane as the chemically active species is reported. The active catalyst is a chiral trifluoromethyl ketone (Tfk) with a pendant carboxylic acid that can be readily incorporated into a peptide. These peptides were capable of epoxidizing alkenes in high yield (up to 89%) and enantiomeric ratios (er) ranging from 69.0:31.0 to 91.0:9.0, depending on the alkene substitution pattern.
The impact of chiral and achiral dioxiranes on the field of organic synthesis has been extensive.1 Of the myriad transformations executed by these highly reactive oxidants, catalytic asymmetric epoxidation is perhaps the best known (Figure 1a).2 In particular, the work of Shi, which employs chiral fructose-based ketone catalysts, has been remarkable for its generality and outstanding levels of enantioselectivity.3 The application of dioxiranes in C-H bond oxidations is also significant.4 In a particularly dramatic illustration of this reaction, Wender reported the use of an isolated dioxirane for the regioselective and stereospecific oxidation of a Bryostatin analogue (Figure 1b).5
Figure 1.
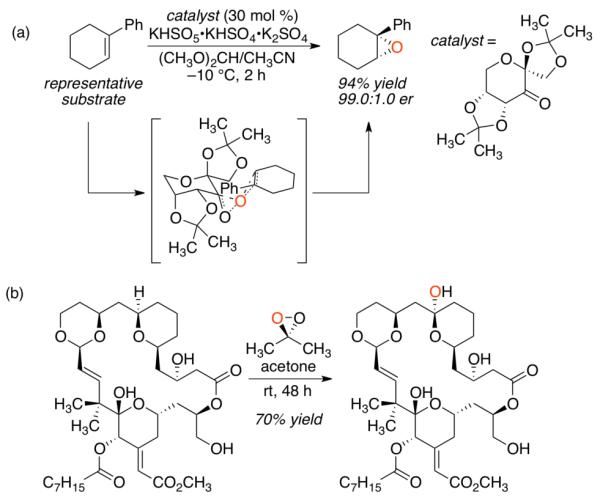
Venerable reactions of dioxiranes. (a) Asymmetric epoxidation with the catalyst of Shi. (b) Regioselective C-H oxidation of a Bryostatin analogue reported by Wender.
With the goal of developing diverse catalyst libraries for the oxidation of complex substrates, we wondered if it might be possible to develop a peptide-embedded ketone that would be competent for catalytic dioxirane-based oxidations. Peptide-based catalysts6 and ligands7 offer inherent modularity and can be tuned for optimization of various properties, such as stereo- and regioselectivity. Peptide-based oxidation catalysts in particular have a venerable history and have also been the subject of more recent studies. The Juliá-Colonna epoxidation of chalcones employs oligomeric peptides to control the reactivity of hydrogen peroxide in a putatively nucleophilic epoxidation reaction (Figure 2a).8 On the other hand, peptide-embedded aspartic acids may be engaged as catalysts for asymmetric electrophilic epoxidation via the intermediate peracid (Figure 2b).9 Our goal in this study was to explore whether or not peptide-embedded ketones such as 1 could be converted to the dioxirane (2) in situ and used as electrophilic oxidants without undergoing intramolecular oxidation (Figure 2c). If so, the stage would be set for a broad study of peptide-based ketones for catalytic oxidation.
Figure 2.
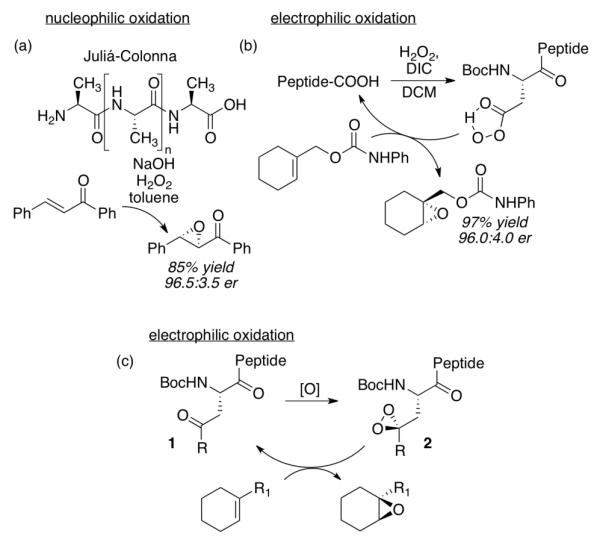
Peptide-based catalysts for asymmetric epoxidation. (a) Nucleophilic epoxidation through the Juliá-Colonna process. (b) Catalytic asymmetric epoxidation with aspartyl peptides. (c) Proposed peptide-embedded ketone catalysts for oxidation.
We thus began with a consideration of the appropriate catalytic monomer to be incorporated into a peptide catalyst. Our design criteria converged on the chiral trifluoromethyl ketone 3 (Scheme 1) for a number of reasons. Firstly, our desire to develop catalysts that would proceed through highly reactive dioxiranes dictated the use of a trifluoromethyl group over simple aliphatic substitution.10 Secondly, we chose to replace the NHBoc group of 1 with the methyl group of 3 to simplify synthesis in this first generation study. The latter decision was inspired by earlier studies of aspartyl peptide-based catalysts, in which we observed that the NHBoc group could be replaced with a methyl substituent without loss of selectivity.11 Our synthesis of 3 is presented below and has been used to make over 5 grams of monomer.
Scheme 1.
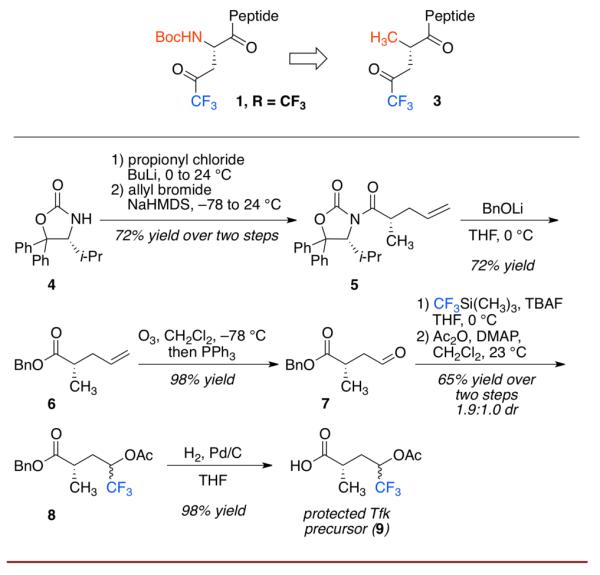
Synthesis of Tfk precursor.
The monomer synthesis began with the chiral auxiliary of Seebach (4),12 which was acylated with propionyl chloride and subsequently alkylated with allyl bromide to give 5 as a single diastereomer after recrystallization. The auxiliary was cleaved with BnOLi to give the enantiopure benzyl ester 6. Ozonolysis of the terminal alkene afforded aldehyde (7), which underwent nucleophilic trifluoromethylation to generate the trifluoromethyl carbinol. This was immediately protected as the acetate ester (8). Finally, the benzyl group was removed through hydrogenolysis to give the free acid 9, which served as a Tfk precursor that could be efficiently incorporated into a peptide and subsequently converted to the ketone to give 3 (Scheme 2).
Scheme 2.
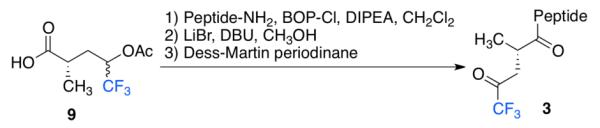
Incorporation of 9 into a peptide.
We proceeded to synthesize a variety of peptide-embedded ketones (Figure 3) in order to test their ability to convert 1-phenylcyclohex-1-ene (10, Table 1) to the corresponding epoxide (11). Our exploration began with peptide 12 (entry 1) because of its structural similarity to a previous epoxidation catalyst9 and because peptides of analogous composition are known to adopt β-type-turns that can potentially control enantioselectivity.13 The reaction conditions were based on those developed by Shi14 and modified to give full conversion of the substrates. Notably, in the absence of ketone, negligible epoxidation occurs (as assessed by 1H NMR).
Figure 3.
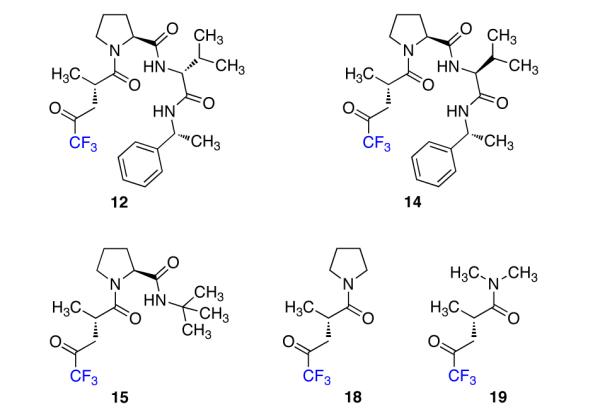
Structures of representative catalysts.
Table 1. Screen of peptide catalysts for epoxidation of 10.
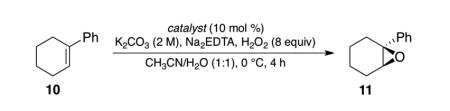 | ||
|---|---|---|
| entry | catalyst | era |
| 1 | Tfk-Pro-D-Val-(R)-Mba (12) | 87.0:13.0 |
| 2 | Tfk-Pro-D-Val-(S)-Mba (13) | 85.0:15.0 |
| 3 | Tfk-Pro-Val-(R)-Mba (14) | 89.5:10.5 |
| 4 | Tfk-Pro-tert-Butylamine (15) | 88.5:11.5 |
| 5 | Tfk-Pro-OCH3 (16) | 88.0:12.0 |
| 6 | Tfk-D-Pro-OCH3 (17) | 86.5:13.5 |
| 7 | Tfk-Pyrrolidine (18) | 84.5:15.5 |
| 8 | Tfk-Dimethylamine (19) | 84.0:16.0 |
Enantioselectivity was determined by HPLC using a chiral stationary phase (Chiralcel OJ-H). Mba = α-methylbenzylamine.
We were gratified to see that under the exploratory reaction conditions, epoxide 11 was formed in an er of 87.0:13.0 (Table 1, entry 1). This ratio decreased when the terminal residue was changed to the S-configuration (catalyst 13, entry 2), but interestingly, when the D-Val residue was changed to L-Val, the er increased to 89.5:10.5 (catalyst 14, entry 3). This result made us question whether or not a β-turn was important to the enantioselectivity of this reaction. To probe this hypothesis, we synthesized catalyst 15, in which the capacity for a β-turn was absent. Surprisingly, this catalyst provided almost the same selectivity as catalyst 14 (entry 4 vs. entry 3). The er was minimally perturbed when the catalyst was simplified further (catalysts 16 and 17, entries 5 and 6).
Finally, we synthesized catalysts 18 and 19, in which the only chiral element was the stereocenter found in Tfk. Although they afforded slightly lower enantioselectivities than the previous catalysts, the asymmetric induction provided by catalysts 18 and 19 (entries 7 and 8) suggests that the substrate interacts mainly with the Tfk residue of the peptide.
In light of these discoveries, we have considered a transition state model in which the substrate approaches from the side of the dioxirane that is opposite the peptide backbone (Figure 4a). We arranged the molecule by minimizing allylic strain15 between the Tfk stereocenter and the amide and then drawing the side-chain in an “all anti” configuration to minimize steric repulsion. The substrate was oriented by matching the smallest alkene substituent (H) with the largest dioxirane substituent (CF3) and by assuming a spiro transition state, which has been shown both experimentally3 and theoretically16 to be favored, ostensibly due to secondary orbital overlap between the lone pairs on oxygen with the π* orbital of the alkene. Additionally, we were able to obtain a crystal structure of 18 in its hydrated form that correlates well with the structure of the dioxirane in our transition state model (Figure 4b).
Figure 4.
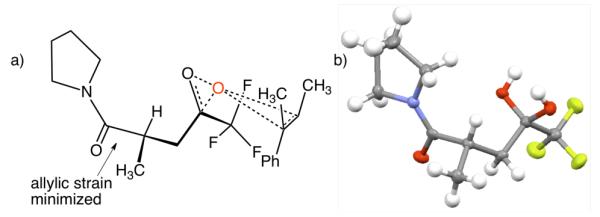
(a) Spiro transition state that gives the observed stereochemistry. (b) X-ray crystal structure of 18•H2O.
Having explored the simplest catalysts, we returned to our initial hit catalyst (14). We found that the selectivity increased slightly when we lowered the concentration of K2CO3 and changed to a solvent mixture of tert-amyl alcohol and water (Scheme 3 and Table 2). These conditions proved operationally straightforward and applicable to a broad set of substrates.17
Scheme 3.
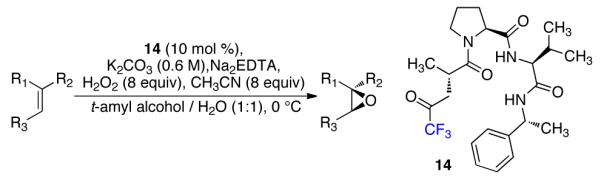
Optimized conditions for epoxidation.
Table 2. Substrate ccope for epoxidation with 14.a.
| entry | substrate | yield (%)b |
erc | configuration of productd |
|---|---|---|---|---|
| 1 |
|
88 | 90.5:9.5 | (−)-S,S18 |
| 2 |
|
83 | 88.5:11.5 | (−)-S,S19 |
| 3 |
|
82e | 87.0:13.0 | S,S19 |
| 4 |
|
88 | 89.5:10.5 | (−)-S,S20 |
| 5 |
|
75 | 91.0:9.0 | (−)-S,S19 |
| 6 |
|
89 | 71.0:29.0 | (−)-S,S21 |
| 7 |
|
75f | 69.5:30.5 | (+)-S22 |
The reaction conditions are shown in Scheme 3.
Products were isolated by flash chromatography.
Enantioselectivity was determined by HPLC with a chiral stationary phase.
Absolute stereochemistry was assigned by measuring the optical rotations and comparing them to the literature.
Yield is for mixture of trans and cis isomers (3.8:1.0). This phenomenon corresponds to the fact that the starting material was employed as a mixture of cis/trans isomers.
For this substrate, 12 equiv of H2O2 and CH3CN were used.
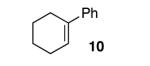
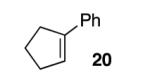
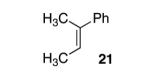
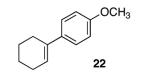
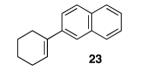
With our optimized conditions, we tested a variety of substrates. We found that our initial substrate, 1-phenylcyclohex-1-ene (10), gave the corresponding epoxide in 88% yield and with an er of 90.5:9.5 (Table 2, entry 1). Similarly, the analogous cyclopentene (20) was also epoxidized in high yield, with only a slight drop in enantioselectivity (entry 2). Acyclic variant 21 showed a greater drop in selectivity, but the high reactivity was maintained (entry 3). Remarkably, the highly reactive 1-(4-methoxyphenyl)cyclohex-1-ene (22) could also be epoxidized with high enantioselectivity (entry 4), suggesting that the background rate for these conditions is minimal. Finally, naphthyl analogue 23, despite its low solubility, could still be epoxidized in 75% yield and even higher enantioselectivity than 10 (entry 5).
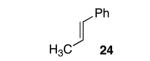
With disubstituted alkenes, the selectivity was only modest, but the yields remained high. Substrate 24 was epoxidized in 89% yield with 71.0:29.0 er (entry 6) and substrate 25 afforded the corresponding epoxide in 75% yield with 69.5:30.5 er (entry 7).
In conclusion, we have developed a trifluoromethyl ketone that can be efficiently incorporated into a peptide and used as a catalyst for the asymmetric epoxidation of alkenes. We were able to tune the selectivity of the catalyst by varying the sequence of the peptide scaffold and this approach may enable us to address classes of substrates for which current methods give poor selectivity. The next phase of this research will involve the evaluation of peptide-embedded ketones (e.g., 1) as a family of catalysts for selective reactions proceeding through transient dioxiranes.
Supplementary Material
Acknowledgment
We are grateful for research support from the National Institute of General Medical Sciences of the National Institutes of Health (GM096403). We also thank N. D. Schley for performing the X-ray crystallography.
Footnotes
Supporting Information Available: Procedures and characterization data. This information is available free of charge at: http://pubs.acs.org.
References
- (1).Adam W, Zhao C-G, Saha-Möller CR, Jakka K. Oxidation of Organic Compounds by Dioxiranes. Wiley; Hoboken: 2009. [Google Scholar]
- (2).Wong O, Shi Y. Chem. Rev. 2008;108:3958–3987. doi: 10.1021/cr068367v. [DOI] [PubMed] [Google Scholar]
- (3).Wang ZX, Tu Y, Frohn M, Zhang JR, Shi Y. J. Am. Chem. Soc. 1997;119:11224–11235. [Google Scholar]
- (4).Curci R, D’Accolti L, Fusco C. Acc. Chem. Res. 2006;39:1–9. doi: 10.1021/ar050163y. [DOI] [PubMed] [Google Scholar]
- (5).Wender PA, Hilinski MK, Mayweg AVW. Org. Lett. 2005;7:79–82. doi: 10.1021/ol047859w. [DOI] [PubMed] [Google Scholar]
- (6).(a) Dalko PI, Moisan L. Angew. Chem., Int. Ed. 2004;43:5138–5175. doi: 10.1002/anie.200400650. [DOI] [PubMed] [Google Scholar]; (b) Berkessel A. Curr. Opin. Chem. Biol. 2003;7:409–419. doi: 10.1016/s1367-5931(03)00065-6. [DOI] [PubMed] [Google Scholar]; (c) Jarvo ER, Miller SJ. Tetrahedron. 2002;58:2481–2495. [Google Scholar]
- (7).(a) Agarkov A, Greenfield S, Xie D, Pawlick R, Starkey G, Gilbertson SR. Biopolymers. 2006;84:48–73. doi: 10.1002/bip.20395. [DOI] [PubMed] [Google Scholar]; (b) Shimizu KD, Snapper ML, Hoveyda AH. Chem. Eur. J. 1998;4:1885–1889. [Google Scholar]
- (8).(a) Juliá S, Masana J, Vega JC. Angew. Chem., Int. Ed. 1980;19:929–931. [Google Scholar]; (b) Kelly DR, Roberts SM. Biopolymers. 2006;84:74–89. doi: 10.1002/bip.20373. [DOI] [PubMed] [Google Scholar]; (c) Berkessel A, Koch B, Toniolo C, Rainaldi M, Broxterman QB, Kaptein B. Biopolymers. 2006;84:90–96. doi: 10.1002/bip.20413. [DOI] [PubMed] [Google Scholar]
- (9).Peris G, Jakobsche C, Miller S. J. Am. Chem. Soc. 2007;129:8710–8711. doi: 10.1021/ja073055a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Blank JT, Miller SJ. Biopolymers. 2006;84:38–47. doi: 10.1002/bip.20392. [DOI] [PubMed] [Google Scholar]
- (11).Jakobsche C, Peris G, Miller S. Angew. Chem., Int. Ed. 2008;47:6707–6711. doi: 10.1002/anie.200802223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hintermann T, Seebach D. Helv. Chim. Acta. 1998;81:2093–2126. [Google Scholar]
- (13).(a) Haque TS, Gellman SH. J. Am. Chem. Soc. 1996;118:6975–6985. [Google Scholar]; (b) Miller SJ. Acc. Chem. Res. 2004;37:601–610. doi: 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]
- (14).(a) Shu L, Shi Y. Tetrahedron Lett. 1999;40:8721–8724. [Google Scholar]; (b) Burke CP, Shu L, Shi Y. J. Org. Chem. 2007;72:6320–6323. doi: 10.1021/jo0708644. [DOI] [PubMed] [Google Scholar]
- (15).Hoffman RW. Chem. Rev. 1989;89:1841–1860. [Google Scholar]
- (16).Bach RD, Andres JL, Owensby AL, Schlegel HB, McDouall JJ. W. J. Am. Chem. Soc. 1992;114:7207–7217. [Google Scholar]
- (17).We note that the results are highly analogous when Oxone is used as the oxidant, under the standard conditions of Shi and coworkers (reference 3).
- (18).Berti G, Macchia B, Macchia F, Monti L. J. Org. Chem. 1968;33:4045–4049. [Google Scholar]
- (19).Bulman Page PC, Buckley BR, Blacker A. J. Org. Lett. 2004;6:1543–1546. doi: 10.1021/ol049782h. [DOI] [PubMed] [Google Scholar]
- (20).Absolute stereochemistry was inferred by analogy to the other substrates.
- (21).Witkop B, Foltz CM. J. Am. Chem. Soc. 1957;79:197–201. [Google Scholar]
- (22).Fujisawa T, Takemura I, Ukaji Y. Tetrahedron Lett. 1990;31:5479–5482. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.