

Abstract
Objective
Mechanical injury induces cell death in cartilage and triggers a remodeling process that ultimately can manifest as osteoarthritis (OA). Autophagy is a process for turnover of intracellular organelles and macromolecules that protects cells during stress responses. This study determined changes and functions of autophagy following mechanical injury to cartilage.
Methods
Bovine and human cartilage explants were subjected to mechanical impact (40% strain, 500 ms). Cell viability, sulfated glycosaminoglyan (sGAG) release and changes in autophagy markers ULK1, Beclin1 and LC3 were evaluated. Cartilage explants were also treated with the mTORC1 inhibitor and autophagy inducer rapamycin and tested for protective effects against mechanical injury, the cell death inducers nitric oxide and TNFα+Actinomycin D and the proinflammatory cytokine IL-1α.
Results
Mechanical injury induced cell death and loss of sGAG in a time-dependent manner. This was associated with significantly decreased ULK1, Beclin1 and LC3 expression in the cartilage superficial zone (P < 0.05) at 48 hours post-injury. The levels of LC3-II were increased at 24 hours post-injury but decreased at 48 and 96 hours. Rapamycin enhanced expression of autophagy regulators and prevented cell death and sGAG loss in mechanically injured explants. Rapamycin also protected against cell death induced by SNP, TNFα+Actinomycin D and prevented sGAG loss induced by IL-1α.
Conclusion
Mechanical injury leads to suppression of autophagy, predominantly in the superficial zone where most of the cell death occurs. Pharmacological inhibition of mTORC1, at least in part by enhancement of autophagy prevented cell and matrix damage suggesting a novel approach for chondroprotection.
Keywords: Autophagy, Cartilage, Cell Death, Mechanical Injury
INTRODUCTION
Cartilage homeostasis is disturbed in osteoarthritis (OA), leading to progressive cartilage destruction. Biomechanical and biochemical factors are responsible for disruption of cell and tissue homeostasis and initiate catabolic pathways (1). Joint injury increases the risk for developing OA. However, the mechanisms by which joint injury leads to OA are not well understood (2–4).
The earliest degenerative changes occur in the cartilage superficial zone (SZ) and include reduced cellularity and structural irregularities (5). This initiates the progressive remodeling and degradation of the cartilage extracellular matrix (ECM) that subsequently affects the mid and deep zones. Mechanical injury has been implicated in initiating the lesions in the SZ and the SZ is more susceptible to cell death induced by mechanical injury (6). Under physiological conditions, mechanical loading of cartilage represents an important anabolic stimulus for chondrocyte metabolism (7). However, abnormal or injurious compression promotes cell death, predominantly by apoptosis (8,9), stimulates release of sulfated glycosaminoglycans (sGAG) from the extracellular matrix (10,11) and reduces chondrocyte anabolic responses to dynamic compression (12).
Autophagy is a cellular response to various types of stress whereby cellular macromolecules, organelles and cytoplasm are engulfed, digested and recycled to sustain cellular metabolism. Constitutive, basal autophagy also has an important homeostatic function, maintaining protein and organelle integrity (13). Although autophagy primarily promotes cellular and organism health, in certain physiological and pathological conditions it can also lead to a form of cell death that is characterized by cytoplasmic vacuolation and termed type II programmed cell death or cell death by autophagy (14–16). However, in most experimental models suppression of autophagy genes leads to cell death, indicating a protective and survival-promoting function of autophagy (17–19).
Atg genes control the autophagy process, leading to the induction and nucleation of autophagic vesicles, their expansion and fusion with lysosomes, allowing enzymatic degradation of macromolecules and recycling of their components (20,21). Among the Atg genes, Atg1, Atg6 and Atg8 (ULK1, Beclin1and LC3 in mammals, respectively) are three major regulators of the autophagy pathway. ULK1 is a key intermediate in the transduction of pro-autophagic signals to autophagosome formation (22). Beclin1 forms a complex with type III PI3-kinase and Vps34 that allows nucleation of the autophagic vesicle (23). Finally, the formation and expansion of the autophagosome requires two protein conjugation systems that involve the Atg proteins LC3, Atg12 and Atg5 (14). LC3 is present in two forms, LC3-I in the cytoplasm and LC3-II bound to the autophagosome membrane. During autophagy, LC3-I is converted to LC3-II through lipidation by a ubiquitin-like system resulting in the association of LC3-II with autophagy vesicles. The amount of LC3-II correlates with the extent of autophagosome formation (15).
Mammalian target of rapamycin (mTOR) is a member of the PI3-kinase-related protein kinase subfamily that plays a critical role in the regulation of various cellular events such as cell growth and proliferation (24,25). mTOR is an important suppressor of autophagy, functioning upstream of the Atg genes and is centrally regulated by multiple upstream signaling pathways involving PI3-kinase/Akt and AMP-activated protein kinase (26). Inhibition of mTOR by rapamycin, a lipophilic macrolide antibiotic, used as an immunosuppressive drug, can induce autophagy in a variety of cell types (27, 28). In addition, recently it was reported that rapamycin treatment extends lifespan in mice (29) and protects against aging-related pathology in various tissues, including brain and heart (30–32).
In articular cartilage, which is characterized by a very low rate of cell turnover autophagy would appear to be essential to maintain cell survival and function. Previously, we demonstrated that autophagy is a constitutively active and apparently protective process for the maintenance of the homeostatic state in normal cartilage (33). A reduction of autophagy was observed in joint aging and OA in humans and mice and this was accompanied by an increase in chondrocyte apoptosis. These results suggested that compromised autophagy might contribute to the development of OA.
The role of autophagy in the response of chondrocytes or other cell types to mechanical stress is unknown. Thus, the overall goal of this study was to evaluate the effect of mechanical injury on autophagy. As we observed an inhibition of autophagy in response to mechanical injury, we also tested potential protective effects of autophagy induction.
MATERIALS AND METHODS
Materials
The following antibodies were used: ULK1 and Beclin1 (Santa Cruz Biotechnology, Santa Cruz, CA), and LC3 (Novus Biologicals, Littleton, CO). Rapamycin was purchased from Calbiochem (San Diego, CA). Sodium nitroprusside (SNP) and Actinomycin D were purchased from Sigma (St. Louis, MO), IL-1α was purchased from Peprotech (Rocky Hill, NJ) and TNFα was purchased from R&D Systems (Minneapolis, MN).
Articular cartilage and chondrocytes
Full thickness articular cartilage explants without subchondral bone were harvested from mature bovine knees (14–30 months of age). Explants (8 mm diameter) were cored out from the weight bearing area of the distal femoral condyle using a dermal punch (Sklar Instruments, West Chester, PA). Prior to mechanical loading, explants were cultured for 48 hours at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% calf serum (CS) and antibiotics. To isolate chondrocytes, cartilage surfaces were rinsed with saline and scalpels were used to cut parallel sections 5 mm apart, vertically from the cartilage surface onto the subchondral bone. These cartilage strips were then resected from the bone. The cartilage tissue was incubated with trypsin at 37°C for 10 minutes. After the trypsin solution was removed, the tissue slices were treated for 12 to 16 hours with type IV clostridial collagenase in DMEM with 5% fetal calf serum. After isolation, the cells were kept in high-density cultures in DMEM supplemented with 10% CS, L-glutamine, and antibiotics and allowed to attach to the surface of the culture flasks. After the cells had grown to confluence, they were split once and grown to confluence again for use in the experiments.
Loading apparatus and mechanical injury
Mechanical injury was applied with an Instron 8511 mechanical testing device (Instron, Norwood, MA) following the protocol described by D’Lima et. al. (8,34). Each explant was centralized on a loading platform and a radially unconfined compressive load was applied through an impermeable stainless steel platen. In this study, after a small preload (0.1 MPa) was applied for two minutes, a 40% strain was applied to the explants for 500ms, conditions that produce reproducible cell death (8,34). Control explants were placed in the loading apparatus but not loaded. Control and loaded explants were cultured at 37°C and 5% CO2 in DMEM supplemented with 2% CS and antibiotics as indicated for the specific experiments and measurements. Experiments with human explants were also performed using similar methods. See supplemental information.
Live/dead assay
Live and dead cells were simultaneously viewed in situ with a confocal microscope (LSM 510; Zeiss, Wetzlar, Germany), using a fluorescent double stain. Calcein AM (1μM; Invitrogen, Carlsbad, CA), a fluorescein derivative that is metabolized by nonspecific esterase present in viable cells, was used to visualize live cells. The use of 8μM ethidium homodimer-1 (Invitrogen), a nucleic acid stain that is excluded by intact cell membranes, enabled visualization of the nuclei of dead cells (35). Cartilage explants were cultured in DMEM supplemented with Calcein AM and ethidium homodimer-1 for 45 minutes. Live and dead cells were counted using a custom designed automated image analysis program based on Matlab software (MathWorks, Natick, MA) to determine percentage of viable cells in the whole explant and separately in the SZ.
Glycosaminoglycan assay
To measure sGAG release we collected culture medium from explants without/with mechanical injury at 0, 24, 48 at 96 hours or after treating with rapamycin at 24, 48 and 96 hours. sGAG was measured using the dimethylmethylene blue method (36). sGAG concentration was normalized to explant weight and the results are reported as μg sGAG per mg cartilage.
Immunohistochemistry
Paraffin-embedded samples were first deparaffinized in the xylene substitute Pro-Par Clearant (Anatech, Battle Creek, MI) and rehydrated in graded ethanol and water. After washing with phosphate buffered saline (PBS), sections were treated with 3% hydrogen peroxide for 10 minutes, washed with PBS and sections were blocked with 5% serum for 30 minutes at room temperature. ULK1, Beclin1 and LC3 antibody (1:100 dilution) were applied and incubated overnight at 4°C. Slides were washed, and sections were incubated with biotinylated goat anti-rabbit secondary antibody for 30 minutes at room temperature, and then incubated using the Vectastain ABC-AP kit (Vector Laboratories, Burlingame, CA) for 30 minutes. Finally, sections were washed and incubated with 3,3-diaminobenzidine tetrahydrochloride (DAB) substrate for 3–10 minutes.
Quantification and localization of positive cells in human cartilage
ULK1, Beclin1 and LC3 localization in each cartilage zone was assessed systematically by counting positive cells in 50 × 50 μm grids starting from the cartilage surface to the deep zone. This was repeated a minimum of three times for each section. The identification of each zone was based on previously reported characteristics that comprise cell shape, morphology, orientation, and pericellular matrix deposition (37). The depth of each zone was recorded for each section for comparative analysis on the frequency of positive cells in each zone. The frequency of positive cells was expressed as a percentage relative to the total number of cells counted in each zone.
Western blotting
Cartilage explants were pulverized in a liquid nitrogen-cooled freezer-mill for two cycles of 1.5 min at the rate of maximum impact frequency. Dry weight of normal and OA cartilage was measured and the same amount of protein was resuspended in SDS gel loading buffer (50mM Tris PH 6.8, 10% glycerol, 4% sodium dodecyl sulfate, 10% 2-mercaptoethanol, and 0.001% bromophenol blue) and mixed for 2 hours at room temperature. Centrifugation at 14000-rpm was performed for 30 minutes, and then supernatants were harvested and heated at 80°C for 10 minutes. The protein concentrations were determined using a bicinchoninic acid (BCA) reagent assay (Pierce Chemical, Rockford, IL). The concentrated samples were then adjusted to equal volumes before resolution on 12% Tris-Glycine gels (Invitrogen). Protein was transferred to nitrocellulose membranes (Invitrogen), blocked with 5% dry milk in Tris-buffered saline–Tween (TBST), and blotted with rabbit polyclonal antibody for LC3 (Novus Biologicals) and β-actin antibody (Abcam, Cambridge, MA) for one hour. The membranes were then incubated with HRP-conjugated anti-mouse IgG (Cell Signalling Technology, Boston, MA) for one hour. Afterwards, the membranes were washed three times with TBST and developed using the enhanced chemiluminescent substrate from Pierce (Rockford, IL).
Flow cytometric assessment of cell viability and apoptosis
Bovine chondrocytes were cultured as described above. Before reaching confluency, the cells were plated in 6-well plates at 5×105 cells per well and cultured in DMEM with 10% FBS for 24 h. Medium was replaced with DMEM 0% FBS and the chondrocytes were preincubated for one hour with rapamicyn (1 μM) and then stimulated with SNP (2 μM) for 18 h. To determine cell viability, the cells were labeled with propidium iodide (Sigma) as described (38). For analysis of apoptosis, the cells were labeled with Annexin-V (Invitrogen). Cells were analyzed on a BD FACSCalibur (Becton Dickinson, Franklin Lakes, NJ). For each condition, 15,000 events were collected.
Statistical analysis
To evaluate normal distribution of the data, we performed the Kolmogorov-Smirnov test. Statistically significant differences between two groups were determined with unpaired Student’s t-test. Statistically significant differences between multiple comparisons were determined by ANOVA with Tukey’s multiple comparisons test. The results are reported as mean ± S.D. P values less than 0.05 were considered significant.
RESULTS
Mechanical injury induces cell death and loss of glycosaminoglycans in articular cartilage
To characterize the effect of mechanical injury on articular cartilage, we measured cell viability and sGAG release. Mature bovine articular cartilage explants were subjected to 40% strain of their original thickness for 0.5 sec. The results showed that mechanical injury induced cell death in a time-dependent manner (Figure 1A), predominantly in the SZ (Figure 1B). This was associated with a disruption of matrix integrity (Figure 1C) and a time-dependent increase in sGAG release (Figure 1D).
Figure 1. Mechanical injury induces cell death and loss of glycosaminoglycans in bovine cartilage explants.

Full-thickness cartilage explants (n=80 explants) were subjected to mechanical injury or placed in the loading machine but not loaded as control. The explants were analyzed by Live/Dead viability assay immediately after mechanical injury (0 hours) or following culture for 24, 48 or 96 hours. In addition, the effect of mechanical stress on cartilage extracellular matrix was evaluated by safranin O staining and by quantification of sulfated glycosaminoglycan (sGAG) release into supernatants. A, Percentage of viable cells in cartilage explants. Values are mean ± SD. ** = P < 0.001 vs control; * = P < 0.01 vs control. B, Percentage of viable cells in superficial zone (SZ) of cartilage explants. Values are mean ± SD. ** = P < 0.001 vs control; * = P < 0.05 vs control. C, Safranin O staining of control explants and after mechanical injury at 0, 24, 48 and 96 hours. Original magnification x40. D, Quantitative analysis of sGAG release into supernatants. Values are mean ± SD. * = P < 0.05 vs control; ** = P < 0.001 vs control. Values represent mean ± SD of five separate experiments each in duplicate.
Autophagy markers are reduced in response to mechanical injury in articular cartilage
To evaluate the effect of mechanical injury on the autophagy pathway, we analyzed the expression of three autophagy markers, ULK1 (autophagy inducer), Beclin1 (autophagy regulator) and LC3 (autophagy executor). After injury, the explants were cultured for 24, 48 and 96 hours. To study the presence of autophagosomes in response to mechanical injury, we analyzed the expression of LC3-II in bovine explants. The results indicated that the expression of LC3-II was increased 24 hours after injury, as an early response to stress, and then decreased at 48 hours. At 96 hours the expression of LC3-I was also decreased, indicating an overall decrease of LC3 expression in response to mechanical injury in a time-dependent manner (Figure 2A).
Figure 2. ULK1, Beclin1 and LC3 expression is reduced in response to mechanical injury.
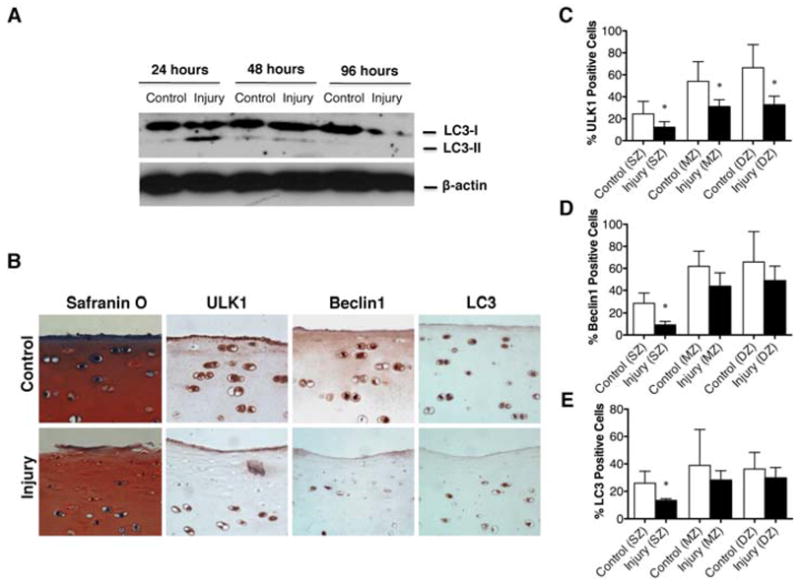
Full-thickness cartilage explants (n=24) were subjected to mechanical injury or cultured without injury to evaluate the effect of mechanical injury on the autophagy pathway. A, Total protein from explants subjected to mechanical injury or cultured without injured at 24, 48 and 96 hours was analyzed by Western blotting using anti-LC3 and β-actin. B, Safranin O staining and immunohistochemical analysis of ULK1, Beclin1 and LC3 from explants subjected to mechanical injury or cultured without injury at 48 hours. Original magnification x40. C–E, Quantitative analysis of ULK1, Beclin1 and LC3 positive cells in superficial zone (SZ), mid zone (MZ) and deep zone (DZ). Values represent mean ± SD of four separate experiments each in duplicate, * = P < 0.05.
To confirm these results, we analyzed the expression of ULK1, Beclin1 and LC3 by immunohistochemistry at 48 hours after injury (Figure 2B). The results showed a significantly decreased expression of all three markers predominantly in the SZ (P < 0.05), (Figure 2C–E). In addition, expression of ULK1 in mid and deep zone was significantly (P < 0.05) decreased compared to control condition without injury.
These results indicate that mechanical stress to articular cartilage decreased the expression of autophagy markers, predominantly in the SZ.
Rapamycin does not affect cell viability and increases expression of autophagy markers in normal cartilage
To study potential protective effects of autophagy, we employed rapamycin, an mTOR inhibitor and autophagy activator (39). Bovine cartilage explants were incubated with increasing doses of rapamycin (1, 5 and 10 μM) for 48 hours and analyzed by live-dead assay or immunohistochemistry for LC3. The results indicated that rapamycin did not affect cell viability (Figure 3A), but increased expression of LC3 (Figure 3B).
Figure 3. Rapamycin does not affect cell viability and increases expression of autophagy regulators.

Full-thickness cartilage explants and bovine chondrocytes were employed to study the potential effect of rapamycin on cell viability by Live-Dead assay. Autophagy activation was assessed by immunohistochemistry. A, Results of Live/Dead viability assay at 48 hours. B, LC3 expression in explants in control conditions and after rapamycin treatment (Rapa; 1, 5 and 10 μM) at 48 hours. Original magnification X 40.
Autophagy induction protects against cell death and loss of glycosaminoglycans after mechanical injury
To elucidate the role of autophagy induction in cell death and cartilage extracellular matrix homeostasis in the mechanical injury model, we induced autophagy by treating bovine cartilage explants with rapamycin (1 μM) and exposed them to mechanical injury. The results indicated that rapamycin treatment prevented mechanical injury-induced cell death (Figure 4A). These results were significant at 48 hours (P < 0.01) compared to injury conditions (Figure 4B and C) and there was a trend towards protection at 96 hours. Furthermore, in response to rapamycin treatment the levels of sGAG in supernatants were significantly decreased at 48 hours and 96 hours (P < 0.05) compared to injury condition without rapamycin treatment (Figure 5A, B).
Figure 4. Autophagy induction by rapamycin protects against cell death in response to mechanical injury.

Full-thickness cartilage explants (n=64 explants) were employed to study the potential effect of rapamycin on response to mechanical injury. Explants were subjected to mechanical injury, treated with rapamycin (Rapa; 1 μM) and analyzed by Live/Dead viability assay at 24, 48, 96 hours. A, Results of Live/Dead assay (original magnification X 40). B, Percentage of viable cells in bovine cartilage explants. Values are mean ± SD. * = P < 0.05 vs control; ** = P < 0.01 vs control. C, Percentage of viable cells in superficial zone (SZ). Values are mean ± SD. * = P < 0.05 vs control; ** = P < 0.001 vs control; & = P < 0.01 vs injury. Values represent mean ± SD of four separate experiments each in duplicate.
Figure 5. Rapamycin inhibits loss of glycosaminoglycans after mechanical injury.

Full-thickness cartilage explants (n=64 explants) were used to study the effect of rapamycin on mechanical injury-induced sulfated glycosaminoglycan (sGAG) release into supernatants based on the dimethylmethylene blue method. Explants were subjected to mechanical injury, treated with rapamycin (Rapa; 1 μM) and analyzed at 24, 48, 96 hours. A, Safranin O staining of control explants and after mechanical injury at 48 hours. Original magnification x40. B, Quantitative analysis of sGAG release into supernatants. Values are mean ± SD. * = P < 0.05 vs injury; ** = P < 0.001 vs control. Values represent mean ± SD of four separate experiments each in duplicate.
Rapamycin protects against biochemically induced cell death and matrix degradation
To test whether the protective effects of rapamycin-induced autophagy extend to other stimuli of cell death and cartilage damage we used a nitric oxide donor, SNP (2 mM), an apoptosis inducer, TNFα (10 ng/ml) + Actinomycin D (1 μg/ml) and a proinflammatory cytokine, IL-1α (10 ng/ml). Rapamycin protected against cell death induced by SNP (P < 0.01) (Figure 6A) and against apoptosis induced by TNFα + Actinomycin D (P < 0.01) (Figure 6B and C). The sGAG loss induced by IL-1α (10 ng/ml) was also prevented dose-dependently by rapamycin (P < 0.01) (Figure 6D).
Figure 6. Rapamycin protects against biochemically induced cell death and matrix degradation.

Chondrocytes and cartilage were preincubated for 1 hour with rapamycin (Rapa; 1, 5, 10 μM ) and then stimulated by sodium nitroprusside (SNP; 2 mM) or TNFα (10 ng/ml) + Actinomycin D (ActD; 1 μg/ml) for 18 hours or IL-1α (10 ng/ml) for 72 hours. A, Quantitative analysis of cell death induced by SNP in chondrocytes at 18 hours was performed by propidium iodide staining. Values are mean ± SD. ** = P < 0.001 vs control; * = P < 0.01 vs SNP 2 mM. B, Quantitative analysis of cell death induced by TNFα + ActD was performed by Annexin-V staining. Values are mean ± SD. ** = P < 0.001 vs control; * = P < 0.01 vs TNFα + ActD. C, Morphological studies using DAPI staining in explants treated or not with TNFα + ActD and rapamycin for 18 h. D, Quantitative analysis of sGAG release into supernatants. Values are mean ± SD. ** = P < 0.001 vs control; * = P < 0.01 vs IL-1α at 72 hours. Values represent mean ± SD of three separate experiments each in duplicate.
Studies on human cartilage and chondrocytes
To confirm the results obtained with the bovine samples, we obtained normal human knee joints. To induce cell death, application of more intense mechanical injury with 40% strain of 500ms, for three times was required. There was a trend towards protection by rapamycin at 96 hours but the results did not reach statistical significance (Suppl. Fig. 1), possibly due to the more intense mechanical injury and the greater variation among the human samples. In response to rapamycin the levels of sGAG in supernatants were decreased and a trend towards protection was observed at 24, 48 and 96 hours (Suppl. Fig. 2). In human chondrocytes, rapamycin protected against apoptotic cell death induced by TNFα + Actinomycin D (P < 0.05) (Suppl. Fig. 3). These results suggest that similar to the findings with bovine tissues, rapamycin has protective effects in human cartilage and chondrocytes.
DISCUSSION
Excessive mechanical load to articular cartilage results in damage to cells and extracellular matrix. During traumatic joint injury, the impact to cartilage causes instant damage but also initiates a pathogenetic process that is due to abnormal cell activation and leads to spreading of cell death and matrix damage beyond the initial area that was exposed to the highest mechanical load (8,10). Understanding mechanisms that are involved in these cellular changes has the potential to identify targets for pharmacological interventions to limit posttraumatic cartilage damage and prevent or attenuate the subsequent development of OA. Recent progress in this field has shown that cellular changes include apoptotic cell death, and increased production of oxygen radicals, matrix degrading enzymes and inflammatory mediators. In addition, there is also an inhibition of anabolic chondrocyte responses (3,4,7).
A topic that is not as well characterized is whether mechanical trauma induces compensatory and protective responses. Autophagy is a fundamental cellular homeostasis mechanism that is activated by different types of stress, including nutrient depletion and hypoxia, improving cellular survival and enhancing cellular fitness, whereas its inhibition precipitates bioenergetic failure and cell death (40). Whether autophagy is activated by mechanical injury has not been investigated in cartilage or in any other tissue or cell type.
In the present study, we used an in vitro model of cartilage injury as seen during single high impact joint trauma (8,34). To assess autophagy, we analyzed the expression of autophagy regulators and the presence of LC3-II, which directly indicates autophagosome formation. The results showed an increase of LC3-II at 24 hours after injury, as an early response to mechanical stress. However, autophagy was suppressed at 48 hours and 96 hours. In addition, the levels of LC3-I were also decreased at 96 hours post-injury, indicating a reduction in the expression of autophagy regulators after mechanical stress in articular cartilage. A uniform suppression of autophagy markers, predominantly in the SZ was observed in response to mechanical injury at 48 hours. Our results demonstrated that mechanical stress to articular cartilage can activate the early mechanisms of autophagy, however this defense mechanism was insufficient to protect articular cartilage against mechanical damage. In addition, the basal expression of ULK1 and Beclin1 was significantly higher in MZ and DZ compared to SZ. By contrast, the expression of LC3, was not significant different between MZ and DZ compared to SZ. It is possible that the relatively lower levels of ULK1 and Beclin1 lead to a lower autophagy activation or an increased susceptibility to autophagy suppression. In fact, the latter was observed in our mechanical injury experiments where autophagy suppression was more profound in the SZ than the middle and deep zone (6).
It is well known that hypoxia, oxidative stress or nutrient deprivation can induce autophagy to maintain homeostatic and survival mechanisms. In terminally differentiated chondrocytes, autophagy regulates maturation and promotes survival under stress and hypoxia conditions (41,42). In fact, silencing the transcription factor hypoxia-inducible factor 1 (HIF-1) in chondrocytes, decreases the expression of Beclin1, suggesting that autophagy could play a protective role against cell death (42).
Compromised autophagy has been observed in various diseases, in animal and in vitro models and contributes to cell dysfunction, cell death and pathogenesis. Conversely, activation of autophagy has been shown to have protective effects. Autophagy can be activated genetically through overexpression of certain autophagy genes (43) and pharmacologically by autophagy inducers, such as rapamycin (44). Rapamycin, a specific and widely used inhibitor of mTOR signaling pathway, is a lipophilic macrolide antibiotic, used as an immunosuppressive drug, induces autophagy in a variety of cell types, extends lifespan and protects against aging-related diseases in mice (29–32). Exposure of normal cartilage explants and chondrocytes to rapamycin had no detrimental effects and activated autophagy. However, it reduced cell death and GAG loss induced by mechanical injury. These findings provide proof-of-concept in vitro that autophagy activation may lead to chondroprotection after joint trauma.
This study used bovine cartilage explants from young animals. Key experiments were performed with normal human knee cartilage explants. Some differences were noted in the magnitude of protection by rapamycin. With human explants there was only a trend towards protection against cell death at 96 hours and there was a trend but no significant difference in sGAG release. These differences between human and bovine tissues may be due to difference in age and in the more intense mechanical load applied. The protective effect of rapamycin against biochemically induced cell death was significant in human chondrocytes.
Although rapamycin is a specific inhibitor of mTORC1 and this is the key regulator of autophagy activation, additional signaling pathways can modulate autophagy and the consequences of mTORC1 inhibition extend beyond autophagy to global changes in cell function and ultimately cell survival. Signaling by mTORC1 can be regulated by various stimuli, including growth factors, nutrients and mechanical signals. The most intensely studied regulatory mechanisms are signaling events such as from growth factor and nutrient signaling via PI3K/AKT, protein kinase B and AMPK. Additional regulators that can modulate mTORC1 are the ER stress response and inflammation signaling via IKKβ (45). A further level of complexity is added by the fact that besides regulation of autophagy activation by mTORC1, the gene and protein expression levels of the autophagy and lysosomal machinery are regulated by diverse transcription factors and microRNAs (46).
Although the direct consequence of mTORC1 inhibition by rapamycin is autophagy activation, the associated changes in protein synthesis via p70s6 kinase will lead to global changes in cell function. There are numerous examples of specific cell functions that are modulated as an indirect consequence of autophagy activation. The actin cytoskeleton and focal adhesion kinase (FAK) activation (47) are regulated by mTORC1 and rapamycin and this may contribute to the observed protective effects in the cartilage injury model. There is also a link between TNF and mTORC1 (48) that has been suggested to contribute to changes in expression of matrix degrading enzymes and osteoclast apoptosis (49). Thus, it is possible that rapamycin/mTOR effects besides regulating autophagy may be involved in the protective effects observed in the cartilage injury model.
In regard to the overall impact on cell and tissue homeostasis, it is apparent that autophagy plays a critical role in preventing the accumulation dysfunctional intracellular long-lived proteins and organelles, and autophagy failure leads to accumulation of toxic, structurally disruptive and energy draining structures and cellular components. Ultimately, cells in which damaged proteins or organelles accumulate die because of bioenergetic and metabolic dysfunction (50).
Previously we reported that autophagy is suppressed in human OA cartilage as well as mouse models of joint aging and mechanical instability-induced OA and this reduction was accompanied with an increase in apoptotic cell death (33). Aging-related or primary OA involve stimuli, such as nitric oxide (NO) and IL-1 that are important activators of cell death and extracellular matrix degradation. In addition, NO and IL-1 are also produced in response to mechanical injury to cartilage (51). Results from the present study indicate that rapamycin also protects against cell death induced by SNP and inhibits sGAG loss induced by IL-1α. Several studies demonstrated that rapamycin decreased inducible NO synthase protein expression in coronary endothelial cells (52) and inhibited IL-1β-induced MMP-9 expression in rat mesangial cells (53). Furthermore, as apoptosis is one of the main features of OA cartilage, we tested effects of rapamycin against the apoptosis inducer TNFα + Actinomycin D (38). The results indicated that rapamycin protects against apoptosis.
In conclusion, the present results indicate that mechanical injury to articular cartilage causes an overall suppression of autophagy, predominantly in the SZ. Rapamycin activates autophagy in cartilage and protects against the damaging effects of mechanical stress and biochemical stimuli, suggesting that enhancement of autophagy is a candidate approach for chondroprotection.
Supplementary Material
Acknowledgments
We are thankful to Jean Valbracht, Lilo Creighton, Diana Brinson, Nick Steklov and Jonathan Netter for technical assistance.
This study was supported by National Institutes of Health Grants AG007996, AR058954, RR027577 and the Sam and Rose Stein Endowment Fund. B. Caramés was supported by Postdoctoral Fellowship “Anxeles Alvariño”, Secretaria Xeral I+D+i, Xunta de Galicia, Spain.
Footnotes
AUTHOR CONTRIBUTIONS
Dr. Lotz had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study design: Lotz, Blanco, Caramés, D’Lima
Acquisition of data: Caramés, Taniguchi, Seino
Analysis and interpretation of data: Caramés, Lotz
Manuscript preparation: Caramés, Lotz, D’Lima
Statistical analysis: Caramés
References
- 1.Yang S, Kim J, Ryu JH, Oh H, Chun CH, Kim BJ, et al. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 2010;16:687–93. doi: 10.1038/nm.2153. [DOI] [PubMed] [Google Scholar]
- 2.Patwari P, Cook MN, DiMicco MA, Blake SM, James IE, Kumar S, et al. Proteoglycan degradation after injurious compression of bovine and human articular cartilage in vitro: interaction with exogenous cytokines. Arthritis Rheum. 2003;48:1292–301. doi: 10.1002/art.10892. [DOI] [PubMed] [Google Scholar]
- 3.Stevens AL, Wishnok JS, White FM, Grodzinsky AJ, Tannenbaum SR. Mechanical injury and cytokines cause loss of cartilage integrity and upregulate proteins associated with catabolism, immunity, inflammation, and repair. Mol Cell Proteomics. 2009;8:1475–89. doi: 10.1074/mcp.M800181-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sui Y, Lee JH, DiMicco MA, Vanderploeg EJ, Blake SM, Hung HH, et al. Mechanical injury potentiates proteoglycan catabolism induced by interleukin-6 with soluble interleukin-6 receptor and tumor necrosis factor alpha in immature bovine and adult human articular cartilage. Arthritis Rheum. 2009;60:2985–96. doi: 10.1002/art.24857. [DOI] [PubMed] [Google Scholar]
- 5.Taniguchi N, Caramés B, Ronfani L, Ulmer U, Komiya S, Bianchi ME, et al. Aging-related loss of the chromatin protein HMGB2 in articular cartilage is linked to reduced cellularity and osteoarthritis. Proc Natl Acad Sci U S A. 2009;106:1181–6. doi: 10.1073/pnas.0806062106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Temple MM, Bae WC, Chen MQ, Lotz M, Amiel D, Coutts RD, et al. Age and site-associated biomechanical weakening of human articular cartilage of the femoral condyle. Osteoarthritis Cartilage. 2007;15:1042–1052. doi: 10.1016/j.joca.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Ramakrishnan P, Hecht BA, Pedersen DR, Lavery MR, Maynard J, Buckwalter JA, et al. Oxidant conditioning protects cartilage from mechanically induced damage. J Orthop Res. 2010;28:914–20. doi: 10.1002/jor.21072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Lima DD, Hashimoto S, Chen PC, Colwell CW, Jr, Lotz MK. Human chondrocyte apoptosis in response to mechanical injury. Osteoarthritis Cartilage. 2001;9:712–9. doi: 10.1053/joca.2001.0468. [DOI] [PubMed] [Google Scholar]
- 9.Thomas CM, Fuller CJ, Whittles CE, Sharif M. Chondrocyte death by apoptosis is associated with cartilage matrix degradation. Osteoarthritis Cartilage. 2007;15:27–34. doi: 10.1016/j.joca.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 10.Otsuki S, Brinson DC, Creighton L, Kinoshita M, Sah RL, D’Lima D, et al. The effect of glycosaminoglycan loss on chondrocyte viability: a study on porcine cartilage explants. Arthritis Rheum. 2008;58:1076–85. doi: 10.1002/art.23381. [DOI] [PubMed] [Google Scholar]
- 11.Ewers BJ, Dvoracek-Driksna D, Orth MW, Haut RC. The extent of matrix damage and chondrocyte death in mechanically traumatized articular cartilage explants depends on rate of loading. J Orthop Res. 2001;19:779–84. doi: 10.1016/S0736-0266(01)00006-7. [DOI] [PubMed] [Google Scholar]
- 12.Kurz B, Jin M, Patwari P, Cheng DM, Lark MW, Grodzinsky AJ. Biosynthetic response and mechanical properties of articular cartilage after injurious compression. J Orthop Res. 2001;19:1140–6. doi: 10.1016/S0736-0266(01)00033-X. [DOI] [PubMed] [Google Scholar]
- 13.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol. 2004;15:231–6. doi: 10.1016/j.semcdb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 18.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 19.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–62. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis. 2008;13:1–9. doi: 10.1007/s10495-007-0154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–74. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 23.Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1:46–52. doi: 10.4161/auto.1.1.1542. [DOI] [PubMed] [Google Scholar]
- 24.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 25.Yang Q, Guan KL. Expanding TOR signaling. Cell Res. 2007;17:666–681. doi: 10.1038/cr.2007.64. [DOI] [PubMed] [Google Scholar]
- 26.Shigemitsu K, Tsujishita Y, Hara K, et al. Regulation of translational effectors by amino acid and mammalian target of rapamycin signaling pathways. Possible involvement of autophagy in cultured hepatoma cells. J Biol Chem. 1999;274:1058–65. doi: 10.1074/jbc.274.2.1058. [DOI] [PubMed] [Google Scholar]
- 27.Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270:815–22. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- 28.Sarkar S, Rubinsztein DC. Huntington’s disease: degradation of mutant huntingtin by autophagy. FEBS J. 2008;275:4263–70. doi: 10.1111/j.1742-4658.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- 29.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience. 2009;164:541–51. doi: 10.1016/j.neuroscience.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 31.Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inuzuka Y, Okuda J, Kawashima T, Kato T, Niizuma S, Tamaki Y, et al. Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation. 2009;120:1695–703. doi: 10.1161/CIRCULATIONAHA.109.871137. [DOI] [PubMed] [Google Scholar]
- 33.Caramés B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62:791–801. doi: 10.1002/art.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D’Lima DD, Hashimoto S, Chen PC, Colwell CW, Jr, Lotz MK. Impact of mechanical trauma on matrix and cells. Clin Orthop Relat Res. 2001;391(Suppl):S90–9. doi: 10.1097/00003086-200110001-00009. [DOI] [PubMed] [Google Scholar]
- 35.Grogan SP, Aklin B, Frenz M, Brunner T, Schaffner T, Mainil-Varlet P. In vitro model for the study of necrosis and apoptosis in native cartilage. J Pathol. 2002;198:5–13. doi: 10.1002/path.1169. [DOI] [PubMed] [Google Scholar]
- 36.Farndale RW, Buttle DJ, Barrett AJ. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim Biophys Acta. 1986;883:173–7. doi: 10.1016/0304-4165(86)90306-5. [DOI] [PubMed] [Google Scholar]
- 37.Guilak F, Alexopoulos LG, Upton ML, Youn I, Choi JB, Cao L, et al. The pericellular matrix as a transducer of biomechanical and biochemical signals in articular cartilage. Ann N Y Acad Sci. 2006;1068:498–512. doi: 10.1196/annals.1346.011. [DOI] [PubMed] [Google Scholar]
- 38.Caramés B, López-Armada MJ, Cillero-Pastor B, Lires-Dean M, Vaamonde C, Galdo F, et al. Differential effects of tumor necrosis factor-alpha and interleukin-1beta on cell death in human articular chondrocytes. Osteoarthritis Cartilage. 2008;16:715–22. doi: 10.1016/j.joca.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 39.Díaz-Troya S, Pérez-Pérez ME, Florencio FJ, Crespo JL. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–65. doi: 10.4161/auto.6555. [DOI] [PubMed] [Google Scholar]
- 40.Madeo F, Tavernarakis N, Kroemer G. Can autophagy induce longevity? Nature Cell Biol. 2010;12:842–6. doi: 10.1038/ncb0910-842. [DOI] [PubMed] [Google Scholar]
- 41.Srinivas V, Bohensky J, Shapiro IM. Autophagy: a new phase in the maturation of growth plate chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells Tissues Organs. 2009;189:88–92. doi: 10.1159/000151428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bohensky J, Shapiro IM, Leshinsky S, Terkhorn SP, Adams CS, Srinivas V. HIF-1 regulation of chondrocyte apoptosis: induction of the autophagic pathway. Autophagy. 2007;3:207–14. doi: 10.4161/auto.3708. [DOI] [PubMed] [Google Scholar]
- 43.Mizushima N, Kuma A. Autophagosomes in GFP-LC3 Transgenic Mice. Methods Mol Biol. 2008;445:119–24. doi: 10.1007/978-1-59745-157-4_7. [DOI] [PubMed] [Google Scholar]
- 44.Rangaraju S, Verrier JD, Madorsky I, Nicks J, Dunn WA, Jr, Notterpek L. Rapamycin activates autophagy and improves myelination in explant cultures from neuropathic mice. J Neurosci. 2010;30:11388–97. doi: 10.1523/JNEUROSCI.1356-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, et al. IKK suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–55. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 46.Jegga AG, Schneider L, Ouyang X, Zhang J. Systems biology of the autophagy-lysosomal pathway. Autophagy. 2011 May 1;7(5) doi: 10.4161/auto.7.5.14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laragione T, Gulko PS. mTOR regulates the invasive properties of synovial fibroblasts in rheumatoid arthritis. Mol Med. 2010;16:352–8. doi: 10.2119/molmed.2010.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, et al. IKK suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–55. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 49.Cejka D, Hayer S, Niederreiter B, Sieghart W, Fuereder T, Zwerina J, et al. Mammalian target of rapamycin signaling is crucial for joint destruction in experimental arthritis and is activated in osteoclasts from patients with rheumatoid arthritis. Arthritis Rheum. 2010;62:2294–302. doi: 10.1002/art.27504. [DOI] [PubMed] [Google Scholar]
- 50.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun HB. Mechanical loading, cartilage degradation, and arthritis. Ann N Y Acad Sci. 2010;1211:37–50. doi: 10.1111/j.1749-6632.2010.05808.x. [DOI] [PubMed] [Google Scholar]
- 52.Parlar A, Can C, Erol A, Ulker S. Posttransplantation therapeutic rapamycin concentration protects nitric oxide-related vascular endothelial function: comparative effects in rat thoracic aorta and coronary endothelial cell culture. Transplant Proc. 2010;42:1923–30. doi: 10.1016/j.transproceed.2010.03.134. [DOI] [PubMed] [Google Scholar]
- 53.Osman B, Akool el-S, Doller A, Müller R, Pfeilschifter J, Eberhardt W. Differential modulation of the cytokine-induced MMP-9/TIMP-1 protease-antiprotease system by the mTOR inhibitor rapamycin. Biochem Pharmacol. 2011;81:134–43. doi: 10.1016/j.bcp.2010.09.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.