

Abstract
The hematopoietic protein tyrosine phosphatase (HePTP) is implicated in the development of blood cancers through its ability to negatively regulate the mitogen-activated protein kinases (MAPKs) ERK1/2 and p38. Small-molecule modulators of HePTP activity may become valuable in treating hematopoietic malignancies such as T cell acute lymphoblastic leukemia (T-ALL) and acute myelogenous leukemia (AML). Moreover, such compounds will further elucidate the regulation of MAPKs in hematopoietic cells. Although transient activation of MAPKs is crucial for growth and proliferation, prolonged activation of these important signaling molecules induces differentiation, cell cycle arrest, cell senescence, and apoptosis. Specific HePTP inhibitors may promote the latter and thereby may halt the growth of cancer cells. Here, we report the development of a small molecule that augments ERK1/2 and p38 activation in human T cells, specifically by inhibiting HePTP. Structure-activity relationship analysis, in silico docking studies, and mutagenesis experiments reveal how the inhibitor achieves selectivity for HePTP over related phosphatases by interacting with unique amino acid residues in the periphery of the highly conserved catalytic pocket. Importantly, we utilize this compound to show that pharmacological inhibition of HePTP not only augments, but also prolongs activation of ERK1/2 and, especially, p38. Moreover, we present similar effects in leukocytes from mice intraperitoneally injected with the inhibitor at doses as low as 3 mg/kg. Our results warrant future studies with this probe compound that may establish HePTP as a new drug target for acute leukemic conditions.
Tyrosine phosphorylation [1] is a key mechanism for signal transduction and the regulation of a broad set of physiological processes characteristic of multicellular organisms. The importance of tyrosine phosphorylation in normal cell physiology is well illustrated by the many inherited or acquired human diseases that stem from abnormalities in protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) [2-5]. Hematopoietic cells have particularly high levels of tyrosine phosphorylation and express more genes for PTKs and PTPs than any other cell type, with the possible exception of neurons [3]. Acute changes in tyrosine phosphorylation mediate antigen receptor-induced lymphocyte activation, leukocyte adhesion and migration, cytokine-induced differentiation, and responses to many other stimuli.
The MAPKs extracellular-signal regulated kinases 1 and 2 (ERK1/2), c-Jun N-terminal kinases 1, 2, and 3 (JNK1/2/3), and the α-, β-, γ-, and δ-isoforms of p38 act as integration points in the signaling cascades of hematopoietic cells [6]. These kinases are ultimately activated via dual phosphorylation of a threonine and a tyrosine residue in their activation loop [7]. The human genome encodes 11 ‘typical’ MAPK phosphatases (MKPs), which inactivate MAPKs by dephosphorylating the phosphotyrosine (pTyr) and phosphothreonine (pThr) residues in their T- X-Y motif [8]. In addition, several ‘atypical’ dual-specific PTPs, including VHR [9] and VHX [10], as well as the Ser/Thr phosphatase PP2A [11,12], dephosphorylate MAPKs. The reason for this abundance of phosphatases relates to the numerous critical roles of MAPKs in the cell and the profound effects of the duration of MAPK activation on cell physiology. To achieve some degree of specificity, MAPK-specific phosphatases 1) reside in different subcellular locations, 2) are subject to different modes of post-translational regulation, 3) use different mechanisms for association, and 4) are expressed in response to different stimuli and in lineage-specific manners. Thus, while MAPK activation is the result of a conserved kinase cascade, several phosphatases serve as negative regulators in a temporal-, spatial-, and cell type-specific manner [6,13].
HePTP (PTPN7) [14,15] is the only pTyr-specific PTP known to dephosphorylate MAPKs in hematopoietic cells. HePTP is a 38-kDa enzyme, consisting of the C-terminal catalytic PTP domain and a short (~45 residues) N-terminal extension, which contains the kinase interaction motif (KIM, residues 15–31). Via its KIM, HePTP tightly associates with its physiological substrates including the MAPKs ERK1/2 and p38 [16-18]. In resting T cells, HePTP dephosphorylates the positive regulatory pTyr residue in the activation loop of these kinases [16,17] and prevents their translocation to the nucleus [17,19]. T cell antigen receptor (TCR) ligation leads to the activation of MAPKs, as well as to the phosphorylation of HePTP at residue Ser23 by cAMP-dependent kinase (also known as protein kinase A, PKA) [20]. This causes a significant fraction of the HePTP/MAPK molecules to dissociate [17], enabling activated, unbound ERK/p38 to translocate to the nucleus and initiate transcription events that are required for T cell activation. Some 30–60 min later, several MKPs accumulate in the nucleus and dephosphorylate ERK/p38 [6]. The inactivated MAPKs subsequently shuttle back to the cytosol and re-associate with HePTP. This dual phosphatase regulation of ERK/p38 is referred to as the “sequential phosphatase model” [6] and is an example of how different PTPs are used in a spatially and temporally ordered manner to control the extent, location, and duration of MAPK activation (Figure 1).
Figure 1. TCR-induced MAPK signaling.
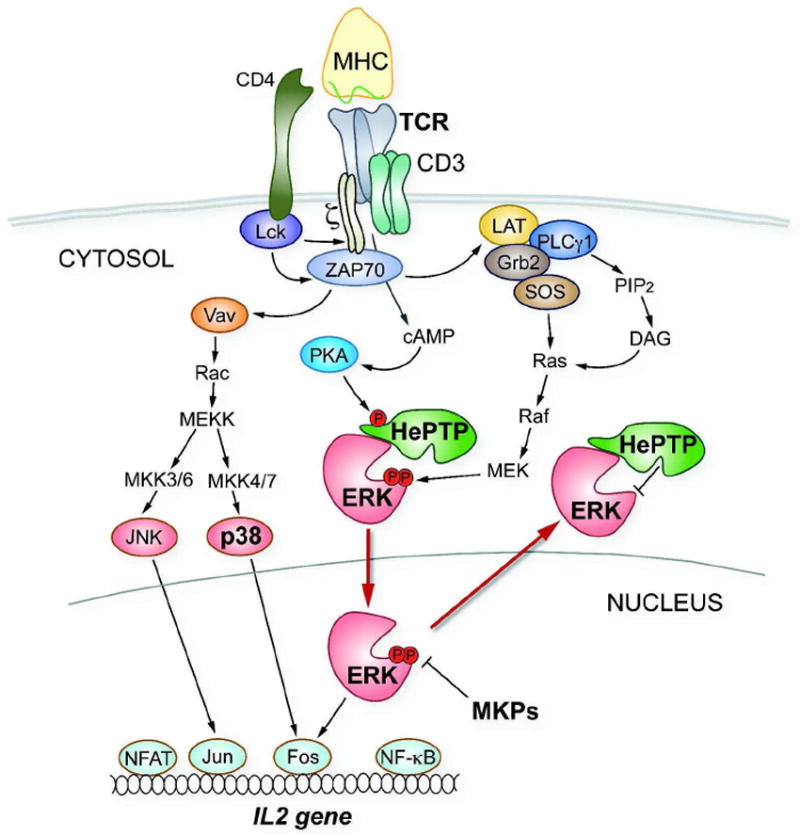
The “sequential phosphatase model” is illustrated for the MAPK ERK1/2. This model also applies to the MAPK p38, which undergoes similar interactions with HePTP.
HePTP is expressed in bone marrow, thymus, spleen, lymph nodes, and in all myeloid and lymphoid lineages and cell lines [14,15,21]. The HePTP gene is located on chromosome 1q32 [22] and is often duplicated in bone marrow cells from patients with myelodysplastic syndrome (MDS) [23,24], which is characterized by disturbed hematopoiesis and an increased risk of acute leukemia. Amplification and over-expression of HePTP is also reported in cases of acute myeloid leukemia (AML) [22]. Searching the Oncomine™ database, we found that HePTP mRNA levels are significantly upregulated in AML and also in T cell acute lymphoblastic leukemia (T-ALL) (Supplementary Figure S1). Because HePTP expression is restricted to hematopoietic lineages, it is a potential target for the development of novel therapeutics directed toward diseases of the blood and the immune system.
Specific small molecule modulators of HePTP activity have only recently been described [25,26]. Such compounds are thought to augment ERK1/2 and p38 activation and may cause a sustained hyperactivation of these MAPKs. In light of recent reports showing that prolonged activation of the Raf-Ras-ERK pathway can result in cell cycle arrest and cell senescence [13], and the fact that p38 can negatively regulate cell cycle progression and activate apoptotic pathways [27], HePTP inhibitors are of interest in the search for new leukemia therapeutics. In addition, such compounds will be useful for further elucidating the mechanisms of MAPK regulation and lymphocyte activation. Here, we report the development of a specific inhibitor of HePTP activity in human T cells. We demonstrate the structural basis for HePTP inhibition and show that interactions of the compound with unique amino acid residues in the HePTP substrate-binding loop and WPD-loop are critical for inhibitor binding and selectivity. Moreover, we show that the compound is active in vivo, augmenting activation of the MAPKs ERK1/2 and p38 in white blood cells from mice injected with a low dose of the inhibitor. Importantly, we demonstrate that pharmacological inhibition of HePTP not only augments, but also prolongs the activation of the MAPKs ERK1/2 and p38.
RESULTS AND DISCUSSION
Potent and selective small molecules that perturb a physiological target in a dose-dependent manner can be used to probe the role of the target in biology. Compared to mutant organisms that are often able to compensate for the loss of a gene, chemical probes can rapidly penetrate into cells, bind to their target proteins, and create loss-of-function (or gain-of-function) phenotypes. Such compounds can be administrated at any time during development and at any desired location of the cell or organism. Thus, chemical probes provide temporal and spatial control over their protein targets, and techniques such as activity-based protein profiling have emerged as powerful strategies [28]. However, the successful generation and application of such molecules remains challenging [29]. This is particularly true when targeting members of enzyme classes with highly conserved catalytic cores, such as PTPs [30-32]. HePTP is an important negative regulator of the MAPKs ERK1/2 and p38 in hematopoietic cells. Protein and mRNA expression levels are upregulated in cells from patients with preleukemic conditions or acute leukemia. HePTP chemical probes can be utilized to study the effects of HePTP inhibition on ERK1/2 and p38 activation in hematopoietic cells. If such compounds are able to augment and sustain MAPK activation levels, they may lead to new strategies in leukemia therapy.
High-throughput screening (HTS) for HePTP inhibitors
HePTP was screened within the Molecular Library Screening Center Network (MLSCN). The primary HTS and dose-response confirmation assays utilized the generic phosphatase substrate para-nitrophenyl phosphate (pNPP) [30]. In our reaction, product phosphate was quantified using the colorimetric malachite green-based Biomol Green reagent. The assay conditions were optimized for HTS in 384-well format (Supplementary Figure S2). Screening parameters were determined and indicated a robust and reliable HTS assay; the average Z’ was 0.8, the signal to background ratio was 6.4, the signal to noise ratio was 300, and the signal window was 12.3. A total of 112,438 compounds were screened at a concentration of 20 μM, and 54 hit compounds were found to dose-dependently inhibit HePTP activity with IC50 values lower than 10 μM. Chemical similarity clustering by a Tanimoto distance [33] of 0.5 resulted in 28 different clusters and singletons, indicating a quite diverse chemical space covered by these molecules (Figure 2). Detailed screening results, including a complete list of the screening hits and the confirmatory assay data, were deposited at the PubChem website (http://pubchem.ncbi.nlm.nih.gov/) under BioAssay ID (AID) 521.
Figure 2. Chemical similarity clustering of the 54 confirmed HePTP screening hits with IC50 values <10 μM.
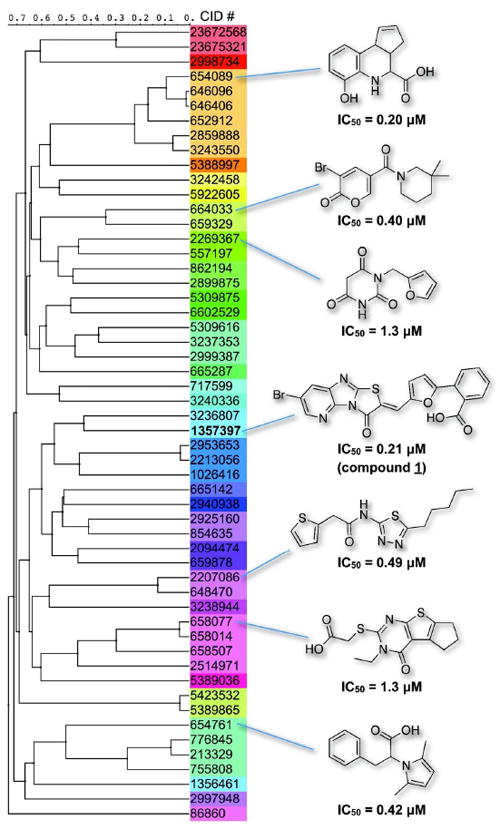
Compounds are listed by their PubChem Chemical ID number (CID), clustered by a Tanimoto distance of 0.5, and linked using the UPGMA method. Representative compounds and their respective IC50 values for HePTP are shown to demonstrate the chemical diversity of the HTS hits. A complete list of the chemical structures can be found on the PubChem website (http://pubchem.ncbi.nlm.nih.gov/), under AID 521.
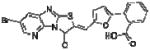
Since the catalytic mechanism of PTPs involves a nucleophilic attack by the thiolate ion of a catalytic cysteine residue, PTPs are prone to inhibition by oxidizing agents or Michael-acceptor compounds. In order to test for this unwanted inhibition, a secondary assay was adapted using 3-O-methylfluorescein phosphate (OMFP), which is hydrolyzed into the fluorescent 3-O-methylfluorescein and phosphate (Supplementary Figure S3). With this continuous assay in place, progress curves of the reaction could be easily acquired and utilized for removing time-dependent inhibitors. Importantly, at this stage compounds were also evaluated for their relative selectivity to inhibit HePTP over the phosphatase MKP-3, which shares with HePTP the physiological targets ERK1/2. In these assays, the majority of compounds demonstrated similar activity against both HePTP and MKP-3 and thus was discarded from further consideration owing to lack of selectivity. However, one compound (compound 1, PubChem CID1357397, NIH MLP probe number ML119) selectively inhibited HePTP over MKP-3 by 25-fold (Table 1). Compound 1 inhibited HePTP catalytic activity with an IC50 value of 0.21 μM. HePTP activity progress curves were linear over 98 min at different concentrations of compound 1, indicating no time-dependent inhibition of the enzyme (Figure 3a). Michaelis-Menten kinetic studies at various inhibitor and substrate concentrations revealed a mixed inhibition pattern for compound 1 with predominantly competitive participation and a competitive inhibition constant of Ki = 0.211 ± 0.250 μM (Figure 3b). Taken together, HTS yielded a potent compound with a 25-fold greater inhibition of HePTP compared to MKP-3 and with a primarily competitive inhibition pattern, suggesting a binding mode that involves the active site/catalytic pocket.
Table 1.
SAR and selectivity analysis for compound 1 and analogs (IC50 values in μM).
| Cmpd # | PubChem CID | Structure | HePTP | MKP-3 | VHR | STEP | PTP-SL | PTP1B | LYP | TCPTP | SHP1 | SHP2 | CD45 | LAR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1* | 1357397* |
|
0.21 | 5.2 | 1.9 | 6.3 | 1.1 | 1.7 | 1.9 | 1 | 2.5 | 1.1 | 3.1 | 73 |
| 2 | 5341943 |
|
1.8 | 17 | 5.8 | ~100 | 20 | 28 | 30 | n/d | n/d | n/d | n/d | n/d |
| 3 | 5341934 |
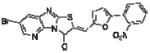
|
4.9 | 57 | 12 | >100 | 41 | ~100 | ~100 | n/d | n/d | n/d | n/d | n/d |
| 4 | 2300608 |
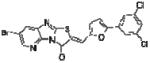
|
5.7 | >100 | 8.8 | >100 | ~100 | >100 | >100 | n/d | n/d | n/d | n/d | n/d |
| 5 | 2258411 |
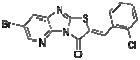
|
35 | >100 | >100 | >100 | >100 | >100 | >100 | n/d | n/d | n/d | n/d | n/d |
| 6 | n/a |
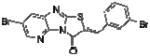
|
>100 | >100 | >100 | >100 | >100 | >100 | >100 | n/d | n/d | n/d | n/d | n/d |
Compound 1 was submitted as a probe to the Molecular Libraries Probe Production Centers (MLPCN) and has the Probe ML# ML119.
Figure 3. Mechanism of action (MOA) and inhibition studies of compound 1 with HePTP and HePTP mutants.
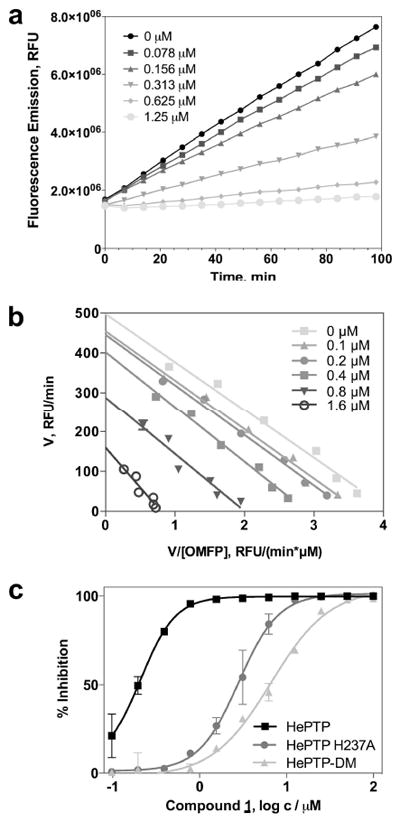
(a) Progress curves of HePTP activity in the presence of different doses of compound 1. No time-dependent inhibition was observed as demonstrated by the linear progress curves of the HePTP phosphatase reaction. (b) Eadie-Hofstee plot of the Michaelis-Menten kinetic studies with compound 1. (c) Dose-response curves for compound 1 with either wt HePTP, HePTP-DM, or HePTP H237A.
Structure-activity relationship (SAR) analysis of compound 1 using chemical analogs
In order to elucidate the molecular basis for inhibition of HePTP, SAR for compound 1 was developed around the thiazolo[2’,3’:2,3]imidazo[4,5-b]pyridin-3(2H)-one scaffold (Table 1). Dose-response phosphatase assays, using OMFP as a substrate, were performed to determine IC50 values for each analog. Since the majority of reported PTP inhibitors contain a structural element that binds to the phosphate-binding loop (P-loop) at the base of the catalytic pocket by mimicking pTyr [30], we expected the negatively charged benzoic acid group of compound 1 to be essential for inhibition. Indeed, HePTP IC50 values of compound 1 analogs clearly supported such a binding mode, in which the benzoic acid functions as a pTyr mimetic. Replacing the carboxylic acid with a nitro group (compound 3) or chlorine substitu ents (compound 4) reduced inhibitory activity by more than 20-fold. Replacing the bromine substituent at the other end of the molecule with methyl groups (compound 2) resulted in an 8.5-fold higher IC50 value (Table 1). Notably, analogs of compound 1 that were significantly smaller because they lacked the furanyl ring and that did not carry a carboxylic acid group, showed moderate or no activity against HePTP (compounds 5 and 6). Also, benzoic acid alone did not inhibit HePTP, even at high micromolar concentrations. Thus, SAR analysis suggests that compound 1 uses an ortho-benzoic acid group to bind into the catalytic pocket of HePTP, while additional interactions between inhibitor and protein surface are necessary for potency.
In silico docking confirms SAR analysis
To investigate the interactions of compound 1 with the HePTP active site on an atomic level, flexible ligand docking with the HePTP crystal structure (PDB code 3D44, ref. 34) was performed using the ICM docking algorithm [41]. The data from the docking studies support a binding mode in which the benzoic acid group in compound 1 functions as a pTyr mimetic. The benzoic acid was found to undergo multiple hydrogen bond interactions with residues of the PTP signature motif (H/V)CxxGxxR(S/T) of the P-loop (Figure 4). Additional hydrogen bond interactions near the catalytic center involved the side chain of the conserved Gln314 of the Q-loop as well as the imidazole group of His237 of the WPD-loop. As expected, compound 1 was found to interact with residues peripheral to the catalytic pocket. These interactions included hydrogen bonds between oxygen and nitrogen atoms of the heterocycle and the side chain of T106 of the substrate-binding loop (SB-loop), as well as favorable polar contacts between the bromine atom of the inhibitor and the ε -amino group of Lys105 in the HePTP SB-loop. We found similar interactions with residues of the SB-loop crucial for the selectivity of previously reported HePTP inhibitors [25]. In silico docking with the crystal structure of MKP-3 revealed a lack of such additional interactions with residues in the periphery of the catalytic pocket, perhaps explaining the substantially lower inhibitory activity of compound 1 against MKP-3. Neither Lys105 nor Thr106 of the HePTP SB-loop is conserved in MKP-3, which is missing a corresponding loop. Together, the in silico docking is in agreement with the SAR analysis and suggests that interactions of compound 1 with Lys105 and Thr106 contribute to the 25-fold selective inhibition of HePTP over MKP-3.
Figure 4. In silico docking of compound 1 into the HePTP active site.

Upper panel: Ribbon and surface representation of the HePTP active site (crystal structure, PDB code 3D44) with docked inhibitor (stick representation). Surface color code: white, neutral; green, hydrophobic; red, hydrogen bond acceptor potential; blue, hydrogen bond donor potential. Lower panel: Stereo ribbon diagram of the docking pose used in the upper panel. Residues that interact with compound 1 (magenta) and the catalytic cysteine (C270 in HePTP) are shown in stick representation.
Mutagenesis studies confirm the proposed binding mode for compound 1
To further explore the binding mode of compound 1, we first used site-directed mutagenesis to mutate Lys105 and Thr106 to alanines and tested compound 1 against the recombinant HePTP double mutant (HePTP-DM). Using OMFP as a relatively small artificial substrate, we showed that kinetic parameters such as Km and Vmax were similar between wild-type (wt) and mutant protein, which allowed us to directly compare IC50 values of the inhibitor. Compound 1 was 31-fold less active against HePTP-DM (IC50 = 6.6 μM, Figure 3c) as compared to wt HePTP. These results suggest that Lys105 and Thr106 are important for the binding of compound 1 to HePTP.
Since His237 had not been implicated for compound binding in previous studies with HePTP inhibitors [25], we also used site-directed mutagenesis to mutate His237 to alanine. We generated the recombinant HePTP H237A mutant and subjected the protein to inhibition studies with compound 1. Again, as predicted by the in silico docking studies, we found His237 of the HePTP WPD-loop important for inhibitor binding. Compound 1 was more than 13-fold less active against the H237A mutant (IC50 = 2.8 μM, Figure 3c) compared to wt HePTP. Thus, the mutagenesis studies confirm the proposed binding mode for compound 1.
Selectivity profiling of compound 1 and analogs against related PTPs
To evaluate the selectivity of compound 1 and analogs more broadly, a large panel of additional related PTPs was employed, including VHR, STEP, PTP-SL, PTP1B, LYP, CD45, SHP1, SHP2, TCPTP, and LAR. VHR, together with MKP-3 and HePTP, shares the common physiological targets ERK1/2. STEP and PTP-SL are the closest relatives of HePTP, and together represent the subfamily of KIM-containing PTPs. PTP1B is the prototypical PTP and a critical regulator of essential pathways such as insulin and leptin signaling. LYP, CD45, SHP1, SHP2, TCPTP, and LAR are critical signaling molecules in lymphocytes, where HePTP is enriched. Using the dose-response OMFP phosphatase assay, we showed that compound 1 was a weaker inhibitor of all additionally tested PTPs compared to HePTP (Table 1). Compound 1 selectively inhibited HePTP over PTP-SL, SHP2, and TCPTP (5-fold), PTP1B (8-fold), VHR (9-fold), LYP (9-fold), SHP1 (12-fold), CD45 (15-fold), STEP (30-fold), and LAR (348-fold). In particular, the high preference of 1 for HePTP over STEP, one of its two most similar relatives (53% sequence identity within the PTP domain), was surprising. HePTP, PTP-SL, and STEP have almost identical active sites, based on sequence similarity. They only differ in the two residues that immediately follow the aspartic acid of their WPD-loops. In HePTP these residues are His237 and Gln238, in PTP-SL His555 and Lys556, and in STEP Gln462 and Lys463. In addition, the crystal structure of STEP (PDB code 2BV5, ref. 26) reveals a different WPD-loop conformation, with the corresponding Gln462 pointing in the opposite direction, compared to His237 or His555 in HePTP and PTP-SL, respectively. Since our in silico docking and mutagenesis studies suggested that His237 contributes to inhibitor binding, selectivity of compound 1 over STEP is likely the result of the missing histidine residue at the corresponding position in STEP, an otherwise highly similar phosphatase.
Interestingly, residues Lys105 and Thr106 in the HePTP SB-loop are not conserved among PTPs. In fact, Thr106 is unique to the three KIM-containing phosphatases HePTP, PTP-SL, and STEP. While a Thr106 corresponding residue is missing in the dual-specificity phosphatases MKP-3 and VHR, the pTyr-specific PTPs typically have an aspartic acid at the equivalent position. The favorable hydrogen bonding interactions between the hydroxyl group of Thr106 and the heterocycle of compound 1 cannot be formed with an aspartic acid residue in that position. Thus, interactions of compound 1 with Thr106 likely confer the observed selectivity for HePTP over the classical pTyr-specific PTPs. Taken together, compound 1 is the most selective HePTP inhibitor reported to date. Given at the appropriate dosage, off-target effects on other phosphatases should be kept to a minimum.
Compound 1 augments ERK1/2 and p38 activation in human T cells, specifically by inhibiting HePTP
For the development of an HePTP chemical probe, essential criteria it must satisfy are that the compound be membrane permeable and inhibit HePTP activity inside intact cells. In T cells, HePTP negatively regulates TCR signaling by dephosphorylating the activation loops of ERK1/2 and p38. Therefore, bona fide HePTP inhibitors should affect TCR-induced activation levels of ERK/p38. In order to probe ERK1/2 and p38 phosphorylation, we incubated Jurkat TAg T cells with selected compounds or vehicle (DMSO) at 37°C for 45 min. Cells were then TCR-stimulated (with OKT3) for 5 min, and reactions were stopped by adding lysis buffer. Immunoblotting was carried out using phospho-ERK1/2 (pERK1/2) and phospho-p38 (pp38) specific antibodies. In order to assess the specificity of the inhibitors, we also included a blot for phosphorylated mitogen activated protein kinase kinase (pMEK), which directly phosphorylates/activates ERK1/2. Therefore, by verifying unchanged MEK activation, augmented pERK1/2 levels should not be the result of increased kinase activity upstream of ERK1/2 within the MAPK signaling cascade.
The immunoblotting results show that compound 1 augmented pERK1/2 and pp38 levels, while activation of MEK was unchanged (Figure 5a). Additionally, we tested three analogs (compounds 3, 4, 5), which also increased pERK1/2 and pp38 levels (Figure 5a). Interestingly, compound 5, which only modestly inhibited HePTP in the in vitro assay (IC50 value of 35 μM), caused substantial augmentation of pERK1/2. However, pMEK levels were also augmented in the presence of compound 5, suggesting off-target effects of compound 5 upstream of ERK1/2. We then subjected compound 1 to a dose-response immunoblot experiment, using a similar assay format (Figure 5b). The effect of compound 1 on ERK1/2 and p38 activation was clearly dose-dependent and peaked between an inhibitor concentration of 1 and 4 μM. Given that pMEK levels were not affected, even at compound concentrations as high as 40 μM, off-target effects of compound 1 on molecules upstream of ERK1/2 can be excluded. MKP-3 is the only other known highly expressed PTP in T cells that acts on ERK1/2 early after TCR stimulation [35]. Compound 1 is 25-fold less potent against recombinant MKP-3 (IC50 = 5.2 μM) compared to HePTP. Thus, compound 1, at concentrations between 1 and 4 μM, is not expected to significantly inhibit MKP-3 in cells. These results suggest that the observed effect of compound 1 on ERK1/2 activation in human T cells can be attributed to inhibition of HePTP.
Figure 5. Screening of HePTP inhibitors in human T cells.
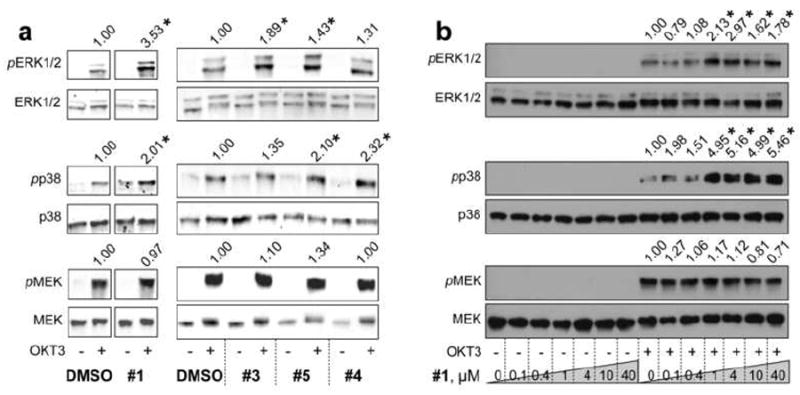
(a) Probing ERK1/2 and p38 phosphorylation in Jurkat TAg T cells in the presence of vehicle (DMSO, 0.2%) or compound 1, 3, 4, or 5 (40 μM), using phospho-specific antibodies against pT202/Y204-ERK1/2 (pERK1/2) and pT180/Y182-p38 (pp38). An anti-phospho-MEK (pMEK) blot was included as a specificity control. (b) Dose-response of compound 1. Cell lysates were probed as in (a). Phospho-protein bands were normalized to total protein levels. Statistical significance was determined by Student’s t-test (*p<0.05). Representative blots are shown from 3 independent experiments.
Inhibition of HePTP causes sustained hyperactivation of ERK1/2 and p38
A prolonged activation of MAPKs could be therapeutically valuable in cancer treatment. Therefore, we tested whether pharmacological inhibition of HePTP in T cells would sustain high levels of ERK1/2 and p38 activation after TCR stimulation. Preincubation of cells with compound 1 at 4 μM led to an increase in basal phosphorylation levels of both pERK1/2 and pp38 compared to the DMSO control (Figure 6; pERK1/2: 22.0 ± 2.5% vs. 13.2 ± 2.7%, p<0.05; pp38: 55.0 ± 5.8% vs. 36.1 ± 3.1%, p<0.05). TCR stimulation with OKT3 led to increases of pERK1/2 and pp38 levels in both DMSO- and compound-treated cells. However, phosphorylation levels were significantly higher in the presence of the inhibitor (pERK1/2: compound 1, 134.3 ± 4.9%; DMSO, 100.0 ± 5.4%, p<0.01; pp38: compound 1, 125.5 ± 4.0%; DMSO: 100.0 ± 4.3%, p<0.01). In the vehicle-treated cells, pERK1/2 levels rapidly decreased starting immediately after stimulation. On the other hand, pERK1/2 levels remained constant up to 1 h after stimulation in cells treated with compound 1, and only then started decreasing. Two hours after stimulation pERK1/2 levels were still significantly higher in the inhibitor-treated samples compared to the DMSO control. Similarly, pp38 levels started decreasing rapidly 15 min after stimulation in vehicle-treated cells, while pp38 levels remained unchanged in cells treated with compound 1, even at 2 h after stimulation. Considering that nuclear MKPs that act on ERK/p38 at later time points (>30 min after TCR stimulation) are structurally similar to MKP-3, i.e. they are missing the loop that corresponds to the SB-loop in HePTP, which is important for inhibitor binding, compound 1 will likely affect these MKPs at levels similar to MKP-3. Thus, at the used concentration of 4 μM, compound 1 is not expected to significantly inhibit other MKPs. Taken together, our results suggest that inhibition of HePTP in T cells not only augments, but also sustains activation of ERK1/2 and p38.
Figure 6. Compound 1 causes sustained hyperactivation of ERK1/2 and p38 in human T cells.
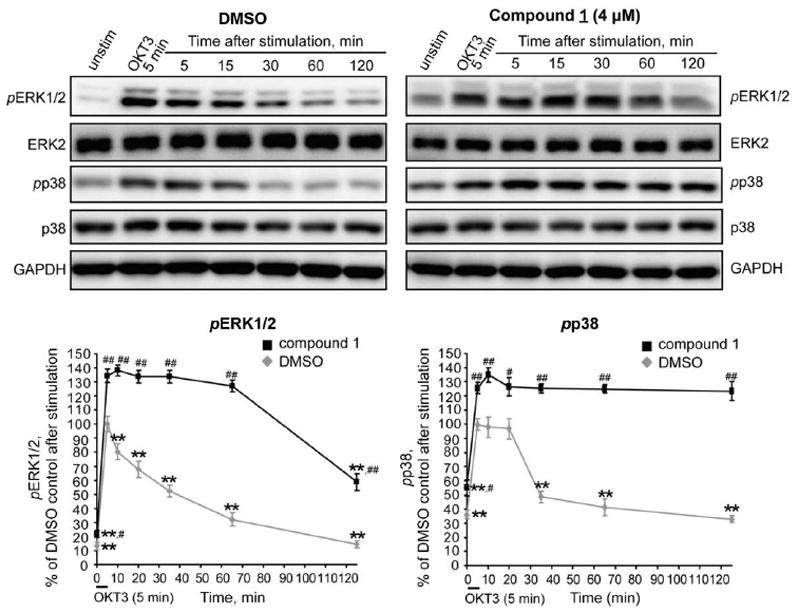
ERK1/2 and p38 phosphorylation in Jurkat TAg T cells, preincubated with vehicle (DMSO, 0.2%) or compound 1 (4 μM), at various time points after TCR stimulation. Phospho-protein bands were normalized to total protein levels and then to GAPDH. Statistical significance was determined by one-way ANOVA with post hoc Tukey’s test (within group (compound 1- or DMSO-treated samples, respectively), **p<0.01), or Student’s t-test (between groups (between compound 1- and DMSO-treated samples), #p< 0.05, ##p< 0.01). Representative blots are shown from 4 independent experiments.
Pharmacokinetic (PK) analysis of compound 1
The goal of these studies was to generate a chemical probe for HePTP that could also be utilized in vivo. Therefore, we assessed metabolic stability of compound 1 by monitoring its depletion over time while incubating it with rat liver microsomes in the presence of NADPH along with a reference compound of known microsomal half-life. LC/MS analysis was used and indicated a stability of 86.2 ± 2.7% after 15 min, 71.0 ± 2.9% after 30 min, and 53.0 ± 1.4% after 45 min. The half-life (t1/2) of the inhibitor was calculated from its decrease with time to t1/2 = 50 min, assuming first order kinetics (Figure 7a). Thus, compound 1 exhibited a metabolic stability that was sufficient for in vivo studies [36-38]. In addition, no major metabolite of compound 1 was identified in these studies. Rather, the compound metabolizes into multiple low-molecular-weight degradation products, as determined by LC/MS.
Figure 7. Pharmacokinetic (PK) analysis of compound 1.
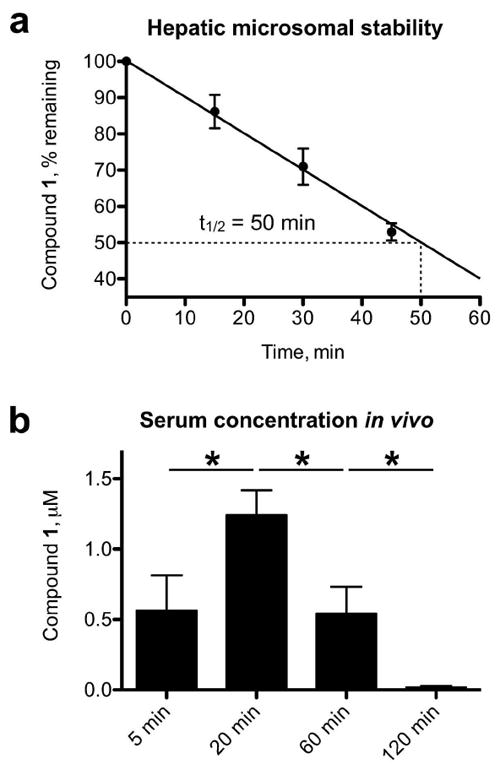
(a) Hepatic microsomal stability of compound 1. The half-life (t1/2) of the inhibitor was calculated from its decrease with time, using linear regression. (b) Serum concentration of compound 1 in mice injected (i.p.) with compound 1 at 3 mg/kg; blood serum was collected after 5, 20, 60, and 120 min.
To determine the achievable concentration of compound 1 in vivo, we injected mice intraperitoneally (i.p.) with the inhibitor at 3 mg/kg and collected blood serum after 5, 20, 60, and 120 min. LC/MS analysis was used to determine serum concentrations, which showed the typical time course for a compound that is injected into the body cavity and must cross tissue in order to get into the systemic circulation (Figure 7b). Inhibitor concentration peaked at 20 min (1.24 ± 0.17 μM), and then decreased over the course of 2 h (0.018 ± 0.010 μM). Thus, given the peak activity of compound 1 in cells at concentrations between 1 and 4 μM, our results suggest that 3 mg/kg or a slightly higher dosage is appropriate for in vivo studies.
Compound 1 augments ERK1/2 and p38 activation in vivo
To test whether compound 1 was able to inhibit HePTP activity in vivo, we injected mice (i.p.) with compound 1 at 3 mg/kg or 5 mg/kg and processed spleen at 3 h or 6 h. Presence of the compound in the splenocytes was verified by LC/MS analysis. The inhibitor was detected in significant amounts 6 h after injection (0.265 ± 0.119 μM), indicating that clearance in the spleen tissue is lower than in the blood. Phosphorylation levels of ERK1/2 and p38 were assessed by immunoblot analysis using phospho-specific antibodies (Figure 8, upper panel). Leukocytes from inhibitor-treated mice exhibited significant higher activation levels of both ERK1/2 and p38 compared to vehicle controls (pERK1/2: 3 mg/kg for 6 h, 2.26 ± 0.46, p<0.05; 5 mg/kg for 6 h, 1.97 ± 0.16, p<0.01; pp38: 3 mg/kg for 3 h, 1.67 ± 0.16, p<0.01; 3 mg/kg for h, 1.75 ± 0.13, p<0.01; 5 mg/kg for 6 h, 2.33 ± 0.33, p<0.05). Given a maximal serum concentration of compound 1 of 1.24 μM (at 3 mg/kg dosage), and an even lower concentration in the spleen tissue (<0.5 μM), it is unlikely that the ERK1/2 and p38 hyperactivation is due to inhibition of PTPs other than HePTP.
Figure 8. Compound 1 augments ERK1/2 and p38 activation in vivo.
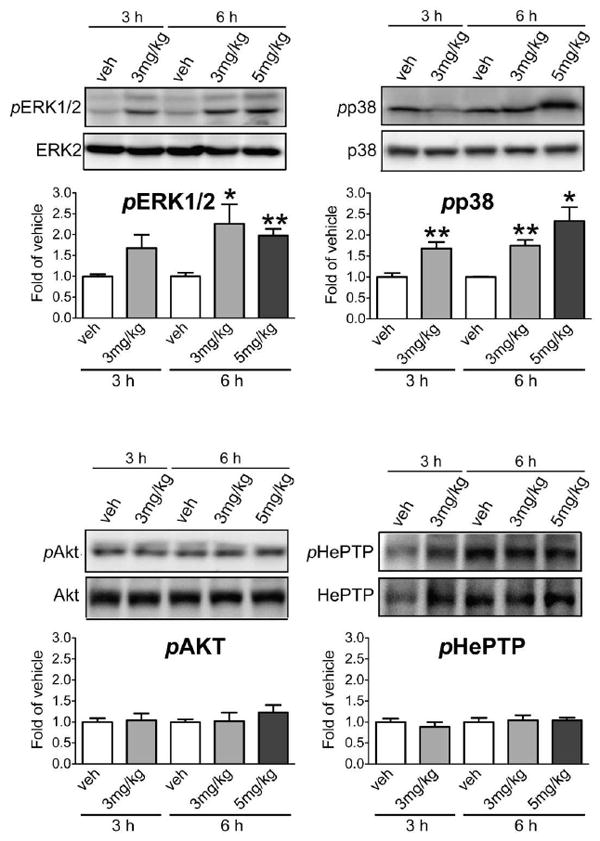
ERK1/2 and p38 phosphorylation in spleenocytes of mice injected (i.p.) with compound 1 or vehicle. Total lysates from spleen were probed with phospho-specific antibodies against pT202/Y204-ERK1/2 (pERK1/2), pT180/Y182-p38 (pp38), pS473-AKT1 (pAKT), and pS23-HePTP (pHePTP). Phosphorylation levels were normalized to total protein, then to GAPDH as a loading control. (*p<0.05, **p<0.01, n=4).
To further address specificity of compound 1, we also determined phosphorylation of AKT, a key-signaling molecule downstream of CD28/PI3K in a pathway that leads to NF-kB activation. There was no change in AKT phosphorylation levels at the doses and time points used (Figure 8, lower panel). Since HePTP activity is regulated by phosphorylation at Ser23 by PKA [17], we also probed this site with a phospho-specific antibody [20]. No changes in phospho-HePTP levels were detected, indicating that the augmented MAPK activation was not due to disruption of the HePTP/MAPK complex as a result of the phosphorylation of the regulatory serine within the KIM (Figure 8, lower panel).
Finally, since the PTPs most similar to HePTP, PTP-SL and STEP, are exclusively expressed in the brain [39], we tested whether compound 1 could cross the blood-brain-barrier (BBB) and potentially inhibit these phosphatases. Brain tissue from mice injected with compound 1 (i.p.) at 3 mg/kg or 5 mg/kg was processed 3 h after injection. Using LC/MS analysis, compound 1 could not be detected in the brain tissue (the detection limit was ~1 nM). Also when we specifically analyzed the cerebellum, the part of the brain where PTP-SL is expressed, compound 1 could not be detected. Thus, compound 1 does not appear to cross the BBB. Taken together, we showed that compound 1 may be used as a chemical probe for HePTP in vivo. Future studies of compound 1 in mouse models of acute leukemia are warranted.
Compound 1 complies with ‘essential criteria’ of a chemical probe
A common understanding of what makes a compound a chemical probe was lacking until recently, when Stephen Frye proposed a set of principles to guide probe qualification [29]. His five requirements for a high quality chemical probe include: 1) molecular profiling, 2) mechanism of action, 3) identity of the active species, 4) proven utility as a probe, and 5) availability. Compound 1 meets all of these criteria. First, we provide comprehensive molecular profiling data. Specifically, we show that compound 1 is highly potent in vitro, in human T cells, and in vivo, and significant effects in cells and in vivo can be achieved with a relatively low dose of the inhibitor, compared to its in vitro IC50 value. Moreover, the compound demonstrates between 5–30 fold selectivity for HePTP over the most similar enzymes in the in vitro assay and shows no signs of an off-target effect in the cell-based or in vivo experiments. Second, we determined the mechanism of action of compound 1 on the atomic level, i.e. the binding mode between the inhibitor and the protein. In addition, the effect of the inhibitor is dose-dependent in all experiments in which it was tested. Third, compound 1 is the active species that inhibits HePTP as no degradation of compound 1 was observed in the in vitro assays, and LC/MS analysis revealed no major metabolites of the compound in the stability assay (only multiple low-molecular-weight degradation products were found. These data suggest that observed effects in vivo are due to compound 1 as the active species. Fourth, the utility of compound 1 as a chemical probe was demonstrated. Namely, txhe compound was utilized to show, for the first time, that pharmacological inhibition of HePTP not only augments, but also sustains ERK1/2 and p38 activation in human T cells and in leukocytes in vivo. And fifth, the compound is readily available to the academic community.
In conclusion, we have developed a potent and relatively specific chemical probe for HePTP, a critical negative regulator of the MAPKs ERK1/2 and p38 in hematopoietic cells. To our knowledge, this is the first time that a small-molecule inhibitor of a protein tyrosine phosphatase has been successfully utilized to control MAPK activation in vivo. Current investigations are underway to determine whether compound 1 can be developed into a treatment strategy for hematopoietic malignancies such as AML or T-ALL.
EXPERIMENTAL PROCEDURES
Materials
Para-nitrophenyl phosphate (pNPP), 3-O-methylfluorescein phosphate (OMFP), dithiothreitol (DTT), and sodium orthovanadate (Na3VO4) were purchased from Sigma-Aldrich. Biomol Green reagent was from Biomol Research Laboratories, Inc. Compound 1 was repurchased from Labotest (#LT00237558) and analogs were ordered from ChemBridge. All compounds had a purity of >95% (verified by LC/MS and 1H-NMR). All other chemicals and reagents were of the highest grade available commercially. All antibodies in the T cell in vitro assays were from Cell Signaling Technology Inc. Antibodies used in the experiments with mouse tissue are listed in Supplementary Table S1. For recombinant proteins see Supplementary Methods.
Chemical library screening and dose-response assays
Dose-response inhibitor studies using OMFP
Phosphatase activity was measured in a 384-well or 96-well format assay system. Compounds were 2-fold serially diluted in DMSO before being added to the reactions for a ten-point dose-response curve. Reactions contained 50 mM Bis-Tris pH 6.0, 1.7 mM DTT, 0.005% Tween 20, and 5% DMSO. OMFP concentrations (corresponding to the Km value of each enzyme) were 117 μM for HePTP (2.75 nM), 50 μM for MKP-3 (1 μg mL-1), 12.5 μM for VHR (3 nM), 35 μM for STEP (5 nM), 28 μM for PTP-SL (5 nM), 99 μM for PTP1B (10 nM), 184 μM for LYP (5 nM), 144 μM for HePTP-DM (5 nM), and 222 μM for HePTP H237A (5 nM). Fluorescence intensity was measured in kinetic mode over 10 min to determine the slopes of progress curves, using an Envision plate reader (PerkinElmer), an excitation wavelength of 485 nm, and an emission wavelength of 528 nm. IC50 values were determined using nonlinear regression (sigmoidal dose-response with variable slope) and the program Prism (v5.0b GraphPad Software, Inc.).
Michaelis-Menten kinetic studies to determine the inhibition mode of compound 1
The HePTP-catalyzed hydrolysis of OMFP was assayed at room temperature in a 60 μL 96-well format reaction system in 50 mM Bis-Tris, pH 6.0 assay buffer containing 1.7 mM DTT, 0.005% Tween-20, and 5% DMSO. Recombinant HePTP (5 nM) was preincubated with various fixed concentrations of inhibitor (0, 0.1, 0.2, 0.4, 0.8, 1.6, 3.2 μM) for 10 min. The reaction was initiated by addition of various concentrations of substrate (0, 12.5, 25, 50, 100, 200, 400 μM) to the reaction mixture. The initial rate was determined using an FLx800 micro plate reader (Bio-Tek Instruments, Inc.) using an excitation wavelength of 485 nm and measuring the emission of the fluorescent reaction product at 528 nm. The nonenzymatic hydrolysis of the substrate was corrected by measuring the control without addition of enzyme. Eadie-Hofstee plots were generated using the program Prism (v5.0b GraphPad Software, Inc.). The inhibition pattern and inhibition constant were determined as described previously [40]. For a comparison of the fitting results the second-order Akaike’s Information Criterion (AICc) was calculated with equation [4], where N is the number of data points, SS the absolute sum of squares and K the number of parameters fit by nonlinear regression plus 1.
| [4] |
The probability to have chosen the right model can be computed by equation [5], where & is the difference between AICc scores.
| [5] |
Generation of HePTP K105A/T106A double mutant and HePTP H237A mutant
In silico docking studies
Flexible ligand docking calculations were performed on a MacPro workstation with the ICM docking algorithm [41] as implemented in the ICM-Pro program (v3.7-1g, Molsoft, LLC.). The coordinates of the three-dimensional (3D) structure of HePTP (PDB code 3D44, ref. 34), MKP-3 (PDB code 1MKP), and STEP (PDB code 2BV5) were converted into ICM objects, charges were assigned, orientations of side chain amides were corrected, and hydrogen atoms added and their positions optimized by energy minimization using MMFF force field. Mutations in the crystal structures were corrected to wt amino acid sequences, and the side chains were optimized using the Optimize Side Chains tool as implemented in ICM-Pro. The binding pockets were defined as follows. HePTP: residues in an 8 Å radius around the pTyr of the peptide ligand; MKP-3 and STEP: residues in an 10 Å radius around the catalytic Cys. Energy-minimized 3D molecular models of the compounds were generated with the implemented routine in ICM.
TCR stimulation, cell lysis, and immunoblotting
Jurkat TAg T cells were treated either with small molecule compound or with DMSO alone (vehicle control) for 45 min at 37°C. Cells were then TCR-stimulated with an optimal dose of OKT3 (500 ng mL-1) for 5 min. Thereafter, cells were either washed with RPMI 1640 medium and incubated in fresh medium for the indicated period of time (experiment in Figure 6) or immediately lysed in lysis buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 1% Triton X-100, 50 mM NaF, 5 mM Na3VO4, 1 mM PMSF, 5 mM EDTA), before proteins were separated using SDS-PAGE, transferred onto PVDF membranes, and blotted with the indicated antibodies. These procedures were carried out as described previously [42]. Densitometric quantifications were performed using either ImageJ (http://rsbweb.nih.gov/ij, Figure 5) or the Genetools program (Syngene, Figure 6). Data are expressed as means ± SEM. Statistical significance (*p< 0.05, ** p< 0.01) was determined by Student’s t-test.
PK analysis
In vivo testing of compound 1
The Yale University Institutional Animal Care and Use Committee approved all animal procedures. C57B6 mice (3 mice at each dose and time, Jackson Laboratory) were injected with compound 1 (i.p., 3 mg kg-1 or 5 mg kg-1) or vehicle and were sacrificed 3 h or 6 h post-injection. Spleen, cerebellum and cortex were processed for biochemical assays. Spleen was lysed in sonication buffer (50 mM Tris pH 7.4, 150 mM NaCl, 5 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 1 mM EGTA and protease inhibitor cocktail (Roche) with brief sonication. Lysates were spun at 800 × g for 10 min and supernatants were kept for western blotting. Cerebellum and cortex were homogenized in TEVP buffer (10 mM Tris pH 7.4, 5 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 1 mM EGTA and protease inhibitor cocktail). Homogenates were centrifuged at 1000 × g for 10 min, and supernatants (S1) was further centrifuged at 10,000 × g for 15 min to obtain a crude synaptosomal fraction (P2). P2 pellets were subsequently resuspended and lysed in TEVP buffer. Protein concentrations were determined with the BCA kit (Thermo Fisher Scientific). For western blotting, samples were prepared in sample buffer (Biorad), resolved by SDS-PAGE, transferred to nitrocellulose membrane, and incubated with primary antibodies listed in Supplementary Table S1 overnight at 4°C. Membranes were washed and incubated in peroxidase-conjugated secondary antibodies. The immunoreactivities were visualized using a Chemiluminescent substrate kit (Thermo Fisher Scientific) and detected using G:BOX with the image program GeneSnap (Syngene). All densitometric quantifications were obtained using the Genetools program (Syngene). Data are expressed as means ± SEM. Statistical significance (p < 0.05) was determined by one-way ANOVA with post hoc Tukey’s test or Student’s t-test. All experiments were repeated at least three times.
Supplementary Material
Acknowledgments
This work was supported by Grants 1R21 CA132121 (to L.T.), R03 MH084230 (to L.T.), 1X01MH077603 (to T.M.), R01 MH52711 (to P.J.L), and U54 HG005033 (to CPCCG) from the National Institutes of Health. This work was also supported by an American Cancer Society Research Scholar Grant (RSG-08-067-01-LIB) to R.P. Compound 1 was submitted as a probe to the Molecular Libraries Probe Production Centers (MLPCN) and has the Probe ML# ML119.
ABBREVIATIONS
- DTT
dithiothreitol
- ERK
extracellular signal-regulated kinase
- HePTP
hematopoietic protein tyrosine phosphatase
- MAPK
mitogen-activated protein kinase
- MKP
MAPK phosphatase
- OMFP
3-O-methylfluorescein phosphate
- pTyr
phosphotyrosine
- pNPP
p-nitrophenyl phosphate
- PTK
protein tyrosine kinase
- PTP
protein tyrosine phosphatase
- SAR
structure activity relationship
- TCR
T cell antigen receptor
Footnotes
Supporting Information Available: Supplementary Methods, Table, and Figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci U S A. 1980;77:1311–1315. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 3.Mustelin T, Vang T, Bottini N. Protein tyrosine phosphatases and the immune response. Nat Rev Immunol. 2005;5:43–57. doi: 10.1038/nri1530. [DOI] [PubMed] [Google Scholar]
- 4.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 5.Tautz L, Pellecchia M, Mustelin T. Targeting the PTPome in human disease. Expert Opin Ther Targets. 2006;10:157–177. doi: 10.1517/14728222.10.1.157. [DOI] [PubMed] [Google Scholar]
- 6.Saxena M, Mustelin T. Extracellular signals and scores of phosphatases: all roads lead to MAP kinase. Semin Immunol. 2000;12:387–396. doi: 10.1006/smim.2000.0219. [DOI] [PubMed] [Google Scholar]
- 7.Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 8.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 9.Denu JM, Zhou G, Wu L, Zhao R, Yuvaniyama J, Saper MA, Dixon JE. The purification and characterization of a human dual-specific protein tyrosine phosphatase. J Biol Chem. 1995;270:3796–3803. doi: 10.1074/jbc.270.8.3796. [DOI] [PubMed] [Google Scholar]
- 10.Alonso A, Merlo JJ, Na S, Kholod N, Jaroszewski L, Kharitonenkov A, Williams S, Godzik A, Posada JD, Mustelin T. Inhibition of T cell antigen receptor signaling by VHR-related MKPX (VHX), a new dual specificity phosphatase related to VH1 related (VHR) J Biol Chem. 2002;277:5524–5528. doi: 10.1074/jbc.M107653200. [DOI] [PubMed] [Google Scholar]
- 11.Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM, Cohen P. Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr Biol. 1995;5:283–295. doi: 10.1016/s0960-9822(95)00059-5. [DOI] [PubMed] [Google Scholar]
- 12.Zhou B, Wang ZX, Zhao Y, Brautigan DL, Zhang ZY. The specificity of extracellular signal-regulated kinase 2 dephosphorylation by protein phosphatases. J Biol Chem. 2002;277:31818–31825. doi: 10.1074/jbc.M203969200. [DOI] [PubMed] [Google Scholar]
- 13.Cerignoli F, Rahmouni S, Ronai Z, Mustelin T. Regulation of MAP kinases by the VHR dual-specific phosphatase: implications for cell growth and differentiation. Cell Cycle. 2006;5:2210–2215. doi: 10.4161/cc.5.19.3267. [DOI] [PubMed] [Google Scholar]
- 14.Zanke B, Suzuki H, Kishihara K, Mizzen L, Minden M, Pawson A, Mak TW. Cloning and expression of an inducible lymphoid-specific, protein tyrosine phosphatase (HePTPase) Eur J Immunol. 1992;22:235–239. doi: 10.1002/eji.1830220134. [DOI] [PubMed] [Google Scholar]
- 15.Adachi M, Sekiya M, Isobe M, Kumura Y, Ogita Z, Hinoda Y, Imai K, Yachi A. Molecular cloning and chromosomal mapping of a human protein-tyrosine phosphatase LC-PTP. Biochem Biophys Res Commun. 1992;186:1607–1615. doi: 10.1016/s0006-291x(05)81592-x. [DOI] [PubMed] [Google Scholar]
- 16.Saxena M, Williams S, Brockdorff J, Gilman J, Mustelin T. Inhibition of T cell signaling by mitogen-activated protein kinase-targeted hematopoietic tyrosine phosphatase (HePTP) J Biol Chem. 1999;274:11693–11700. doi: 10.1074/jbc.274.17.11693. [DOI] [PubMed] [Google Scholar]
- 17.Saxena M, Williams S, Tasken K, Mustelin T. Crosstalk between cAMP-dependent kinase and MAP kinase through a protein tyrosine phosphatase. Nat Cell Biol. 1999;1:305–311. doi: 10.1038/13024. [DOI] [PubMed] [Google Scholar]
- 18.Gronda M, Arab S, Iafrate B, Suzuki H, Zanke BW. Hematopoietic protein tyrosine phosphatase suppresses extracellular stimulus-regulated kinase activation. Mol Cell Biol. 2001;21:6851–6858. doi: 10.1128/MCB.21.20.6851-6858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pettiford SM, Herbst R. The protein tyrosine phosphatase HePTP regulates nuclear translocation of ERK2 and can modulate megakaryocytic differentiation of K562 cells. Leukemia. 2003;17:366–378. doi: 10.1038/sj.leu.2402767. [DOI] [PubMed] [Google Scholar]
- 20.Nika K, Hyunh H, Williams S, Paul S, Bottini N, Tasken K, Lombroso PJ, Mustelin T. Haematopoietic protein tyrosine phosphatase (HePTP) phosphorylation by cAMP-dependent protein kinase in T-cells: dynamics and subcellular location. Biochem J. 2004;378:335–342. doi: 10.1042/BJ20031244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gjorloff-Wingren A, Saxena M, Han S, Wang X, Alonso A, Renedo M, Oh P, Williams S, Schnitzer J, Mustelin T. Subcellular localization of intracellular protein tyrosine phosphatases in T cells. Eur J Immunol. 2000;30:2412–2421. doi: 10.1002/1521-4141(2000)30:8<2412::AID-IMMU2412>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 22.Zanke B, Squire J, Griesser H, Henry M, Suzuki H, Patterson B, Minden M, Mak TW. A hematopoietic protein tyrosine phosphatase (HePTP) gene that is amplified and overexpressed in myeloid malignancies maps to chromosome 1q32.1. Leukemia. 1994;8:236–244. [PubMed] [Google Scholar]
- 23.Fonatsch C, Haase D, Freund M, Bartels H, Tesch H. Partial trisomy 1q. A nonrandom primary chromosomal abnormality in myelodysplastic syndromes? Cancer Genet Cytogenet. 1991;56:243–253. doi: 10.1016/0165-4608(91)90177-v. [DOI] [PubMed] [Google Scholar]
- 24.Mamaev N, Mamaeva SE, Pavlova VA, Patterson D. Combined trisomy 1q and monosomy 17p due to translocation t(1;17) in a patient with myelodysplastic syndrome. Cancer Genet Cytogenet. 1988;35:21–25. doi: 10.1016/0165-4608(88)90116-1. [DOI] [PubMed] [Google Scholar]
- 25.Bobkova EV, Liu WH, Colayco S, Rascon J, Vasile S, Gasior C, Critton DA, Chan X, Dahl R, Su Y, Sergienko E, Chung TDY, Mustelin T, Page R, Tautz L. Inhibition of the Hematopoietic Protein Tyrosine Phosphatase by Phenoxyacetic Acids. ACS Medicinal Chemistry Letters. 2011;2:113–118. doi: 10.1021/ml100103p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eswaran J, von Kries JP, Marsden B, Longman E, Debreczeni JE, Ugochukwu E, Turnbull A, Lee WH, Knapp S, Barr AJ. Crystal structures and inhibitor identification for PTPN5, PTPRR and PTPN7: a family of human MAPK-specific protein tyrosine phosphatases. Biochem J. 2006;395:483–491. doi: 10.1042/BJ20051931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 28.Barglow KT, Cravatt BF. Activity-based protein profiling for the functional annotation of enzymes. Nat Methods. 2007;4:822–827. doi: 10.1038/nmeth1092. [DOI] [PubMed] [Google Scholar]
- 29.Frye SV. The art of the chemical probe. Nat Chem Biol. 2010;6:159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- 30.Tautz L, Mustelin T. Strategies for developing protein tyrosine phosphatase inhibitors. Methods. 2007;42:250–260. doi: 10.1016/j.ymeth.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 31.Barr AJ, Ugochukwu E, Lee WH, King ON, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Muller S, Knapp S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell. 2009;136:352–363. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vintonyak VV, Antonchick AP, Rauh D, Waldmann H. The therapeutic potential of phosphatase inhibitors. Curr Opin Chem Biol. 2009;13:272–283. doi: 10.1016/j.cbpa.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 33.Willett P. Similarity-based virtual screening using 2D fingerprints. Drug Discov Today. 2006;11:1046–1053. doi: 10.1016/j.drudis.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 34.Critton DA, Tortajada A, Stetson G, Peti W, Page R. Structural basis of substrate recognition by hematopoietic tyrosine phosphatase. Biochemistry. 2008;47:13336–13345. doi: 10.1021/bi801724n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arimura Y, Yagi J. Comprehensive expression profiles of genes for protein tyrosine phosphatases in immune cells. Sci Signal. 2010;3:rs1. doi: 10.1126/scisignal.2000966. [DOI] [PubMed] [Google Scholar]
- 36.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27:1350–1359. [PubMed] [Google Scholar]
- 37.McGinnity DF, Soars MG, Urbanowicz RA, Riley RJ. Evaluation of fresh and cryopreserved hepatocytes as in vitro drug metabolism tools for the prediction of metabolic clearance. Drug Metab Dispos. 2004;32:1247–1253. doi: 10.1124/dmd.104.000026. [DOI] [PubMed] [Google Scholar]
- 38.Yang Z, Zadjura LM, Marino AM, D’Arienzo CJ, Malinowski J, Gesenberg C, Lin PF, Colonno RJ, Wang T, Kadow JF, Meanwell NA, Hansel SB. Utilization of in vitro Caco-2 permeability and liver microsomal half-life screens in discovering BMS-488043, a novel HIV-1 attachment inhibitor with improved pharmacokinetic properties. J Pharm Sci. 2010;99:2135–2152. doi: 10.1002/jps.21948. [DOI] [PubMed] [Google Scholar]
- 39.Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tautz L, Bruckner S, Sareth S, Alonso A, Bogetz J, Bottini N, Pellecchia M, Mustelin T. Inhibition of Yersinia tyrosine phosphatase by furanyl salicylate compounds. J Biol Chem. 2005;280:9400–9408. doi: 10.1074/jbc.M413122200. [DOI] [PubMed] [Google Scholar]
- 41.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 42.Alonso A, Bottini N, Bruckner S, Rahmouni S, Williams S, Schoenberger SP, Mustelin T. Lck dephosphorylation at Tyr-394 and inhibition of T cell antigen receptor signaling by Yersinia phosphatase YopH. J Biol Chem. 2004;279:4922–4928. doi: 10.1074/jbc.M308978200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.