

Primary aldosteronism (PA) is defined as a group of endocrine disorders in which aldosterone production is inappropriately high, relatively autonomous and independent of the renin-angiotensin system and extracellular potassium (K+) [the major normal aldosterone stimulators], and in which aldosterone secretion is not suppressed by sodium loading. Patients with PA exhibit hypertension, high plasma aldosterone concentration, low plasma renin activity and varying degrees of hypokalemia and metabolic alkalosis (1). The two most common causes of PA are unilateral aldosterone-producing adenoma (APA) and idiopathic hyperaldosteronism (IHA) with bilateral adrenal hyperplasia.
PA is the most common cause of secondary (remediable) hypertension, estimated to occur in approximately 8% of the general hypertensive population and in about 20% of patients with pharmacologically resistant hypertension. Because the cardiovascular morbidity and mortality rates are much higher in PA than in primary (essential) hypertension, intense research efforts have focused on understanding the pathophysiology of these disorders. While APA, estimated to account for up to 50% of PA, is potentially curable with unilateral laparoscopic adrenalectomy, IHA must be treated with life-long mineralocorticoid receptor blockers and other antihypertensive agents. For both APA and IHA, an important unresolved question has been “What drives autonomous adrenal zona glomerulosa (ZG) cell proliferation and the production of aldosterone in these most common forms of PA?”
For IHA, this question is now beginning to be addressed by studies in knockout mice, several of which have been reported as models of PA with bilaterally increased aldosterone production. The first approach has been using TASK channel knockouts. TASK (TWIK-related acid sensitive K) channels are members of the KCNK family of 2-pore domain/4 transmembrane K+ channels. The channel subunits expressed in the adrenal, TASK-1 and TASK-3, form homo- and hetero-dimeric “leak” channels that provide the background hyperpolarizing conductance important for setting the negative membrane voltage of aldosterone-producing ZG cells (2). Aldosterone production from ZG cells is calcium-dependent, and elevation in intracellular free calcium is critical for increased production to occur. Low voltage-activated T-type calcium channels, the major calcium channel of the ZG, are generally closed in the resting state but are available for opening during membrane depolarization. Recent studies have shown that TASK channels are predominant K conductance channels of the ZG cell, serving to clamp the membrane at hyperpolarized voltages, thereby restraining aldosterone production.
In 2008, it was demonstrated that double deletion of the channel subunits (TASK-1 and TASK-3 double knockout mice) produces a ~20 mV depolarization of ZG plasma membrane potential that permits autonomous overproduction of aldosterone characteristic of the IHA phenotype (Figure) (3). TASK-1 and TASK-3 double knockout mice have hypertension, increased urinary aldosterone excretion, decreased plasma renin, increased aldosterone-to-renin ratio, failure to suppress aldosterone secretion with salt-loading and failure to normalize aldosterone production with angiotensin AT1 receptor blockade, demonstrating aldosterone autonomy (3). Interestingly, single subunit TASK-1 channel knockout mice have a completely different phenotype resembling human glucocorticoid-remediable primary aldosteronism, but not associated with documented membrane depolarization due to presumed concurrent up-regulation of TASK-3 (4). TASK-3 single subunit knockout mice have yet to be reported.
Figure.
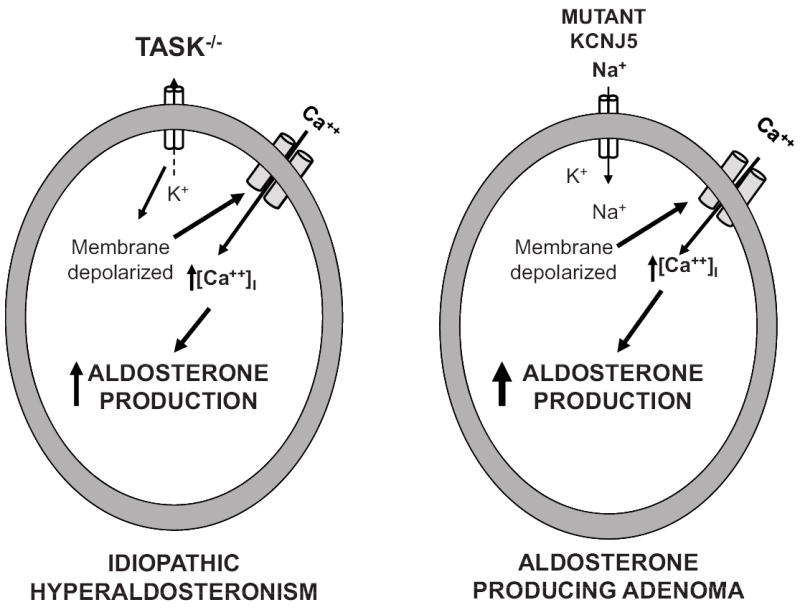
Schematic representation of proposed mechanisms controlling aldosterone hypersecretion in idiopathic hyperaldosteronism (IHA; left) and aldosterone-producing adenoma (APA; right). In the TASK-1 and-2 knockout mouse (TASK-/-), the absence of background K+ conductance from the zona glomerulosa cell leads to persistent membrane depolarization, activating T-type calcium channels and increasing intracellular Ca++ concentration [Ca++]i. In human aldosterone-producing adenomas, the mutated inwardly rectifying K+ channel encoded by KCNJ5 loses its selectivity for K+ and permits Na+ to enter the cell inducing chronic membrane depolarization which activates voltage-gated calcium channels and raises [Ca++]i. Increased [Ca++]I increases aldosterone production and drives cell proliferation.
In addition to TASK channel knockout mice, the deletion in mice of circadian clock genes cryptochrome-1 (Cry1) and -2 (Cry2), which regulate circadian biological activity, also results in PA (5). These animals have increased aldosterone production and salt-sensitive hypertension due to constitutive ZG over-expression of 3-β-hydroxysteroid dehydrogenase isoform 6, which is normally under negative transcriptional control by the circadian clock (5).Whether any of these mouse models of PA with increased bilateral aldosterone production will provide insight into the pathogenesis of human IHA and related disorders awaits further investigation, including association of single nucleotide polymorphisms of the appropriate genes.
For APA, the pathophysiology remained an enigma until 2011 when Choi et al. (6) reported that somatic mutations of the gene for KCNJ5, encoding an inwardly rectifying K+ channel expressed in human APAs, induce autonomous adrenal cell proliferation and aldosterone hypersecretion. Inwardly rectifying K+ channels are structurally different from TASK channels. Major pharmacological differences exist between KCNJ5 and TASK channels: KCNJ5 channels are voltage-dependent and are activated by G protein βγ subunits whereas TASK channels have open pores, are weakly voltage-dependent and are inhibited by extracellular H+ ions and Gα/q. The Choi et al. seminal discovery demonstrated two different mutations in or near the KCNJ5 channel selectivity filter permitting the channel to conduct Na+ (Figure). Na+ entry into the cell induces chronic membrane depolarization leading to activation of voltage-gated calcium channels. The consequent increase in intracellular calcium concentration increases the expression of enzymes involved in aldosterone biosynthesis and the activation of signals for increased cell proliferation. Thus, patients with these KCNJ5 mutations have increased constitutive aldosterone production and ZG cell proliferation resulting in APA.KCNJ5 mutations were found in 8 of 22 human APAs, all of which were relatively large (greater than 2 cm) (6). An additional mutation in KCNJ5 was also described in a father and 2 daughters (ages 4 & 7) with severe hypertension, Mendelian aldosteronism and massive adrenal hyperplasia necessitating bilateral adrenalectomy (6).The findings of this study demonstrated a role for K+ channel mutations in endocrine neoplasia and suggested that the mechanism of these KCNJ5 mutations might be related to the benign nature of these tumors.
Important questions remained, however, concerning the relative frequency of somatic KCNJ5 mutations in the overall population of APAs and the relationship of these mutations to tumor size and the phenotypic characteristics of the patients. In this issue of Hypertension, Murthy et al. (7) provide answers to these questions. First, they confirm the findings of Choi et al. (6) in a much larger sample (73 patients) from Australia and the United Kingdom. In the present study, 41 percent of sporadic APAs were found to have somatic mutations in the selectivity filter of KCNJ5.In addition, this study identifies a new mutation in the region of the selectivity filter, defines a new phenotype in which aldosterone responses to activation of the renin-angiotensin system are absent in patients with mutations, and demonstrates an association of KCNJ5 with tumor size, albeit with substantial size overlap between the genotypes (7).
The observation that APAs with KCNJ5 mutations are associated with phenotypic unresponsiveness of plasma aldosterone to upright posture in these patients is intriguing (7).This stands in marked contrast to aldosterone stimulation with upright posture in normal subjects, patients with IHA and the subset of APA without mutations. Since upright posture activates the renin-angiotensin system, aldosterone responsiveness to this stimulus has been attributed to adrenal angiotensin II sensitivity. The simplest explanation for failure of this mechanism in APA is that the cells are already maximally activated and cannot be further activated by Ang II.
The recent identification of KCNJ5 mutations in the pathophysiology of APA raises a number of intriguing questions: (1) Why do some patients with these mutations develop bilateral adrenal hyperplasia, while the majority develop APAs? (2) Why does the common pathway of increased intracellular calcium that results from ZG cell membrane depolarization lead to APA in humans but to IHA with bilateral adrenal hyperplasia in mice? (3) Why do ZG cells become hyper-responsive to Ang II in IHA and in some, but not all, patients with APA? Answers to these questions await future research.
During the past 4 years, new discoveries in the molecular biology and genetics of PA have led to greater understanding of the pathogenesis of both IHA and APA. The studies reported here by Murthy et al. (7) enrich our knowledge of the role of KCNJ5 in the etiology, phenotypic characteristics, epidemiology of APA and help refine the modern diagnostic approach to this relatively common and important form of human hypertension.
Acknowledgments
Sources of Funding This work was supported by grants from the National Institutes of Health (R01-HL-095796, R01-HL-087998 and P01-HL-074940).
Footnotes
Disclosures None
References
- 1.Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M, Young WF, Jr, Montori VM. Case detection, diagnosis and treatment of patients with Primary aldosteronism: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93:3266–3281. doi: 10.1210/jc.2008-0104. [DOI] [PubMed] [Google Scholar]
- 2.Bayliss DA, Barrett PQ. Emerging roles of two-pore domain potassium channels and their potential therapeutic impact. Trends Pharmacol Sci. 2008;29:566–575. doi: 10.1016/j.tips.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies LA, Hu C, Guagliardo NA, Sen N, Chen X, Talley EM, Carey RM, Bayliss DA, Barrett PQ. Task channel deletion in mice causes primary hyperaldosteronism. Proc Natl Acad Sci U S A. 2008;105:2203–2208. doi: 10.1073/pnas.0712000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heitzmann D, Derand R, Jungbauer S, Bandulik S, Sterner C, Schweda F, El Wakil A, Lalli E, Guy N, Mengual R, Reichold M, Tegtmeier I, Bendahhou S, Gomez-Sanchez CE, Aller MI, Wisden W, Weber A, Lesage F, Warth R, Barhanin J. Invalidation of TASK1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. Embo J. 2008;27:179–187. doi: 10.1038/sj.emboj.7601934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, Emoto N, Okuno Y, Tsujimoto G, Kanematsu A, Ogawa O, Todo T, Tsutsui K, van der Horst GT, Okamura H. Salt-sensitive hypertension in circadian clock-deficient Cry-null mices involves dysregulated adrenal Hsd3b6. Nat Med. 2010;16:67–74. doi: 10.1038/nm.2061. [DOI] [PubMed] [Google Scholar]
- 6.Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, Lolis E, Wisgerhof MV, Geller DS, Mane S, Hellman P, Westin G, Akerstrom G, Wang W, Carling T, Lifton RP. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331:768–772. doi: 10.1126/science.1198785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murthy M, Azizan EAB, Stowasser M, Gordon R, Kowalski B, Xu S, Brown MJ, O’Shaughnessy KM. Somatic mutations affecting the selectivity filter of KCNJ5 are frequent in two large unselected collections of adrenal aldosteronomas. Hypertension. 2012 doi: 10.1161/HYPERTENSIONAHA.111.186239. [DOI] [PubMed] [Google Scholar]