

Abstract
The carboxypeptidase A enzyme from Metarhizium anisopliae (MeCPA) has broader specificity than the mammalian A-type carboxypeptidases, making it a more useful reagent for the removal of short affinity tags and disordered residues from the C-termini of recombinant proteins. When secreted from baculovirus-infected insect cells, the yield of pure MeCPA was 0.25 mg per liter of conditioned medium. Here, we describe a procedure for the production of MeCPA in the cytosol of Escherichia coli that yields approximately 0.5 mg of pure enzyme per liter of cell culture. The bacterial system is much easier to scale up and far less expensive than the insect cell system. The expression strategy entails maintaining the proMeCPA zymogen in a soluble state by fusing it to the C-terminus of maltose-binding protein (MBP) while simultaneously overproducing the protein disulfide isomerase DsbC in the cytosol from a separate plasmid. Unexpectedly, we found that the yield of active and properly oxidized MeCPA was highest when coexpressed with DsbC in BL21(DE3) cells that do not also contain mutations in the trxB and gor genes. Moreover, the formation of active MeCPA was only partially dependent on the disulfide-isomerase activity of DsbC. Intriguingly, we observed that most of the active MeCPA was generated after cell lysis and amylose affinity purification of the MBP-proMeCPA fusion protein, during the time that the partially purified protein was held overnight at 4 °C prior to activation with thermolysin. Following removal of the MBP-propeptide by thermolysin digestion, active MeCPA (with a C-terminal polyhistidine tag) was purified to homogeneity by immobilized metal affinity chromatography (IMAC), ion exchange chromatography and gel filtration.
Keywords: MeCPA, carboxypeptidase, maltose-binding protein, polyhistidine tag, thermolysin, His-tag, DsbC, DsbA, protein disulfide isomerase, trxB, gor
Introduction
Affinity tags have become indispensable tools for the purification of recombinant proteins [1–3], yet the negative impact of tags on protein structure and function has been well documented [4–15] and many more instances are likely to have gone unreported. Accordingly, reliable methods for the removal of affinity tags are needed.
N-terminal affinity tags can be removed by highly specific endoproteases (e.g. TEV protease or enteropeptidase) that are able to produce digestion products with native N-termini in many cases [16]. This is possible because all or nearly all of the specificity determinants for these enzymes are situated on the N-terminal side of the scissile bond. However, this feature of endoproteases renders them less desirable for the removal of C-terminal affinity tags. For example, endoproteolytic removal of a hexahistidine tag from the C-terminus of a protein with TEV protease, by digestion of the sequence ENLYFQ↓GHHHHHH, would leave behind six residues from the TEV protease recognition site, effectively negating any advantage that might be gained by removing the six histidines.
An alternative means of removing short C-terminal tags is to utilize an exoprotease instead of an endoprotease. Histidine and most other amino acids are substrates for bovine carboxypeptidase A [17,18]. Recently, we described the production of an A-type carboxypeptidase from the entomopathogenic fungus Metarhizium anisopliae (MeCPA) in the baculovirus expression system and its purification to homogeneity [17]. This recombinant MeCPA has the C-terminal tag HHHHHHPR. The hexahistidine sequence was designed to facilitate the purification of the enzyme from conditioned medium as well as its removal from the products of a carboxypeptidase digest, while the C-terminal PR dipeptide was intended to prevent autodigestion of the polyhistidine tag (A-type carboxypeptidases do not digest C-terminal lysine, arginine, or proline residues [17,18]). The recombinant MeCPA-His6PR was highly active and had even broader specificity than bovine CPA [17]. For instance, amino acids that are cleaved very slowly by BoCPA, such as the acidic residues and glycine, are removed far more efficiently by MeCPA-His6PR [17]. However, the relatively low yield of pure enzyme from the baculovirus system (ca. 0.25 mg/liter) prompted us to explore alternative ways of producing it. The presence of two disulfide bonds in MeCPA and the essential role played by the prodomain in its folding present significant challenges for the production of this enzyme in heterologous expression systems. Here, we describe a strategy for the expression of MeCPA-His6PR in the cytosol of Escherichia coli and a relatively simple procedure to purify it to homogeneity. The bacterial system yields about 0.5 mg of pure enzyme per liter of cell culture and is more convenient and less expensive than is the production of MeCPA in insect cells.
Materials and methods
Bacterial strains
E. coli BL21(DE3) (fhuA2 [lon] ompT gal (λ DE3) [dcm] ΔhsdS; λ DE3 = λ sBamHIo ΔEcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 Δnin5) and Origami™ B(DE3) cells (F− ompT hsdSB(rB− mB−) gal dcm lacY1 ahpC (DE3) gor522:: Tn10 trxB (KanR, TetR) were obtained from Novagen (Madison, WI). E. coli SHuffle T7 Express™ cells (fhuA2 lacZ::T7 gene1 [lon] ompT ahpC gal λatt::pNEB3-r1-cDsbC (SpecR, lacIq) ΔtrxB sulA11 R(mcr-73::miniTn10--TetS)2 [dcm] R(zgb-210::Tn10 --TetS) endA1 Δgor Δ(mcrC-mrr)114::IS10) were obtained from New England Biolabs (Ipswich, MA).
Construction of the MBP-proMeCPA-His6PR fusion protein expression vector pBA2257
The open reading frame (ORF) encoding proMeCPA-His6PR was amplified by polymerase chain reaction (PCR), using the baculovirus expression vector 1177-X1-8 [17] as the template, with primers PE-2353 (5′-GGGG ACA AGT TTG TAC AAA AAA GCA GGC TCG ATG CCG GCT GAG AGC CC-3′) and PE-2264 (5′-GGGG ACC ACT TTG TAC AAG AAA GCT GGG TTA TTA GCG AGG ATG GTG ATG GTG ATG GTG ACT CAT CTG C-3′). An attB1 recombination site preceded codon 17 of pre-proMeCPA at one end of the resulting PCR amplicon (eliminating the N-terminal signal peptide). At the other end, the C-terminus of the MeCPA ORF (codon 418) was extended by the addition of DNA encoding a HHHHHHPR tag (hereafter denoted His6PR) and a pair of tandem termination codons, followed by an attB2 recombination site. This PCR amplicon was recombined into pDONR201 (Invitrogen, Carlsbad, CA) via the Gateway BP reaction to create pBA2256 and the construct was verified by DNA sequencing. The proMeCPA-His6PR ORF was subsequently recombined into pKM596 [19] via the Gateway LR reaction to create the expression vector pBA2257, encoding the MBP-proMeCPA-His6PR fusion protein.
Construction of the DsbC expression vector pBA2219
First, the small XbaI/BamHI fragment of pDW387 [20], a p15A derivative, was replaced with the corresponding fragment of pET3d (Novagen) to construct pDW402. Second, pDW402 was linearized with BamHI and the 5′ overhangs filled in with the Klenow fragment of DNA polymerase I (New England Biolabs). The 5′ phosphates were then removed from the blunt ends by Calf Intestinal Alkaline Phosphatase (New England Biolabs) after which the Gateway cloning cassette RfA (Invitrogen) was ligated between them using T4 DNA ligase (New England Biolabs). The orientation of the inserted Gateway cloning cassette was confirmed by DNA sequencing. This Gateway destination vector was named pBA2220. Third, the E. coli DsbC ORF (except for the DNA encoding its signal peptide) was amplified by PCR from pDW395 [21] using primers PE-2297 (5′-GGGG ACA ACT TTG TAC AAA AAA GTT GTG GAC GAC GCG GCA ATT CAA CAA ACG-3′) and PE-2276 (5′-GGGG ACA ACT TTG TAC AAG AAA GTT GCA TTA TTA TTT ACC GCT GGT CAT TTT TTG G-3′). This PCR amplicon was recombined into pDONR221 (Invitrogen) via the BP reaction to create pBA2258. Finally, the DsbC ORF was moved from pBA2258 into the destination vector pBA2220 via the Gateway LR reaction to create the DsbC expression vector pBA2219.
Construction of the mutant DsbC (C118A/C121A) expression vector pBA2283
The plasmid pBA2283, A double mutant (C118A/C121A) derivative of the DsbC expression vector pBA2219 was constructed by overlap extension PCR [22], using primers PE-2297 and PE-2276 in conjunction with the mutagenic primers PE-2394 (5′-GTT TAC TGA TAT TAC CGC GGG TTA CGC GCA CAA ACT GCA TGA GC-3′) and PE-2395 (5′-GCT CAT GCA GTT TGT GCG CGT AAC CCG CGG TAA TAT CAG TAA AC-3′) followed by a Gateway BP reaction with pDONR221 to produce pBA2259. The plasmid pBA2259 was then recombined with pBA2220 via the Gateway LR reaction to generate the DsbC mutant expression vector pBA2283.
Construction of DsbC-DsbA fusion vector pBA2285
The plasmid pBA2285, encoding a DsbC-DsbA fusion protein, was constructed by overlap extension PCR [22]. The DsbC ORF was amplified from pBA2219 using primers PE-2297 and PE-2413 (5′-ACC GCC ACC CTG GAA GTA CAG GTT CTC ACC GCC ACC TTT ACC GCT GGT CAT TTT TTG GTG TTC G-3′). The DsbA ORF was amplified from pDW394 [21] using primers PE-2364 (5′-GGGG ACC ACT TTG TAC AAG AAA GCT GGG TTA TTA TTT TTT CTC GGA CAG ATA TTT C-3′) and PE-2412 (5′-GGT GGC GGT GAG AAC CTG TAC TTC CAG GGT GGC GGT GCG CAG TAT GAA GAT GGT AAA CAG-3′). The two ORFs were fused together during a third PCR, using the amplicons from the first two PCRs as template, with primers PE-2297 and PE-2364. The final PCR product encoded a DsbC-DsbA fusion protein with the linker peptide GGGENLYFQGGG between the two domains. This PCR amplicon was recombined into pDONR221 (Invitrogen) via the BP reaction to create pBA2284 and the DNA sequence was confirmed. Finally, the DsbC-DsbA ORF was moved into the destination vector pBA2220 to create pBA2285.
Transformation and growth of bacteria
Competent E. coli cells were purchased and transformed with pBA2257 plasmid DNA in accordance with the instructions of the vendor. Transformants were selected on Luria-Bertani (LB) agar plates containing 125 mg/L of ampicillin. In some cases, cells were simultaneously transformed with pBA2257 and the DsbC expression vector pBA2219, the otherwise identical plasmid pBA2283 encoding catalytically inactive DsbC, or the DsbC-DsbA fusion protein expression vector pBA2285. Co-transformants were selected on LB agar plates containing ampicillin (125 mg/L) and kanamycin (35 mg/L).
LB medium (Quality Biological, Gaithersburg, MD) containing the appropriate antibiotic(s) was inoculated with a single colony of freshly transformed cells and incubated with shaking at 250 rpm in a New Brunswick Scientific I2500 incubator shaker (Edison, NJ) at 30°C for 16 hours (overnight). The following day, 1 L of LB containing the appropriate antibiotic(s) along with 0.2% dextrose and 100 μM ZnCl2 was inoculated with an overnight culture to achieve an initial OD600 of 0.1. The cells were grown at 30°C and 250 rpm until the OD600 reached 0.3–0.5. (In experiments where multiple cultures were grown at the same time, each culture was transferred to an incubator set at 18°C without shaking until all cultures reached the target density range.) At that time, 5 mL of 200 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to each flask and the cultures were grown overnight at 18°C with shaking.
Amylose affinity purification and activation of MBP-proMeCPA-His6PR
Cells from 1 L cultures grown overnight at 18°C were pelleted by centrifugation and resuspended in 75 mL of ice-cold 50 mM sodium phosphate (pH 7.5) and 150 mM NaCl (buffer A). The cells were broken by an APV-1000 homogenizer (Invensys APV Products, Albertslund, Denmark) at 69 MPa and centrifuged at 30,000 g for 30 min. The supernatant was filtered through a 0.45 μm cellulose acetate membrane. The filtrate was applied to a 5 mL MBP-trap column (GE Healthcare Bio-Sciences Corp., Piscataway, New Jersey), washed with ten column volumes of buffer A, and then eluted with buffer A containing 30 mM maltose. Fractions containing the MBP-proMeCPA-His6PR fusion protein were pooled. When multiple affinity purifications were performed in rapid succession, the protein concentrations of all samples were adjusted to be the same, based upon their absorbance at 280 nm, after amylose affinity chromatography by the addition of buffer A when necessary. Next, 10 mL of 5X concentrated thermolysin buffer (100 mM Tris-HCl pH 8.0, 1.5 M NaCl, 20 mM CaCl2, 50% glycerol) was added to 40 mL of each protein sample. A 5 mg/mL stock solution of thermolysin (EMB Life Sciences, La Jolla, CA) was prepared in 1X thermolysin buffer and an equal volume was added to each sample. Two different thermolysin digestion conditions were employed. In analytical scale experiments (1 L cultures), an enzyme to substrate ratio of 1:30 was used and the reactions were incubated at 37 °C for 1.5 hours. However, for the large scale purification of MeCPA-His6PR (6 × 1 L cultures), an enzyme:substrate ratio of 1:300 was used and the reactions were incubated for 8 hrs at 25 °C. In either case, the reactions were then clarified by centrifugation at 4500 × g and filtration through a 0.22 μm polyethersulfone membrane.
Purification of MeCPA-His6PR
The clarified thermolysin digest was adjusted with 50 mM phosphate-buffered saline (PBS) pH 7.5 (buffer B) containing 500 mM imidazole to a final imidazole concentration of 25 mM and immediately applied to a 5 mL HisTrap column (GE Healthcare BioSciences Corp.) equilibrated with buffer B containing 25 mM imidazole. The column was washed with 10 column volumes of buffer B containing 25 mM imidazole, and then the bound protein was eluted with a gradient of 25–500 mM imidazole in buffer B over 3 column volumes. The peak fractions were pooled, concentrated in an Amicon stirred cell using a YM-10 membrane (Millipore, Billerica, MA 01821 USA) and then diluted 1:100 with 20 mM Tris-HCl pH 7.3 (buffer C). The sample was then applied to an 5 mL SP HP cation exchange column (GE Healthcare) equilibrated with buffer C. The column was washed with 10 volumes of buffer C and then the bound protein was eluted over 30 column volumes of buffer C with a gradient of 0–1 M NaCl. The peak fractions corresponding to activated MeCPA-His6PR were pooled and applied to a Sephacryl S100 16/60 preparative gel filtration column equilibrated with buffer B. The peak fractions were pooled, concentrated to 2 mg/mL, divided into 20 mL aliquots, flash frozen under liquid nitrogen, and stored at −80 °C until use. Data pertaining to the yield and purity of protein at each step in the process are provided in Table 1.
Table 1.
Purification of MeCPA-His6PR
| Purification step | Total protein/La | Purification-fold | Purityb | Yield/L |
|---|---|---|---|---|
| Bacterial lysate | 2.7 g | 1 | 0 | 2.7 g |
| Amylose | 2.7 g | 19.3 | 45.2 % | 0.14 g |
| Thermolysin digestion | 0.12 g | 1 | 1.4 % | 0.12 g |
| IMAC | 0.12 g | 171.4 | 96.5 % | 0.70 mg |
| Ion-exchange | 0.70 mg | 1.43 | >98.0 % | 0.49 mg |
| Gel filtration | 0.49 mg | 1.40 | >98.0 % | 0.34 mg |
Protein concentration and total amount of protein was determined by measuring its absorbance at 280 nm.
Purity of MBP-proMeCPAHis6PR (up until thermolysin digestion) and MeCPA- His6PR (after thermolysin digestion) was estimated by SDS-PAGE using ImageQuant software.
Spectrophotometric assay for MeCPA-His6PR activity
Activity assays were conducted essentially as described [23]. In brief, approximately 1–3 mg of N-(3-[2-Furyl]acryloyl)-Phe-Phe (FAPP) (Sigma-Aldrich, St. Lous, MO) was dissolved in 50 μL of dimethyl sulfoxide (DMSO) and always prepared fresh. Assay buffer (94 μL of 25 mM Tris-HCl pH 7.5, 100 mM NaCl) was used as a “blank” for spectrophotometeric measurements at a wavelength of 340 nm in a 100 μL quartz cuvette. One mL of FAPP was added to the assay buffer and diluted if necessary to give an initial A330 reading of 0.5–0.7. The assay was initiated by addition of 5 μL of protein (either partially purified protein after thermolysin treatment or samples of greater purity obtained after additional chromatography steps). Spectrophotometric measurements were recorded at 15 sec intervals over 3 min.
Results
Production of MeCPA in the baculovirus expression system
Seeking to emulate a published procedure for the production of active MeCPA from baculovirus-infected insect cells [24], we engineered a modified form of MeCPA (MeCPA-His6PR) with a C-terminal polyhistidine tag followed by a Pro-Arg dipeptide (to prevent autodigestion of the His-tag) and succeeded in secreting it as described [17]. However, in contrast to the previous report, in which the enzyme was observed to accumulate in the mature, enzymatically active form, we detected only the inactive zymogen (proMeCPA-His6PR) in the medium [17]. Consequently, we needed to find a means of activating the zymogen in vitro, following partial purification by IMAC. By trial and error, we discovered that thermolysin very efficiently cleaved the zymogen to yield active MeCPA-His6PR, which proved to be remarkably resistant to further digestion by thermolysin. However, in our hands, the yield of pure MeCPA-His6PR from the baculovirus expression system was only about 0.25 mg per liter of conditioned medium, making it prohibitively expensive to produce in this manner for the purpose of using it as a reagent to remove affinity tags from recombinant proteins.
Alternative strategies for the production of MeCPA-His6PR
A number of strategies were subsequently investigated to increase the yield of MeCPA-His6PR and thereby make it less expensive to produce. These included efforts to secrete the zymogen from the yeasts Pichia pastoris and Kluyveromyces lactis, from the Gram-positive bacterium Brevibacillus choshinensis, and from mammalian Chinese hamster ovary cells. We also attempted to produce proMeCPA-His6PR in the periplasm and cytoplasm of E. coli, in both fused and unfused formats. Additionally, both traditional and hyperbaric refolding protocols were explored, using inclusion bodies purified from E. coli. To detect MeCPA-His6PR activity, crude lysates, refolding reactions, culture supernatants, and/or partially purified fusion proteins were digested with thermolysin and subjected to a standard spectrophotometric assay to measure carboxypeptidase activity [23]. Unfortunately, no carboxypeptidase activity could be detected in any of these samples (data not shown).
A novel method for the production of active MeCPA-His6PR in E. coli
We next experimented with a strain of E. coli developed at New England Biolabs called T7 SHuffle Express™. These cells are derived from the BL21 strain first popularized by Studier and colleagues for protein expression using the bacteriophage T7 promoter [25]. T7 SHuffle Express™ cells have been modified to inactivate the thioredoxin reductase (trxB) and glutathione reductase (gor) genes. Together, these mutations have the effect of generating a more oxidizing environment in the cytoplasm, which has been shown to promote the formation of disulfide bonds in this cellular compartment [26,27]. Additionally, T7 SHuffle Express™ cells produce the (normally periplasmic) E. coli protein disulfide isomerase DsbC in the cytoplasm, from a single chromosomal copy of the gene lacking its signal peptide.
Although not yet explicitly demonstrated for MeCPA, the propeptides of closely related carboxypeptidases have been shown to act as chaperones that are required to promote the proper folding of the enzymes [28]. Accordingly, we proceeded under the assumption that MeCPA-His6PR must initially be produced as the zymogen, with its propeptide attached (Fig 1). However, when proMeCPA-His6PR is expressed in E. coli, it accumulates exclusively as insoluble aggregates (data not shown). Therefore, our strategy was to fuse proMeCPA-His6PR to the C-terminus of E. coli maltose-binding protein, a highly effective solubilizing agent [29], to maintain it in a soluble form so that it might have the opportunity to fold and form its two disulfide bonds in T7 SHuffle Express cells (Fig 1). Indeed, as shown in Fig. 2, significant carboxypeptidase activity was detected after amylose affinity purification of the soluble MBP-proMeCPA-His6PR fusion protein from T7 SHuffle Express™ cells and removal of the propeptide by thermolysin digestion (Fig. 2, ○). Thermolysin itself does not digest the FAPP substrate (Fig. 2, ■).
Fig 1.

(A) Schematic diagram of the recombinant proteins used in this study. Top, wild-type and mutant DsbC produced by pBA2219 and pBA2283, respectively. Middle, DsbC/DsbA fusion protein produced by pBA2285. Bottom, MBP-proMeCPAHis6RP fusion protein produced by pBA2257. In-frame amino acid extensions and interdomain linkers arising from Gateway cloning artifacts are indicated by lines. (B) Amino acid sequence of proMeCPA-His6RP. The thermolysin cleavage site is indicated by an arrow. The two pairs of cysteine residues that form disulfide bonds [24] are enclosed by boxes and linked together by lines. The C-terminal His6PR tag is underlined.
Fig. 2.

Comparison of MeCPA activity in partially purified enzyme preparations from different strains of E. coli. All samples were prepared in an identical manner, as detailed in the Materials and methods. Briefly, cultures expressing the MBP-proMeCPA-His6PR fusion protein, and in some cases also expressing wild-type or mutant DsbC or a DsbC/DsbA fusion protein, were induced with IPTG and grown overnight at 18 °C. The cells were pelleted by centrifugation, resuspended in buffer A, lysed, and subjected to amylose affinity chromatography. Equal amounts of the partially pure fusion proteins were treated with thermolysin and then assayed for carboxypeptidase activity by monitoring the decrease in absorbance at 330 nm that accompanies the hydrolysis of FAPP. (■), thermolysin only (no protein); (△), BL21(DE3) cells; (×), Origami B(DE3) cells; (○), T7 SHuffle Express cells; (●), T7 SHuffle Express cells + pBA2219; (▲), BL21(DE3) cells + pBA2219; (□), BL21(DE3) cells + pBA2219; (◆), BL21(DE3) cells + pBA2285.
This discovery prompted us to compare the yield of active MeCPA-His6PR obtained from three closely related strains of bacteria: BL21(DE3), Origami™ B(DE3), and T7 SHuffle Express™ (Fig. 2). We found that T7 SHuffle Express™ produced the greatest amount of active MeCPA-His6PR (Fig. 2, ○), followed by Origami™ B (DE3) (Fig. 2, ×), but essentially no carboxypeptidase activity could be detected when the fusion protein was purified by amylose affinity chromatograpny from BL21(DE3) cells and treated with thermolysin (Fig. 2, △). The major difference between Origami™ B cells and T7 SHuffle Express™ cells is that the latter strain expresses DsbC in the cytosol; both strains contain the trxB and gor mutations. We therefore wondered if overprodution of DsbC in the cytosol, beyond the level achieved in T7 SHuffle Express™ cells, might further improve the yield of active MeCPA-His6PR. To this end, a separate plasmid vector (pBA2219) was constructed to co-express DsbC with the MBP-proMeCPA-His6PR fusion protein (Fig 1). Transcription of the dsbC gene in pBA2219 is regulated by the bacteriophage T7 promoter and, like the MBP-proMeCPA-His6PR fusion protein, the production of DsbC is induced by IPTG.
When we utilized the DsbC plasmid expression vector in conjunction with the MBP-proMeCPA-His6PR expression vector in T7 SHuffle Express™ cells, we observed a marked increase in the amount of active MeCPA that was produced (Fig. 2, ●), consistent with the idea that overproduction of DsbC leads to higher yields of properly oxidized proteins in the cytosol of E. coli [27]. Next, we investigated what would happen if the MBP-proMeCPA-His6PR fusion protein was co-expressed with DsbC in BL21(DE3) cells (which do not have the trxB and gor mutations). Remarkably, this resulted in a dramatic increase in the yield of properly folded and oxidized MeCPA-His6PR (Fig. 2, ▲). This unexpected result was an important breakthrough because trxB/gor mutants grow much more slowly than their wild-type counterparts and do not achieve as high a post-induction cell density.
When samples of the total intracellular protein from the strains analyzed in the experiment depicted in Fig. 2 were examined by SDS-PAGE, it became apparent that, for an unknown reason, BL21(DE3) cells containing the DsbC overproducing plasmid were making much higher levels of DsbC than were T7 SHuffle Express™ cells containing the same DsbC plasmid (Fig. 3). Thus, we observed a correlation between the level of DsbC that was produced and the amount of active MeCPA-His6PR that was recovered from the cells, and the production of properly oxidized MeCPA-His6PR was not impeded by the absence of the trxB and gor mutations in BL21(DE3). Therefore, we chose to use BL21(DE3) cells containing the MBP-proMeCPA-His6PR expression vector and the DsbC overproducing plasmid as a starting point for the large-scale purification of MeCPA-His6PR from E. coli.
Fig. 3.

Expression of MBP-proMeCPA-His6PR in various strains of E. coli, as indicated, with or without the DsbC expression vector pBA2219. Samples of the total (T) and soluble (S) intracellular protein are shown. M indicates molecular weight markers (kDa).
The protein disulfide isomerase activity of DsbC contributes in part to the yield of active MeCPA-His6PR
It seemed reasonable to imagine that the protein disulfide isomerase activity of DsbC contributed to the accumulation of active MeCPA-His6PR, which has two disulfide bonds. However, DsbC has been observed to exhibit chaperone-like behavior that is independent of its catalytic activity [30]. To investigate this question, we constructed pBA2283, a DsbC expression vector otherwise identical to pBA2219 that produces a mutant DsbC in which both redox-active cysteines have been replaced by alanine residues (Fig 1). The DsbC mutant was co-expressed with the MBP-proMeCPA-His6PR fusion protein in BL21(DE3) cells, after which the fusion protein was subjected to amylose affinity chromatography, treated with thermolysin, and assayed for carboxypeptidase activity. Intriguingly, co-expression of MBP-proMeCPA-His6PR with the DsbC mutant gave rise to an intermediate level of MeCPA activity (Fig. 2, □); less than that obtained upon co-expression of wild-type DsbC but significantly more than what was detected in the absence of DsbC.
Co-expression of DsbA along with DsbC in the cytosol does not lead to a further improvement in the yield of active MeCPA-His6PR
Whereas DsbC is involved mainly in the rearrangement (shuffling) of disulfide bonds in E. coli, the strongly oxidizing enzyme DsbA is thought to catalyze their initial formation [31]. Therefore, we next sought to investigate whether co-expression of both DsbC and DsbA in the cytosol would further improve the yield of active MeCPA-His6PR. To accomplish this, we constructed a plasmid vector to produce a DsbC-DsbA fusion protein (Fig. 1), thereby ensuring that the cells would contain the same amount of each enzyme (although DsbC is a homodimer and DsbA is a monomer). The DsbC-DsbA fusion protein was co-expressed with the MBP-proMeCPA-His6PR fusion protein in BL21(DE3) cells, after which the latter fusion protein was purified by amylose affinity chromatography, treated with thermolysin, and assayed for carboxypeptidase activity. The result was definitive: co-expression of the DsbC-DsbA fusion did not improve the yield of proMeCPA-His6PR fusion protein beyond what was achieved by co-expression of DsbC alone (Fig. 2, ◆).
The post-lysis incubation effect
Our initial purification protocol (employed in all of the experiments described so far) entailed breaking the E. coli cells in a cell homogenizer, capturing the MBP-proMeCPA-His6PR fusion protein on amylose resin, eluting it with maltose, and letting the eluate sit overnight at 4 °C prior to removal of the propeptide by thermolysin the next morning. However, one day we ran the amylose column and performed the thermolysin digest immediately afterwards. Under these conditions, very little carboxypeptidase activity was detected (Fig. 4). This prompted us to conduct the following experiment. A culture of E. coli BL21(DE3) cells co-expressing the MBP-proMeCPA-His6PR fusion protein and DsbC was pelleted, resuspended in amylose purification buffer, lysed, and clarified by centrifugation. One third of the supernatant was immediately subjected to amylose affinity chromatography, digested with thermolysin, and assayed for carboxypeptidase activity (Fig. 4, ■). Another third of the cleared lysate was incubated overnight at 4 °C, then subjected to amylose affinity chromatography, digested by thermolysin, and assayed for activity (Fig 4, ▲). The final portion of the cleared lysate was immediately subjected to amylose affinity chromatography, after which the eluate was incubated overnight at 4 °C and then activated by thermolysin and assayed for activity (Fig. 4, ●). The material that was affinity purified immediately after lysis but allowed to stand overnight at 4 °C before treatment with thermolysin (the original protocol) yielded much more active MeCPA-His6PR than did the other protocols.
Fig 4.

Comparison of MeCPA activity obtained from three different protocols. (◆), Buffer only control; (■), Clarified cells lysate was immediately subjected to amylose affinity chromatography, digested with thermolysin and assayed; (▲), Clarified cell lysate was incubated overnight at 4 C, then subjected to amylose affinity chromatography, digested immediately afterward by thermolysin and assayed; (●), Clarified cell lysate was immediately subjected to amylose affinity chromatography and then incubated overnight prior to thermolysin digestion and activity assay.
Large-scale purification of MeCPA-His6PR from E. coli
On the basis of the experiments described above, we devised a simple and scalable protocol for the purification of active MeCPA-His6PR from the cytosol of E. coli. A step-wise SDS-PAGE analysis of the steps in the process is shown in Fig. 5, and the yield at each step is reported in Table 1. The optimum yield is obtained from BL21(DE3) cells that are co-expressing the MBP-proMeCPA-His6PR fusion protein and DsbC in the cytosol. The fusion protein is first purified by amylose affinity chromatography, which yields a major band corresponding to the fusion protein, but also a ladder of contaminants that may represent proteolytic fragments of the fusion protein. As discussed above, it is important to incubate the affinity-purified fusion protein overnight at 4 °C prior to cleavage of the fusion protein by thermolysin to yield the mature, active MeCPA-His6PR. There is a massive loss of material upon treatment of the amylose-purified material with thermolysin; the active MeCPA-His6PR is barely visible on an SDS gel as a very faint band running just below the prominent digestion product, which is MBP (Fig. 5, lane 4). The active MeCPA-His6PR, now separated from its prodomain, is subsequently purified by IMAC, utilizing its C-terminal His-tag. Unfortunately, the thermolysin digestion conditions employed here, which we made an extensive effort to optimize, still result in a minor amount of endoproteolytic cleavage within the C-terminal His-tag, leading to autodigestion of the remainder of the tag by MeCPA-His6PR to yield MeCPA-His2. The amino acid sequence of the truncated carboxypeptidase was deduced from the results of LC-electrospray mass spectrometry analysis (data not shown). Consequently, a small portion (ca. 10%) of the MeCPA is rendered “tagless”. Alternatively, if the thermolysin digest is incomplete, then a second protein, corresponding to proMeCPA-His6PR attached to part of the N-terminal linker peptide that originally joined it to MBP will also be present after IMAC and can not be removed by ion exchange or gel filtration chromatography. However, the minor amount of C-terminally truncated MeCPA-His2 is readily separated from the full-length MeCPA-His6PR on an ion exchange column, as shown in Fig. 6. We used gel filtration chromatography as a final polishing step (Fig 5), obtaining a final yield of 0.34 mg per liter of cell culture (Table 1). However, the protein is pure, free of thermolysin, and ready to use after ion exchange chromatography at which point the yield was 0.49 mg per liter of cell culture.
Fig. 5.
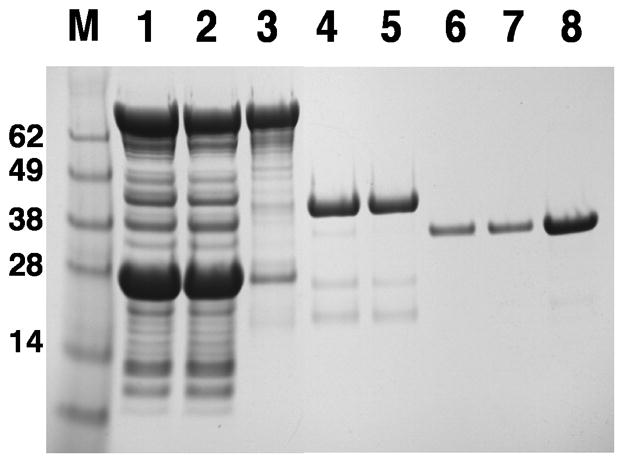
SDS-PAGE analysis of steps in the large-scale purification of MeCPA-His6PR. Lane 1, soluble intracellular protein; lane 2, flow-through from amylose column; lane 3, protein eluted from the amylose column; lane 4, protein after digestion with thermolysin; lane 5, flow-through from Ni-NTA column; lane 6, protein eluted from Ni-NTA column; lane 7, protein after ion exchange chromatography; lane 8, protein after gel filtration. M indicates molecular weight markers (kDa).
Fig. 6.

Separation of MeCPA-His2 from MeCPA-His6PR by ion exchange chromatography. Peaks 1 and 2 correspond to MeCPA-His2 and MeCPA-His6PR, respectively. The identity of the peaks was determined by LC electrospray mass spectrometry.
Discusson
In this report, we describe a novel means of producing active and fully oxidized MeCPA-His6PR from the cytosol of E. coli. The zymogen is maintained in a soluble form by fusing it to MBP, thereby enabling the disulfide isomerase DsbC to assist with the formation of the proper disulfide bonds and evidently also with the folding of proMeCPA. The yield of pure product exceeds that obtained from baculovirus-infected insect cells and significantly reduces the cost of producing this reagent, which has been shown to be an effective enzyme for removing short affinity tags from the C-termini of recombinant proteins [17]. The economic advantages are realized by obviating the need for expensive insect cell medium and avoiding other complications associated with baculovirus expression. Additionally, it is more convenient to work from a cell pellet as starting material rather than a large volume of conditioned medium.
The final yield of MeCPA-His6PR (~ 0.5 mg per liter of cell culture) is still rather low for a bacterial expression system. One explanation for this may be that the majority of the soluble MBP fusion protein did not bind to the amylose column (Fig. Lane 2). The unbound fusion protein did not bind to a fresh amylose column either (data not shown), indicating that the original column was not saturated. Rather, the unbound material probably consists of soluble aggregates in which the MBP moiety is incapable of adhering to amylose resin.
An important aspect of the purification protocol described here is the use of thermolysin to activate MBP-proMeCPA-His6PR by removing the MBP and prodomain from the MeCPA-His6PR. As shown in Fig. 5, thermolysin also obliterates practically all of the contaminants that are present after amylose affinity chromatography, which is a major advantage for subsequent purification steps. Unfortunately, however, nearly all of the MeCPA is also degraded by thermolysin, indicating that only a very small fraction of the MeCPA that is fused to MBP is properly folded and oxidized. The sub-stoichiometric yield of thermolysin-resistant MeCPAHis6RP, along with inefficient binding of the fusion protein to amylose resin, are the principal reasons for the low overall yield of active enzyme. We have identified by mass spectrometry and N-terminal amino acid sequencing the largest stable thermolysin digestion product (Fig. 5, lane 4) as MBP with a portion of the inter-domain linker peptide attached (data not shown). It is easily removed during subsequent IMAC. The identity of the smaller thermolysin-resistant bands on the SDS gel is uncertain, but one of them migrates in a position that is consistent with the expected size of the prodomain of MeCPA.
The finding that the yield of active MeCPA-His6PR was highest in BL21(DE3) cells without the trxB and gor mutations but overproducing cytosolic DsbC was unanticipated. In a study of parameters affecting the production of the normally periplasmic E. coli phytase AppA (which contains four disulfide bonds) in the cytosol of BL21(DE3), co-expression of DsbC did not improve the yield of properly folded and oxidized AppA [32]. Also unexpected was the observation that co-expression of a DsbC mutant with no redox-active cysteines was partially effective at increasing the yield of MeCPA, although it should be noted that the mutant DsbC was expressed at a significantly lower level in BL21(DE3) (~ 50% as judged by SDS-PAGE) than its wild-type counterpart, which complicates the interpretation of this result. Nevertheless, it can be concluded that the beneficial effect of DsbC on the yield of active MeCPA-His6PR cannot be attributed to its catalytic activity alone, but presumably also depends on its chaperone-like qualities to some degree.
Another unanticipated result was our observation that incubation of the amylose-purified MBP-proMeCPA-His6PR fusion protein overnight was important for the formation of the active enzyme (Fig. 4) because this suggests that the vast majority of properly folded and oxidized MeCPA-His6PR was formed after cell lysis. Yet based on the experiments depicted in Fig. 2, DsbC must somehow be involved. It may be noteworthy in this regard that some DsbC co-purifies with the MBP-proMeCPA-His6PR fusion protein during amylose affinity chromatography (Fig. 5, lane 3 at approximately 28 kDa) and therefore might have an opportunity to interact with it during overnight incubation. Curiously, however, incubation of the cleared lysate overnight, which had a much higher concentration of DsbC than the affinity purified material, actually yielded less of the active MeCPA-His6PR. Further experiments will be needed to more precisely elucidate the roles of DsbC and spontaneous oxidation in the folding of MeCPA.
As mentioned above, despite the relatively high yield and solubility of the MBP-proMeCPA-His6PR fusion protein in the cytosol of E. coli (Fig. 3), only a very small fraction of the MeCPA appears to properly folded and oxidized; the vast majority of it is degraded by thermolysin (Fig. 5). A similar phenomenon has been documented for the human papilloma virus E6 oncoprotein fused to MBP in E. coli [33]. The soluble MBP-E6 fusion protein was shown to exist in two forms: a small population of monodisperse material in which the E6 is properly folded, and a much larger population of soluble aggregates in which it is not [34]. If some means of untangling the soluble MBP-proMeCPA-His6PR aggregates could be devised, so that they could then be acted upon by DsbC, then this might result in a dramatically higher yield of active MeCPA-His6PR. The fact that oxidized and active MeCPA forms after cell lysis and partial purification by amylose affinity chromatography (Fig. 4) presents an opportunity to experiment with additives that might untangle the aggregated fusion proteins (without interfering with the activity of DsbC or thermolysin), such as a low concentration of guanidine-HCl [35] or detergents [36,37]. Alternatively, as shown for several eukaryotic proteins, “pre-expression” of chaperones and/or oxidoreductases prior to overproduction of the disulfide-containing target as an MBP fusion protein may also lead to greater yields of properly oxidized protein in this case [38].
In summary, we have devised an effective strategy to produce active MeCPA-His6PR from E. coli at a yield of about 0.5 mg/liter of culture medium. The critical advance was to make the aggregation-prone zymogen soluble by fusing it to MBP, and in so doing, allow the DsbC disulfide isomerase to act on the soluble fusion protein in the cytosol. Yet as discussed above, the majority of oxidized MeCPA-His6PR appeared after cell lysis and partial purification, but did not do so in the absence of DsbC. Thus, while it seems clear that DsbC increases the yield of active MeCPA-His6PR, exactly how it does so remains somewhat of a mystery.
HIGHLIGHTS.
We describe a novel method for the production of a disulfide-containing fungal carboxypeptidase in the cytosol of E. coli.
The zymogen is maintained in a soluble form by fusing it to the C-terminus of E. coli maltose-binding protein.
Once rendered soluble in this manner, normally periplasmic DsbC co-expressed in the cytosol promotes the formation of the proper disulfide bonds.
Catalytically inactive DsbC is still partially effective, indicating a chaperone-like effect may be involved.
The zymogen is activated by digestion with thermolysin and the active carboxypeptidase is purified by metal-chelate, ion exchange, and gel filtration chromatography.
Acknowledgments
We thank Dr. Joseph E. Tropea for the suggestion to try co-expression of the MeCPA fusion protein and DsbC in BL-21(DE3) cells lacking the trxB and gor mutations. This project was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products or organizations imply endorsement by the US government. We thank the Biophysics Resource in the Structural Biophysics Laboratory, NCI-Frederick, for use of the LC/ESMS instrument.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arnau J, Lauritzen C, Petersen GE, Pedersen J. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expr Purif. 2006;48:1–13. doi: 10.1016/j.pep.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2003;60:523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- 3.Waugh DS. Making the most of affinity tags. Trends Biotechnol. 2005;23:316–320. doi: 10.1016/j.tibtech.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 4.Sun-Lailam BB, Hevel JM. Efficient cleavage of problematic tobacco etch virus (TEV)-protein arginine methyltransferase constructs. Anal Biochem. 2009;387:130–132. doi: 10.1016/j.ab.2008.12.031. [DOI] [PubMed] [Google Scholar]
- 5.Wu J, Filutowicz M. Hexahistidine (His6)-tag dependent protein dimerization: a cautionary tale. Acta Biochim Pol. 1999;46:591–599. [PubMed] [Google Scholar]
- 6.Woestenenk EA, Hammarström M, van den Berg S, Härd T, Berglund H. His tag effect on solubility of human proteins produced in Escherichia coli: a comparison between four expression vectors. J Struct Funct Genom. 2004;5:217–229. doi: 10.1023/b:jsfg.0000031965.37625.0e. [DOI] [PubMed] [Google Scholar]
- 7.Chant A, Kraemer-Pecore CM, Watkin R, Kneale GG. Attachment of a histidine tag to the minimal zinc finger protein of the Aspergillus nidulans gene regulatory protein AreA causes a conformational change at the DNA-binding site. Protein Expr Purif. 2005;39:152–159. doi: 10.1016/j.pep.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 8.Amor-Mahjoub M, Suppini JP, Gomez-Vrielyunck N, Ladjimi M. The effect of the hexahistidine-tag in the oligomerization of HSC70 constructs. J Chromatogr B. 2006;844:328–334. doi: 10.1016/j.jchromb.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 9.Renzi F, Panetta G, Vallone B, Brunori M, Arceci M, Bozzoni I, Laneve P, Caffarelli E. Large-scale purification and crystallization of the endoribonuclease XendoU: trouble-shooting with His-tagged proteins. Acta Crystallogr F. 2006;62:298–301. doi: 10.1107/S1744309106006373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaberc-Porekar V, Menart V, Jevsevar S, Vidensek A, Stalc A. Histidines in affinity tags and surface clusters for immobilized metal-ion affinity chromatography of trimeric tumor necrosis factor alpha. J Chromatogr A. 1999;852:117–128. doi: 10.1016/s0021-9673(99)00374-x. [DOI] [PubMed] [Google Scholar]
- 11.Horchani H, Sabrina L, Régine L, Sayari A, Gargouri Y, Verger R. Heterologous expression and N-terminal His-tagging processes affect the catalytic properties of staphylococcal lipases: a monolayer study. J Colloid Interface Sci. 2010;350:586–594. doi: 10.1016/j.jcis.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 12.Carson M, Johnson DH, McDonald H, Brouillette C, Delucas LJ. His-tag impact on structure. Acta Crystallogr D. 2007;63:295–301. doi: 10.1107/S0907444906052024. [DOI] [PubMed] [Google Scholar]
- 13.Freydank AC, Brandt W, Drager B. Protein structure modeling indicates hexahistidine-tag interference with enzyme activity. Proteins. 2008;72:173–183. doi: 10.1002/prot.21905. [DOI] [PubMed] [Google Scholar]
- 14.Xu CG, Fan XJ, Fu YJ, Liang AH. Effect of location of the His-tag on the production of soluble and functional Buthus martensii Karsch insect toxin. Protein Expr Purif. 2008;59:103–109. doi: 10.1016/j.pep.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 15.Klose J, Wendt N, Kubald S, Krause E, Fechner K, Beyermann M, Bienert M, Rudolph R, Rothemund S. Hexa-histidine tag position influences disulfide structure but not binding behavior of in vitro folded N-terminal domain of rat corticotropin-releasing factor receptor type 2a. Protein Sci. 2004;13:2470–2475. doi: 10.1110/ps.04835904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waugh DS. An overview of reagents for the removal of affinity tags. Protein Expr Purif. 2011;80:283–293. doi: 10.1016/j.pep.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Austin BP, Tözsér J, Bagossi P, Tropea JE, Waugh DS. The substrate specificity of Metarhizium anisopliae and Bos taurus carboxypeptidases A: insights into their use as tools for the removal of affinity tags. Protein Expr Purif. 2011;77:53–61. doi: 10.1016/j.pep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardell SJ, Craik CS, Clauser E, Goldsmith EJ, Stewart CB, Graf M, Rutter WJ. A novel rat carboxypeptidase, CPA2: characterization, molecular cloning and evolutionary implications on substrate specificity in the carboxypeptidase gene family. J Biol Chem. 1988;263:17828–17836. [PubMed] [Google Scholar]
- 19.Fox JD, Waugh DS. Maltose-binding protein as a solubility enhancer. Methods Mol Biol. 2003;205:99–117. doi: 10.1385/1-59259-301-1:99. [DOI] [PubMed] [Google Scholar]
- 20.Tsao KL, Waugh DS. Balancing the production of two recombinant proteins in Escherichia coli by manipulating plasmid copy number: high-level expression of heterodimeric Ras farnesyltransferase. Protein Expr Purif. 1997;11:233–240. doi: 10.1006/prep.1997.0794. [DOI] [PubMed] [Google Scholar]
- 21.Tsao KL, DeBarbieri B, Michel H, Waugh DS. A versatile plasmid expression vector for the production of biotinylated proteins by site-specific, enzymatic modification in Escherichia coli. Gene. 1996;169:59–64. doi: 10.1016/0378-1119(95)00762-8. [DOI] [PubMed] [Google Scholar]
- 22.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 23.Peterson LM, Holmquist B, Bethune JL. A unique activity assay for carboxypeptidase A in human serum. Anal Biochem. 1982;125:420–426. doi: 10.1016/0003-2697(82)90024-0. [DOI] [PubMed] [Google Scholar]
- 24.Joshi L, St Leger RJ. Cloning, expression, and substrate specificity of MeCPA, a zinc carboxypeptidase that is secreted into infected tissues by the fungal entomopathogen Metarhizium anisopliae. J Biol Chem. 1999;274:9803–9811. doi: 10.1074/jbc.274.14.9803. [DOI] [PubMed] [Google Scholar]
- 25.Studier FW, Rosenberg AH, Dunn JJ, Dubendorf JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 26.Prinz WA, Ashlund F, Holmgren A, Beckwith J. The role of thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J Biol Chem. 1997;272:15661–15667. doi: 10.1074/jbc.272.25.15661. [DOI] [PubMed] [Google Scholar]
- 27.Bessette PH, Aslund F, Beckwith J, Georgiou G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc Natl Acad Sci USA. 1999;96:13703–13708. doi: 10.1073/pnas.96.24.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips MA, Rutter WJ. Role of the prodomain in folding and secretion of rat pancreatic carboxypeptidase A1. Biochemistry. 1996;35:6771–6776. doi: 10.1021/bi960113o. [DOI] [PubMed] [Google Scholar]
- 29.Kapust RB, Waugh DS. Escherichia coli malotose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J, Song JL, Zhang S, Wang Y, Cui DF, Wang CC. Chaperone activity of DsbC. J Biol Chem. 1999;274:19601–19605. doi: 10.1074/jbc.274.28.19601. [DOI] [PubMed] [Google Scholar]
- 31.Depuydt M, Messens J, Collet JF. How proteins form disulfide bonds. Antioxid Redox Signal. 2011;15:49–66. doi: 10.1089/ars.2010.3575. [DOI] [PubMed] [Google Scholar]
- 32.Hatahet F, Nguyen VD, Salo KEH, Ruddock LW. Disruption of reducing pathways is not essential for efficient disulfide bond formation in the cytoplasm of E. coli. Microb Cell Fact. 2010;9:67. doi: 10.1186/1475-2859-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nominé Y, Ristriani T, Laurent C, Lefèvre JF, Weiss E, Travé G. Formation of soluble inclusion bodies by HPV E6 oncoprotein fused to maltose-binding protein. Protein Expr Purif. 2001;23:22–32. doi: 10.1006/prep.2001.1451. [DOI] [PubMed] [Google Scholar]
- 34.Nominé Y, Ristriani T, Laurent C, Lefèvre JF, Weiss E, Travé G. A strategy for opttimizing the monodispersity of fusion proteins: application to purification of recombinant HPV E6 oncoprotein. Protein Eng. 2001;14:297–305. doi: 10.1093/protein/14.4.297. [DOI] [PubMed] [Google Scholar]
- 35.Czepczynska-Krzel H, Czerwinski M, Krezel A, Krop-Watorek A. Isolation of carcinoembryonic antigen N-terminal domains (N-A1) from soluble aggregates. Protein Expr Purif. 2011;78:78–85. doi: 10.1016/j.pep.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 36.Tao H, Liu W, Simmons BN, Harris HK, Cox TC, Massiah MA. Purifying natively folded proteins from inclusion bodies using sarkosyl, Triton X-100, and CHAPS. Biotechniques. 2010;48:61–64. doi: 10.2144/000113304. [DOI] [PubMed] [Google Scholar]
- 37.Park DW, Kim SS, Nam MK, Kim GY, Kim J, Rhim H. Improved recovery of active GST-fusion proteins from insoluble aggregates: solubilization and purification conditions using PKM2 and HtrA2 as model proteins. BMB Rep. 2011;44:279–284. doi: 10.5483/BMBRep.2011.44.4.279. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen VD, Hatahet F, Salo KEH, Enlund E, Zhang C, Ruddock LW. Pre- expression of a sylfhydryl oxidase significantly increases the yields of eukaryotic disulfide bond containing proteins expressed in the cytoplasm of E. coli. Microb Cell Fact. 2011;10:1. doi: 10.1186/1475-2859-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]