

Conspectus
The combustion of organic matter is perhaps the oldest and most common chemical transformation utilized by mankind. The generation of a C–O bond at the expense of a C–H bond during this process may be considered the most basic form of C–H functionalization. This illustrates the extreme generality of the term ‘C–H functionalization,’ as it can describe the conversion of literally any C–H bond into a C–X bond (X being anything except H). Therefore, it may be of use to distinguish between what, in our view, are two distinct categories of C–H functionalization logic: ‘guided’ and ‘innate.’ Guided C–H functionalizations, as the name implies, are guided by external reagents or directing groups (covalently or fleetingly bound) to install new functional groups at the expense of specifically targeted C–H bonds. Conversely, innate C–H functionalizations may be broadly defined as reactions that exchange C–H bonds for new functional groups based solely on natural reactivity patterns in the absence of other directing forces.
Two substrates that illustrate this distinction are dihydrojunenol and isonicotinic acid. The C–H functionalization processes of hydroxylation or arylation, respectively, can take place at multiple locations on each molecule. Innate functionalizations lead to substitution patterns that are dictated by the inherent bias (steric or electronic) of the substrate undergoing C–H cleavage, whereas guided functionalizations lead to substitution patterns that are controlled by external directing forces such as metal complexation or steric bias of the reagent. Although the distinction between guided and innate C–H functionalizations may not always be clear in cases that do not fit neatly into a single category, it is a useful convention to consider when analyzing reactivity patterns and strategies for synthesis. We must emphasize that although a completely rigorous distinction between guided and innate C–H functionalization may not be practical, we have nonetheless found it to be a useful tool at the planning stage of synthesis.
In this Account, we trace our own studies in the area of C–H functionalization in synthesis through the lens of ‘guided’ and ‘innate’ descriptors. We show how harnessing innate reactivity can be beneficial for achieving unique bond constructions between heterocycles and carbonyl compounds, enabling rapid and scalable total syntheses. Guided and innate functionalizations were used synergistically to create an entire family of terpenes in a controlled fashion. We continue with a discussion of the synthesis of complex alkaloids with high nitrogen content, which required the invention of a uniquely chemoselective innate C–H functionalization protocol. These findings led us to develop a series of innate C–H functionalization reactions for forging C–C bonds of interest to the largest body of practicing organic chemists: medicinal chemists. Strategic use of C–H functionalization logic can have a dramatically positive effect on the efficiency of synthesis, whether guided or innate.

Introduction
As ubiquitous structural motifs in organic chemistry, C–H bonds have been the focus of intense study throughout the history of chemical research. Free-radical halogenation, electrophilic aromatic substitution, and deprotonation of acidic C–H bonds are all early examples of C–H functionalization. The most alluring aspect of C–H functionalization logic in organic synthesis — indeed contributing to the dramatic resurgence of interest in this area — is the potential to simplify and shorten synthetic routes and expand retrosynthetic trees.1 Our laboratory has been intrigued and inspired by the great potential of C–H functionalization, particularly in the area of total synthesis. Over the years it has become apparent that two different forms of C–H functionalization logic exist: ‘innate’ and ‘guided’ (vide supra). For example, as shown in Scheme 1, simple arenes can undergo a wealth of direct functionalizations.2,3 In the case of benzoic acid 4, electrophilic substitutions can take place to deliver 6 or 7, or directed metallations can afford 1 or 2. The former process can be considered innate and the latter considered guided, while both accomplish C–H functionalization with useful regiocontrol. The same can be said of m-xylene (5) and its innate (8) and guided (3) products. It is our contention that from the vantage point of a molecule maker engaged in retrosynthetic analysis, neither should be viewed as superior, but rather as complementary strategies. Because our contributions to the field of C–H functionalization have been modest, a comprehensive review is beyond the scope of this account and has been reported elsewhere in detail.4 What follows in this account are some vignettes of how innate and guided C–H functionalization strategies have positively impacted our research.
Scheme 1.

Guided2 and innate3 C–H functionalization. Bonds formed by guided transformations are indicated in red, and those formed by innate transformations are indicated in blue throughout this account.
Enolate Coupling
Enolate and Indole/Pyrrole
Indoles are ubiquitous structural motifs found in a variety of pharmaceuticals, synthetic materials, and natural products.5 The marine-derived fischerindoles (9–11), welwitindolinones (12), ambiguines (13), and hapalindoles (14, 15), are especially intriguing indole-containing natural products given their biological activity and unique structural features (Scheme 2A).6,7 A commonality in this natural product family is the C–C bond linking the C3 position of the indole subunit and the six-membered ring containing an adjacent nitrogen atom. Because of this structural characteristic, many members of this family may be traced back to a common scaffold (16). Although a more “programmed” approach to forming the key indole–carvone bond is feasible via cross-coupling reactions,8 this typically requires several steps to establish the necessary functional groups on each coupling partner. A C–H functionalization approach would instead allow for a more concise formation of this pivotal C–C bond.
Scheme 2.

A) A family of marine natural products (9–15) that could all be retrosynthetically traced back to a common indole–carvone intermediate (16); B) Innate indole–enolate coupling yielding the common indole–carvone intermediate 16.6,7
The strategy subsequently invented was based on the innate reactivity of the two components, namely indole (17) and carvone (18), which needed to be merged (Scheme 2B).6,9 This was accomplished by deprotonating both reacting partners and reacting them in an oxidative fashion using a copper(II) source. This coupling reaction is applicable to a broad range of carbonyl compounds, generating coupling products from ketones (20a–20q, 20s–20w, 20ab, 20ac), esters (20r), and amides (20x–20aa) (Scheme 3). Methylketones were the only substrates that have shown to undergo extensive homodimerization, making this reaction especially suitable for more complex carbonyl compounds and formation of quaternary carbon centers. Numerous reactive functional groups were tolerated under the reaction conditions such as chlorides (20ab), epoxides (20w), alkenes (20a–20m, 20t, 20w, 20ab, 20ac), α,β-unsaturated carbonyls (20a–20l, 20t, 20ac), alcohols (20s), esters (20r), and sulfonamides (20x–20aa). With respect to the indole coupling partner, various substitution patterns and functional groups including halogens (20d–20f, 20i–20k), methyl groups (20c, 20h), and alkoxyl groups (20b, 20g) were tolerated. Although this reaction contains interesting mechanistic features, an extensive discussion is beyond the scope of this account and has been reported elsewhere.9 This indole–enolate coupling reaction showcases an example of an innate, double, selective C–H functionalization method: it is ‘innate’ in that indoles react with electrophiles at its nitrogen atom or at C3, and enolates react at its oxygen atom or at its α carbon; it is a double functionalization, since it is replacing two C–H bonds; and finally, it is selective, as the C–C bond formation selectively takes place over C–N or C–O bond formation.
Scheme 3.

Innate C–H coupling between indoles and enolates (% yields are shown in parenthesis).9
This simplifying transformation enabled short, scalable, and protecting group-free syntheses of several members of the hapalindole family (9–15), including complex structures such as fischerindole I (10), welwitindolinone A (12), and ambiguine H (13).6,7,9,10 Furthermore, enantioselective syntheses of natural products acremoauxin A (21) and oxazinin 3 (22) were realized.9
As an extension of the reaction between indoles and enolates, the analogous coupling between a pyrrole and an enolate was also developed (Scheme 4).9,11 As with the indole–enolate coupling, numerous functional groups (24a–24i) and pyrrole systems (24a–24d) were tolerated under the reaction conditions. This reaction was then applied to the enantioselective synthesis of the chemotherapeutic (S)-ketorolac (25).
Scheme 4.
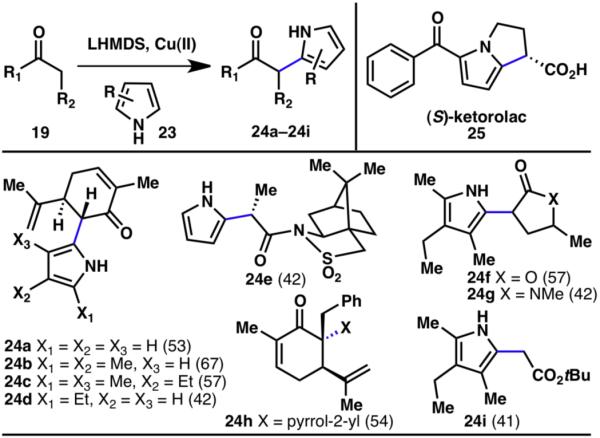
Innate C–H coupling between pyrroles and enolates (% yields are shown in parenthesis).9,11
Both indoles and pyrroles were also successfully coupled with 2-carboxymethyl-3-oxindoles (26; Scheme 5).12 This reaction was shown to be compatible with methyl (31f, 31h, 31i), methylether (31a, 31c), bromo (31d), and cyano (31b) substitutions for the heterocyclic coupling partner.
Scheme 5.
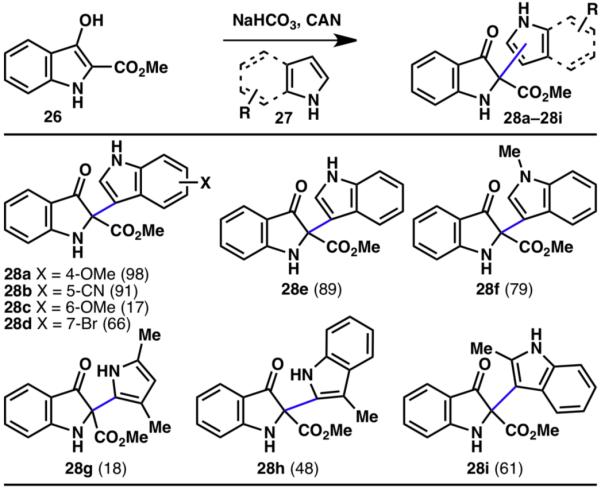
Innate C–H coupling of 2-carboxymethyl-3-oxindoles and indoles (% yields are shown in parenthesis).12
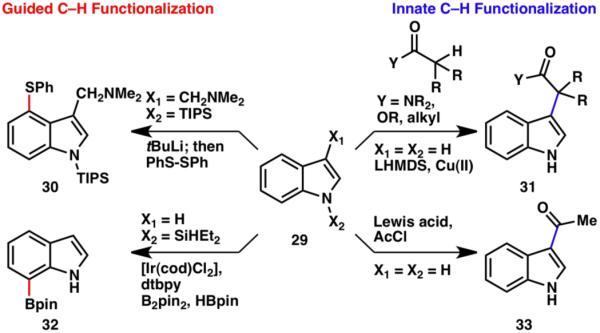
In retrospect, the C–H functionalization of indoles, guided or innate, could easily be the subject of an entire book due to this heterocycle’s rich reactivity. A small representative sampling is depicted in Scheme 6. Guided C–H functionalizations have been used for the modification of the indole C4 (30) and C7 (32) positions,13 whereas for the desired bond formation at C3, the innate reactivity of indole can be exploited. The well-known Friedel–Crafts acylation3c (33), as well as the enolate–indole coupling (31),6,9 is highly C3-selective.
Scheme 6.
Enolate–Enolate Coupling
Unsymmetrical 2,3-disubstituted-1,4-dicarbonyl compounds are present in numerous pharmaceuticals and natural products. Since the 1,4-connectivity of two carbonyl groups clashes with their natural electronic properties, these structural motifs are especially challenging to make via the direct coupling of two carbonyl compounds.14 Several methods for this transformation have been established, but limitations including multistep sequences, pre-installation of functional groups, and poor selectivities, diminish the utility of these strategies. The direct heterocoupling of enolate pairs to form dicarbonyl compounds poses many challenges including homodimerization, Claisen or aldol condensation, overoxidation, dehydration, or α-oxidation.
Nonetheless, reaction conditions for the oxidative coupling of oxazolidinones with ketones were developed using either Cu(II) or Fe(III) (Scheme 7).14,15 Intriguingly, the two metals found to be suitable for the coupling strategy exhibited complementary reactivities. This oxidative coupling strategy is also applicable to the reaction of oxindoles with ketones as outlined in Scheme 8.14,15
Scheme 7.
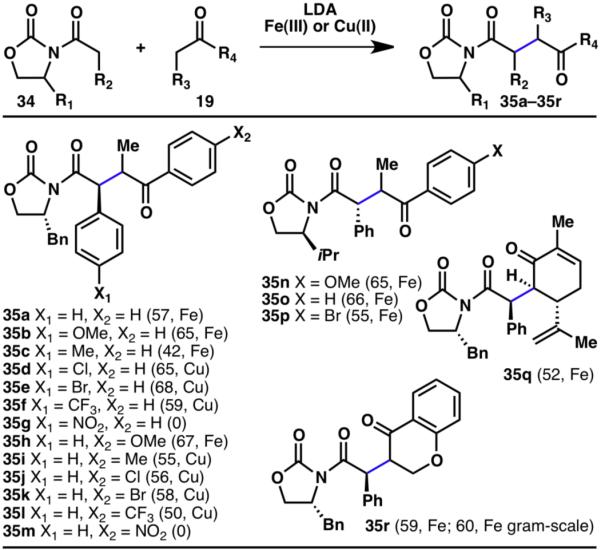
Innate C–H coupling between oxazolidinones and ketones (% yields are shown in parenthesis).14,15
Scheme 8.
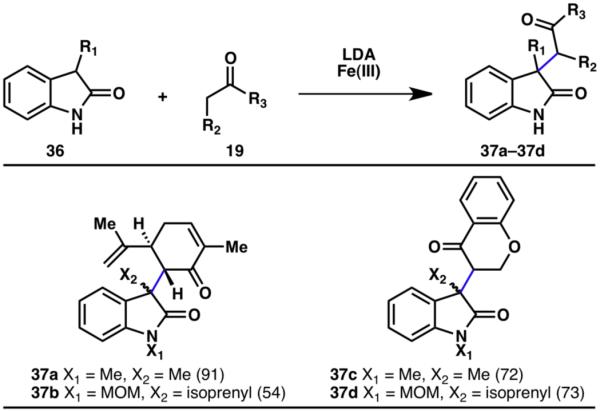
Innate C–H coupling between oxindoles and ketones (% yields are shown in parenthesis).14,15
Oxazolidinone amides (34) utilized in the coupling with ketones were also amenable to cross-coupling with esters (38), although the reaction conditions were slightly modified, with the addition of LiCl (Scheme 9).14 In the case of esters, only Cu(II) was a suitable oxidant for the desired transformation, and a slight excess of the ester was required due to homodimerization problems. Numerous functional groups were compatible with the reaction conditions including alkenes (39e), ethers (39f, 39k, 39q, 39r), CF3 groups (39g, 39n), electron-rich (39f, 39i–39k, 39q, 39r), and -deficient arenes (39l, 39m) as well as an indole moiety (39j). The mechanism of this transformation has been studied in detail by our laboratory14 and others16 and the explanation of the observed diastereoselectivities is published elsewhere.14
Scheme 9.

Innate C–H coupling between oxazolidinones and esters.14
The direct coupling of two carbonyl compounds has been applied to the synthesis of (−)-bursehernin (42), yielding the desired lignan lactone in enantio- and stereoselective fashion after just three steps (Scheme 10A).14,15 However, the true synthetic power of this coupling strategy was demonstrated in the short, enantioselective synthesis of highly complex natural products, stephacidin A (43) and B (45) and avrainvillamide (44; Scheme 10B).17 Double deprotonation of diketopiperazine 46 gave the corresponding dianion, which was coupled intramolecularly to furnish the penultimate ring with diastereoselective formation of the central quaternary carbon (Scheme 10C).
Scheme 10.

A) Synthesis of bursehernin (42) via innate C–H enolate coupling;14,15 B) Structures of avrainvillamide (44), stephacidin A (43) and B (45); C) Innate C–H enolate coupling in the synthesis of 44, 43 and 45.17
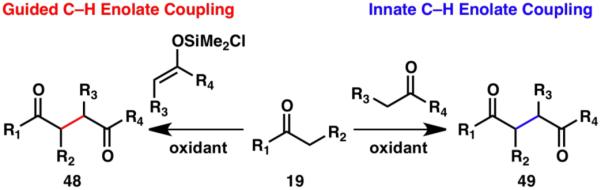
The direct coupling of enolate pairs to form dicarbonyl compounds poses many synthetic challenges. These challenges may be overcome through the use of a silyl linker between the reacting carbonyl units to guide the desired reactivity (48; Scheme 11).18 Alternatively, the disparate oxidation potentials of different carbonyl compounds may be exploited for the synthesis of unsymmetrical 2,3-disubstituted-1,4-dicarbonyl compounds via an innate C–H functionalization approach (49).14
Scheme 11.
1,3-Diol Synthesis
The selective oxidation of a single C–H bond to an alcohol moiety is a common reaction in Nature. Development of such a procedure in the laboratory is hampered by the typical issues of site-selectivity and chemoselectivity. When attempting to establish a protocol for the oxidation of the β-C– H bond of an alcohol, inspiration was taken from the Hofmann–Loeffler–Freytag reaction (Scheme 12A).3c In our approach, the desired regioselectivity was achieved using a directing group to place a highly reactive intermediate in close proximity (Scheme 12B).19
Scheme 12.
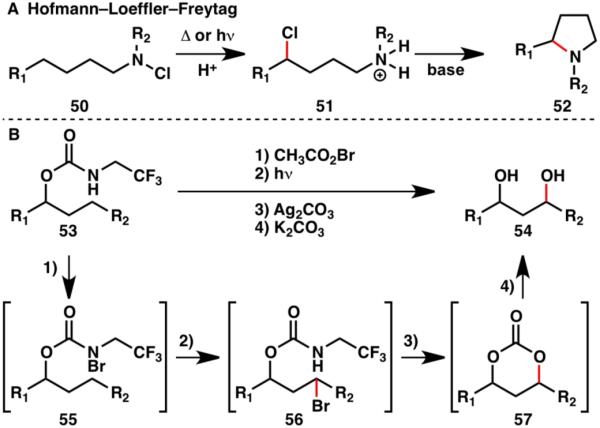
A) Hofmann–Loeffler–Freytag reaction;3c B) Guided C–H oxidation to form 1,3-diols.19
In this guided C–H functionalization approach, a carbamate was used as a directing group (53), and was converted to a reactive N-bromide by halogenation with CH3COOBr. Bromocarbamate 55 then facilitated a 1,6-hydrogen transfer to form alkyl bromide 56. The directing group held the reactive species in close proximity to the desired reaction site and thus ensured formation of a single regioisomer. Furthermore, the directing group also acted as an internal nucleophile, displacing the bromine and forming carbonate 57. After hydrolysis, the conversion of an alcohol into a 1,3-diol (54) by guided C–H functionalization was accomplished.19 This strategy was shown to be applicable to numerous alcohols (Scheme 13). It should be noted however, that this transformation is currently limited to benzylic (54a–54d) and tertiary (54e–54k) C–H bonds. Nonetheless, its implementation in the efficient synthesis of several natural products (58–60) shows its applicability in target-oriented synthesis.19
Scheme 13.
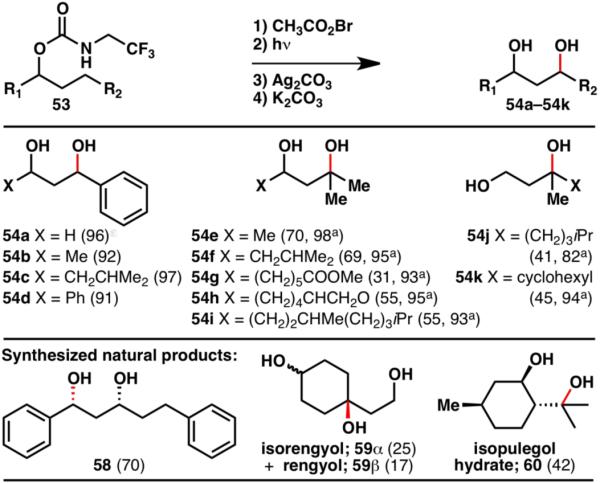
Guided C–H 1,3-diol formation from various alcohols (% yields are shown in parenthesis; ayields based on recovered starting material).19
This guided C–H functionalization reaction was combined with Curci’s TFDO oxidation,20 an innate C–H functionalization method, to provide the foundation for the divergent synthesis of five eudesmane terpenes (64–68) at different oxidation levels.21 The ability to selectively oxidize C–H bonds within a complex setting allowed for a unique retrosynthetic approach to these natural products. Inspired by Nature’s terpene synthesis, which can be divided into cyclase and oxidase phases, the carbon skeleton containing a single functional group was constructed in gram quantities using a nine-step synthetic sequence, resulting in the natural product dihydrojunenol (64; Scheme 14A).21-22 In the second phase, this lowly oxidized terpene was subjected to several C–H oxidation events. A combination of innate TFDO oxidation (blue) and guided 1,3-diol formation (red) gave rise to four different eudesmane terpenes (65–68, Scheme 14B).21 High site-selectivity of the TFDO oxidation was observed; this and other mechanistic features have been studied in detail and reported elsewhere.23
Scheme 14.
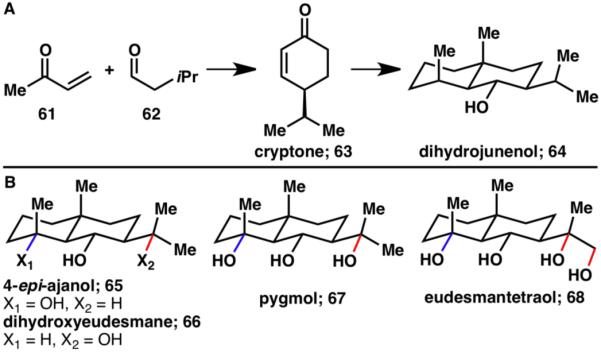
A) Cyclase phase synthesis resulting in the formation of sesquiterpene dihydrojunenol (64);21,22 B) Synthesis of eudesmane natural products (65–68) from dihydrojunenol (64) by innate and guided C–H oxidation.21
Comparison with iron-catalyzed24 and dioxirane-mediated methods for C–H oxidation20 revealed the orthogonality of those innate approaches to the guided carbamate strategy. The logic of combining innate and guided C–H functionalizations illustrates the power of their complementary use in divergent target-oriented synthesis. Scheme 15 shows how the use of guided (70)20 and innate (71)21 C–H functionalization allows the installment of the required oxidation pattern of two different natural products in only one step each from a common precursor. Similarly, application of the innate iron oxidation method24 and our guided 1,3-diol protocol leads to two different products from 72 (74 and 73, respectively).19
Scheme 15.

Pyrrole–Imidazole Alkaloids
The pyrrole–imidazole alkaloid family comprises some of the most complex and vigorously pursued marine natural products known to date.25 This is certainly evident when considering the structural complexities of palau’amine (77), massadine (78), and axinellamines A (82) and B (83). The total synthesis of these targets, as well as several biosynthetic precursors, has recently been reported.26 Retrosynthetic analysis of 77, 78, 82 and 83 revealed that oxidation of the C20 hemiaminal position would simplify the synthetic route tremendously while simultaneously allowing for the preparation of a late-stage common precursor. Several oxidation conditions were screened before silver(II) picolinate was identified as a suitable oxidant for the desired transformation (Scheme 16). Notably, this innate C– H functionalization demonstrated both great position selectivity and redox control (halting at the hemiaminal oxidation state). With this key oxidation strategy validated, the synthesis of palau’amine (77), massadine (78), and axinellamines A (82) and B (83) was completed.26
Scheme 16.

Synthesis of palau’amine (77), massadine (78), axinellamine A (82) and B (83) (% yields are shown in parenthesis).26
Two aspects of this enabling silver-promoted innate C–H oxidation reaction were its remarkable chemoselectivity (oxidation in H2O/TFA in the presence of ca. 10 nitrogen atoms) and ligand-controlled redox selectivity (Ag2O alone led to extensive over-oxidation). This inspired us to explore other silver-catalyzed reactions, potentially of more broad applicability and utility.
Arylation of Nitrogen-Containing Heterocycles
A classical, yet underappreciated example of innate C–H functionalization is the Minisci reaction, coupling alkyl radicals generated from carboxylic acids to heterocyclic substrates.27 However, this reaction suffers from limitations including harsh conditions necessary to generate the alkyl radical, the use of the heterocyclic coupling partner in excess or as a solvent, and poor levels of regioselectivity.
Inspired by the Minisci reaction, we reported the first example of a mild, room-temperature, catalytic generation of aryl radical species from boronic acids and their subsequent addition to heteroarenes, wherein arylboronic acids were converted into their corresponding aryl radicals via treatment with catalytic silver in the presence of a persulfate oxidant (Scheme 17).28 The radicals thus generated can be subsequently trapped by heteroarenes (84), forming a new C–C bond after an oxidation/re-aromatization sequence. This scalable reaction features exceptionally practical conditions including inexpensive commercial reagents, as well as ambient temperature and atmospheric conditions.
Scheme 17.
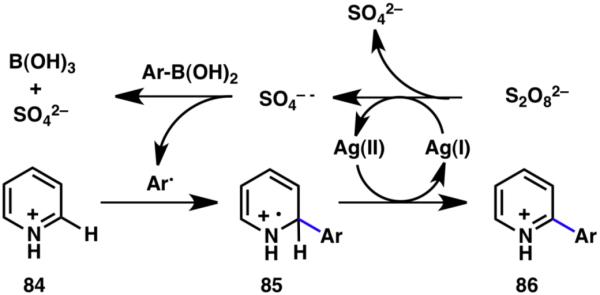
Proposed mechanism of the innate C–H arylation.28
Various boronic acid substitution patterns were tolerated in the arylation protocol, including both electron-donating (89b, 89f, 89h–89l, 89n, 89o) and -withdrawing substituents (89c–89e, 89g, 89m; Scheme 18). Whereas meta- and para-substitution (89a–89m) did not adversely impact the coupling reaction, ortho-substitution (89n, 89o) was only effective in the context of electron-rich boronic acids. Numerous functional groups including halogens (89g–89i, 89k), esters (89c), nitriles (89m), nitrates (89e), ethers (89b, 89o) and trifluoromethyl groups (89d) were tolerated in the reaction due to the mild conditions involved.28
Scheme 18.
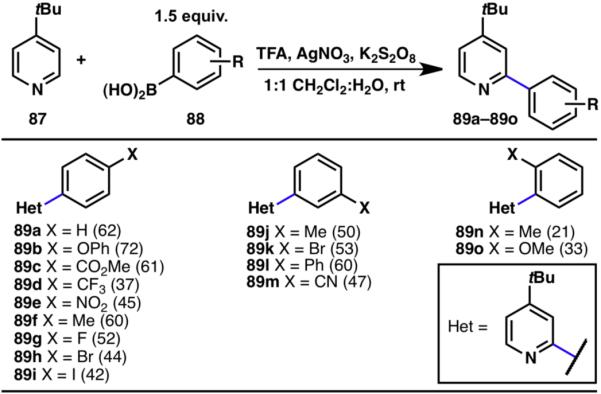
Innate C–H arylation of 4-t-butylpyridine (% yields are shown in parenthesis).28
Pyridines and quinolines (90) react with aryl radicals predominantly at their C2 position, and a second, minor regioisomer may be obtained if the 4-position is also available for substitution (Scheme 19). As expected, electron-withdrawing substituents were not able to override the natural reactivity pattern of pyridines (92e, 92f). In addition to pyridine, other electron-poor 6-membered heteroarenes such as pyrimidine (92b), pyridazine (92c), and pyrazine (92d) readily underwent arylation. Quinolines (92j, 92k), isoquinolines (92l, 92m), quinoxalines (92n), and phthalazines (92o) are also competent substrates, with functionalization occurring at the more electron-deficient heteroarene rings.28
Scheme 19.
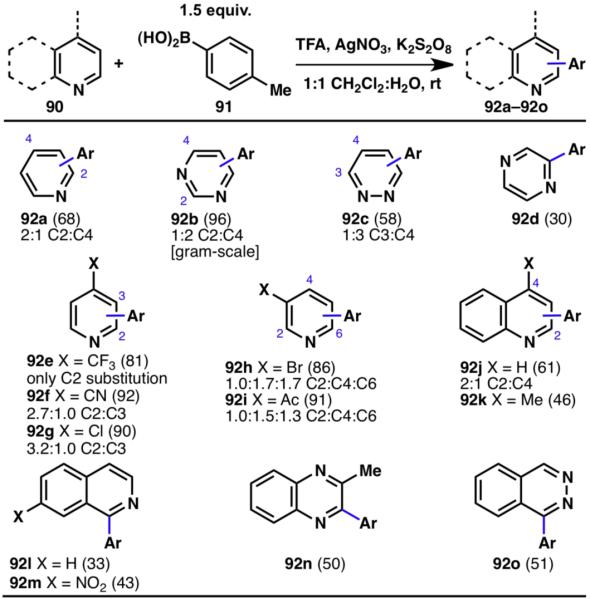
Innate C–H arylation of various N-containing heterocycles (% yields are shown in parenthesis).28
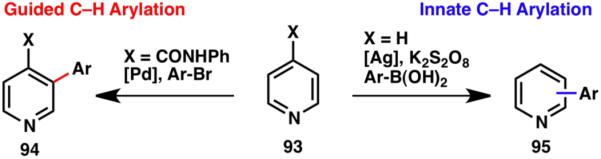
The formation of an aryl–aryl bond is one of the most important transformations in chemistry, especially in medicinal chemistry. Tremendous advances have been made in C–H arylation of arenes and heterocycles in recent years,29 including the challenging guided approach to the C–H functionalization of pyridine (94; Scheme 20).30 An innate C–H arylation of heterocycles has been reported to provide an orthogonal method for the C–H arylation of pyridine (95).28
Scheme 20.
Arylation of Quinones
The protocol for innate C–H arylation of heterocycles was expanded to include the direct arylation of quinones, which are an important class of compounds in chemistry, material science, nanotechnology, and medicine.31 Quinones are especially susceptible to arylation by the procedure described above, as they are inherently reactive with radicals, and the intermediate dihydroquinones can be reoxidized under the oxidative reaction conditions. Treatment of tolylboronic acid (91) and quinones (96) with potassium persulfate and a silver catalyst under ambient conditions yielded the desired arylquinones (97, Scheme 21).32a
Scheme 21.
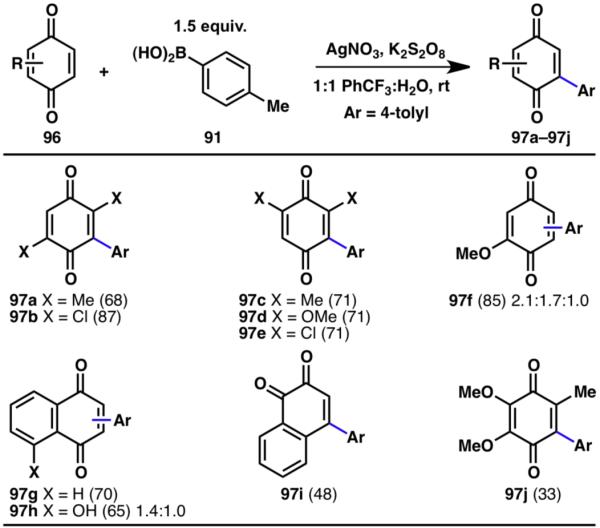
Innate C–H arylation of quinones (% yields are shown in parenthesis).32a
Substituents of quinones tolerated in this reaction included methyl (97a, 97c), hydroxymethyl (97d, 97f), chloride (97b, 97e), and hydroxyl groups (97h) as well as combinations thereof (97j). Furthermore, trisubstituted quinones (97j) and benzannulated quinones (97g–97i) were also shown to be suitable substrates for C–H arylations. Both electron-deficient and electron-rich arylboronic acids (99) can be coupled to quinone (Scheme 22). Ortho- (100r–100t), meta- (100n–100q, 100u–100y) and para-substitution (100a–100q) of the boronic acids were tolerated, as well as numerous functional groups such as ethers (100f, 100g, 100o–100q, 100t, 100w), halides (100d, 100e, 100o, 100p, 100s), ketones (100l), nitriles (100x), nitro (100k), and trifluoromethyl (100h) groups. In addition to arylboronic acids, alkylboronic acids (100z–100ad) were also successfully employed in this reaction.32a
Scheme 22.

Innate C–H arylation of benzoquinone with various arylboronic acids (% yields are shown in parenthesis).32a
In addition to the simple arylated quinones described above, large and structurally complex molecules of medicinal and biological interest can be quinonylated via the described strategy. Estroneboronic acid and farnesyl-BF3K were both successfully converted into the corresponding quinonylated compounds (100ae, 100af). This clearly shows the applicability of this approach in complex systems, justifying its potential for late-stage use in a synthetic sequence. The farnesol example (100af) also illustrates the use of allylic radicals in the coupling strategy, although isomerization of the double bond was observed.32a Subsequent to the reporting of the quinonylation and arylation strategies, Molander and co-workers have shown that alkyl trifluoroborate potassium salts can be used as precursors in radical alkylation of heterocycles.32b
Trifluoromethylation of Nitrogen-Containing Heteroarenes
The field of trifluoromethylation has received much attention in recent years due to the unique chemical and physical properties of the CF3 functional group. Its widespread use in pharmaceuticals, agrochemicals, liquid crystals, dyes, and polymers has led to the demand for development of robust reagents and procedurally simple chemical strategies for trifluoromethylation reactions.33 Specifically, the trifluoromethylation of heterocycles is of particular interest to medicinal and material chemists. This demand has led us to investigate the applicability of the Langlois reagent (NaSO2CF3),34 a bench-stable solid, as a surrogate for gaseous CF3I in the innate trifluoromethylation of nitrogen-containing heterocycles. We have shown that heteroarene compounds can be directly trifluoromethylated using NaSO2CF3 under oxidative conditions at ambient temperature without the requirement of an inert atmosphere or added metals (Scheme 23).35 The reaction thus proceeds under exceptionally practical conditions and is operationally simple to perform.
Scheme 23.

Innate trifluoromethylation of N-heterocycles (% yields are shown in parenthesis).35
Many functional groups are tolerated in the trifluoromethylation method, including unprotected alcohols (102k), amines (102c, 102m), ketones (102a, 102d), esters (102o), and halides (102a). In addition, this reaction has proven to be scalable, and may even be carried out under purely aqueous conditions without the requirement of an organic workup. The reaction provides a prototypical example of an innate C–H functionalization in which the regioselectivity of CF3 incorporation is determined solely by the innate reactivity of the substrate.35
Several substituted pyridines (102a–102c) were competent participants in the radical promoted C–H trifluoromethylation reaction. 2-Acetylpyrrole was shown to be selectively functionalized at the C5-position (102d). For melatonin, in which the C3-position is blocked, functionalization occurs within the five-membered ring at the remaining available position (102f). Phthalazine and quinoxalines are most reactive within their carbocyclic rings towards the CF3 radical (102g–102i). These and additional substrates, including uridine, caffeine, and pyrazoles, smoothly underwent trifluoromethylation (102j–102n).35
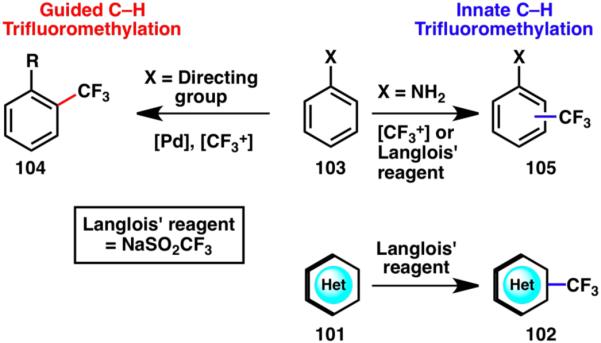
Because of the high demand for trifluoromethylation methods from the medicinal chemistry community, much research effort in recent years has been devoted to the development of powerful trifluoromethylation strategies for arenes or heterocycles. An example of groundbreaking work in the area is the palladium-catalyzed cross-coupling of aryl chlorides and Et3SiCF3, developed by Buchwald and coworkers.36 In the area of C–H functionalization, the Yu group has pioneered a guided approach that provides easy access to trifluoromethylated arenes (104),37 whereas CF3I38 or Langlois’ reagent35 allow for C–H trifluoromethylations that are controlled by the innate reactivity of the participating arenes and heterocycles (105 and 102, respectively; Scheme 24).
Scheme 24.
Complementary Reactivities
Innate C–H trifluoromethylation and arylation was also shown to be effective in the context of complex molecular scaffolds containing multiple potential reactive sites. As described above, reactivity patterns may be rationalized based on the electronics and sterics inherent to the reacting substrate. However, the observed reactivity also necessarily depends on the characteristics of the reacting partner. As such, complementary reactivity may be observed depending on the electronic nature of the incoming reagent. In the context of the C–H arylations (vide supra), the incoming aryl species is considered nucleophilic in nature.28 Conversely, the CF3 radical generated from NaSO2CF3 is considered electrophilic. The opposing reactivity of these two reacting species is fully illustrated in Scheme 25, in which several substrates (106, 108–110, 113) are subjected to both arylation28 and trifluoromethylation conditions.35 These results may be broadly valuable to the medicinal chemistry community as a tool to rapidly diversify libraries of existing analogs.
Scheme 25.

Complementary reactivity of electron-deficient CF3 and electron-rich arene radicals (% yields are shown in parenthesis).28,35
Conclusion
The last century, and particularly the last several decades, has witnessed the emergence of powerful strategies and methods for the functionalization of C–H bonds even in the most complex settings.4 As a research group geared toward the total synthesis of natural products, we embrace this development, because when aiming for efficiency and ideality39 in total synthesis, one cannot ignore the economic value40 that C–H functionalization logic has to offer. To demonstrate the increased efficiency of a synthetic route when applying C–H functionalization logic, three past and recent routes toward targets discussed in this account are compared in terms of step count (Scheme 26). An intermediate in the synthesis of hapalindoles (118) was previously generated in six steps when using a programmed approach with pre-functionalized coupling partners,41 whereas exploiting the innate reactivity of the two coupling partners by making use of a direct C–H/C–H coupling allowed the same intermediate to be constructed in only three steps (Scheme 26A).6 The total synthesis of natural products isorengyol (59α) and rengyol (59β) previously required nine steps,42 however, this was shortened to three steps by means of a guided C–H functionalization approach (Scheme 26B).19 Finally, while the synthesis of a trifluoromethylated derivative of varenicline (112) would likely require a multistep transformation involving protecting and functional group manipulations from varenicline (Chantix®; 110), potentially requiring a de novo synthesis, the recent trifluoromethylation method obviates this possibility. A one-step transformation from 110 to 112 is possible when taking advantage of the innate reactivity of the targeted C–H bond (Scheme 26C).35
Scheme 26.

Increased efficiency in synthetic routes when applying innate and/or guided C–H functionalization logic, in the context of: A) intermediate 118 in the synthesis of hapalindoles;6,41 B) natural products isorengyol (59α) and rengyol (59β);19,42 c) trifluoromethylated derivative of varenicline (112).35
This account has traced some of our contributions to this vibrant area from the vantage point of the two categories of C–H functionalization logic: innate and guided. The target itself will often dictate which one is enlisted and in some cases a synergistic use of the two can be beneficial. Both types of logic hold potential to enhance efficiency40 (as Breslow refers to it “liberating chemistry from the tyranny of functional groups”)43 in organic chemistry and it is clear that they will continue to permeate the fabric of modern retrosynthetic analysis.
Acknowledgements
The authors thank DAAD for a postdoctoral fellowship (T. B.), NSERC for a doctoral fellowship ( Y. I.), and the NIH/NIGMS (GM-073949) for financial support.
Biographical Sketch
Dr. Tobias Brückl was born in Regensburg, Germany, in 1979 and received his diploma in chemistry at the Ludwig-Maximilians-University Munich in 2005, which included a research internship at the University of Oxford with Prof. David Hodgson. He completed his Ph.D. in Chemistry under the supervision of Professor Thomas Carell and is currently a DAAD postdoctoral fellow studying innate trifluoromethylation of N-containing heterocycles with Professor Phil S. Baran at TSRI.
Dr. Ryan Baxter was born in Muscoda, Wisconsin in 1982. He received his B.Sc. from the University of Wisconsin - Madison in 2005 under Professor Samuel H. Gellman before moving to the University of Michigan where he received his M.Sc. in 2007 and Ph.D. in 2010 under the guidance of Professor John Montgomery. He is currently a postdoctoral research associate at TSRI working with Professors Donna G. Blackmond and Phil S. Baran.
Yoshihiro Ishihara was born in Kyoto, Japan in 1984 but was raised in Montreal, Canada. He received his B.Sc. in Chemistry at McGill University in 2005, and remained there until 2007 to complete his M.Sc. in Chemistry under the supervision of Professor Hanadi Sleiman. He is currently a graduate student studying the total synthesis of terpenoids with Professor Phil S. Baran at TSRI.
Prof. Phil S. Baran was born in New Jersey in 1977 and received his undergraduate education from NYU with Professor David I. Schuster in 1997. After earning his Ph.D. with Professor K. C. Nicolaou at Scripps in 2001, he pursued postdoctoral studies with Professor E. J. Corey at Harvard until 2003, at which point he began his independent career at TSRI, rising to the rank of Professor in 2008. His laboratory is dedicated to the study of fundamental organic chemistry through the auspices of natural product total synthesis.
References
- (1).Gutekunst WR, Baran PS. C-H Functionalization Logic in Total Synthesis. Chem. Soc. Rev. 2011;40:1976–1991. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- (2).a) Nguyen T-H, Chau NTT, Castanet A-S, Nguyen KPP, Mortier J. First General, Direct, and Regioselective Synthesis of Substituted Methoxybenzoic Acids by Ortho Metalation. J. Org. Chem. 2007;72:3419–3429. doi: 10.1021/jo070082a. [DOI] [PubMed] [Google Scholar]; b) Wang D-H, Mei T-S, Yu J-Q. Versatile Pd(II)-Catalyzed C−H Activation/Aryl−Aryl Coupling of Benzoic and Phenyl Acetic Acids. J. Am. Chem. Soc. 2008;130:17676–17677. doi: 10.1021/ja806681z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Murphy JM, Liao X, Hartwig JF. Meta Halogenation of 1,3-Disubstituted Arenes Via Iridium-Catalyzed Arene Borylation. J. Am. Chem. Soc. 2007;129:15434–15435. doi: 10.1021/ja076498n. [DOI] [PubMed] [Google Scholar]
- (3).a) Onda K, Shiraki R, Ogiyama T, Yokoyama K, Momose K, Katayama N, Orita M, Yamaguchi T, Furutani M, Hamada N, Takeuchi M, Okada M, Ohta M, Tsukamoto S-I. Design, Synthesis, and Pharmacological Evaluation of N-Bicyclo-5-Chloro-1H-Indole-2-Carboxamide Derivatives as Potent Glycogen Phosphorylase Inhibitors. Bioorg. Med. Chem. 2008;16:10001–10012. doi: 10.1016/j.bmc.2008.10.021. [DOI] [PubMed] [Google Scholar]; b) Leonard F, Wajngurt A, Tschannen W, Block FB. Unnatural Amino Acids. I. 3-Carboxytyrosine Derivatives. J. Med. Chem. 1965;8:812–815. [Google Scholar]; c) Smith MB, March J. March’s Advanced Organic Chemistry. John Wiley & Sons; Hoboken: 2007. [Google Scholar]
- (4).C-H Activation. In Topics in Current Chemistry; Yu J-Q, Shi Z, editors. Vol. 292. Springer Berlin; Heidelberg: 2010. p. 380.
- (5).Sundberg RJ. Indoles. Academic Press; San Diego: 1996. [Google Scholar]
- (6).Richter JM, Ishihara Y, Masuda T, Whitefield BW, Llamas T, Pohjakallio A, Baran PS. Enantiospecific Total Synthesis of the Hapalindoles, Fischerindoles, and Welwitindolinones Via a Redox Economic Approach. J. Am. Chem. Soc. 2008;130:17938–17954. doi: 10.1021/ja806981k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Baran PS, Maimone TJ, Richter JM. Total Synthesis of Marine Natural Products without Using Protecting Groups. Nature. 2007;446:404–408. doi: 10.1038/nature05569. [DOI] [PubMed] [Google Scholar]
- (8).Metal-catalyzed Cross-coupling reactions; Diederich F, Stang PJ, editors. Wiley-VCH Verlag GmbH; Weinheim: 1998. p. 517.
- (9).Richter JM, Whitefield BW, Maimone TJ, Lin DW, Castroviejo MP, Baran PS. Scope and Mechanism of Direct Indole and Pyrrole Couplings Adjacent to Carbonyl Compounds: Total Synthesis of Acremoauxin A and Oxazinin 3. J. Am. Chem. Soc. 2007;129:12857–12869. doi: 10.1021/ja074392m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Baran PS, Ambhaikar NB, Guerrero CA, Hafensteiner BD, Lin DW, Richter JM. Oxidative C-C Bond Formation in Heterocyclic Chemistry. Arkivoc. 2006:310–325. [Google Scholar]
- (11).Baran PS, Richter JM, Lin DW. Direct Coupling of Pyrroles with Carbonyl Compounds: Short Enantioselective Synthesis of (S)-Ketorolac. Angew. Chem., Int. Ed. 2005;44:609–612. doi: 10.1002/anie.200462048. [DOI] [PubMed] [Google Scholar]
- (12).Jessing M, Baran PS. Oxidative Coupling of Indoles with 3-Oxindoles. Heterocycles. 2011;82:1739–1745. [Google Scholar]
- (13).a) Iwao M. Directed Lithiation of 1-Triisopropylsilylgramine. A Short Access to 3,4-Disubstituted Indoles. Heterocycles. 1993;36:29–32. [Google Scholar]; b) Robbins DW, Boebel TA, Hartwig JF. Iridium-Catalyzed, Silyl-Directed Borylation of Nitrogen-Containing Heterocycles. J. Am. Chem. Soc. 2010;132:4068–4069. doi: 10.1021/ja1006405. [DOI] [PubMed] [Google Scholar]
- (14).DeMartino MP, Chen K, Baran PS. Intermolecular Enolate Heterocoupling: Scope, Mechanism, and Application. J. Am. Chem. Soc. 2008;130:11546–11560. doi: 10.1021/ja804159y. [DOI] [PubMed] [Google Scholar]
- (15).Baran PS, DeMartino MP. Intermolecular Oxidative Enolate Heterocoupling. Angew. Chem., Int. Ed. 2006;45:7083–7086. doi: 10.1002/anie.200603024. [DOI] [PubMed] [Google Scholar]
- (16).Casey BM, Flowers RA. On the Nature of the Oxidative Heterocoupling of Lithium Enolates. J. Am. Chem. Soc. 2011;133:11492–11495. doi: 10.1021/ja205017e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Baran PS, Hafensteiner BD, Ambhaikar NB, Guerrero CA, Gallagher JD. Enantioselective Total Synthesis of Avrainvillamide and the Stephacidins. J. Am. Chem. Soc. 2006;128:8678–8693. doi: 10.1021/ja061660s. [DOI] [PubMed] [Google Scholar]
- (18).Clift MD, Taylor CN, Thomson RJ. Oxidative Carbon-Carbon Bond Formation Via Silyl Bis-Enol Ethers: Controlled Cross-Coupling for the Synthesis of Quaternary Centers. Org. Lett. 2007;9:4667–4669. doi: 10.1021/ol702306z. [DOI] [PubMed] [Google Scholar]
- (19).Chen K, Richter JM, Baran PS. 1,3-Diol Synthesis Via Controlled, Radical-Mediated C-H Functionalization. J. Am. Chem. Soc. 2008;130:7247–7249. doi: 10.1021/ja802491q. [DOI] [PubMed] [Google Scholar]
- (20).Mello R, Fiorentino M, Fusco C, Curci R. Oxidations by Methyl(trifluoromethyl)dioxirane. 2. Oxyfunctionalization of Saturated Hydrocarbons. J. Am. Chem. Soc. 1989;111:6749–6757. [Google Scholar]
- (21).Chen K, Baran PS. Total Synthesis of Eudesmane Terpenes by Site-Selective C-H Oxidations. Nature. 2009;459:824–828. doi: 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- (22).Chen K, Ishihara Y, Morón Galán M, Baran PS. Total Synthesis of Eudesmane Terpenes: Cyclase Phase. Tetrahedron. 2010;66:4738–4744. [Google Scholar]
- (23).Chen K, Eschenmoser A, Baran PS. Strain Release in C-H Bond Activation? Angew. Chem., Int. Ed. 2009;48:9705–9708. doi: 10.1002/anie.200904474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chen MS, White MC. A Predictably Selective Aliphatic C-H Oxidation Reaction for Complex Molecule Synthesis. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- (25).Köck M, Grube A, Seiple IB, Baran PS. The Pursuit of Palau’amine. Angew. Chem., Int. Ed. 2007;46:6586–6594. doi: 10.1002/anie.200701798. [DOI] [PubMed] [Google Scholar]
- (26).a) O’Malley DP, Li K, Maue M, Zografos AL, Baran PS. Total Synthesis of Dimeric Pyrrole-Imidazole Alkaloids: Sceptrin, Ageliferin, Nagelamide E, Oxysceptrin, Nakamuric Acid, and the Axinellamine Carbon Skeleton. J. Am. Chem. Soc. 2007;129:4762–4775. doi: 10.1021/ja069035a. [DOI] [PubMed] [Google Scholar]; b) Seiple IB, Su S, Young IS, Nakamura A, Yamaguchi J, Jørgensen L, Rodriguez RA, O’Malley DP, Gaich T, Köck M, Baran PS. Enantioselective Total Syntheses of (−)-Palau’amine, (−)-Axinellamines, and (−)-Massadines. J. Am. Chem. Soc. 2011 doi: 10.1021/ja2047232. 10.1021/ja2047232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).a) Minisci F, Fontana F, Vismara E. Substitutions by Nucleophilic Free-Radicals - A New General Reaction of Heteroaromatic Bases. J. Heterocycl. Chem. 1990;27:79–96. [Google Scholar]; b) Minisci F, Vismara E, Fontana F. Recent Developments of Free-Radical Substitutions of Heteroaromatic Bases. Heterocycles. 1989;28:489–519. [Google Scholar]
- (28).Seiple IB, Su S, Rodriguez RA, Gianatassio R, Fujiwara Y, Sobel AL, Baran PS. Direct C-H Arylation of Electron-Deficient Heterocycles with Arylboronic Acids. J. Am. Chem. Soc. 2010;132:13194–13196. doi: 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).a) Ackermann L. Carboxylate-Assisted Transition-Metal-Catalyzed C-H Bond Functionalizations: Mechanism and Scope. Chem. Rev. 2011;111:1315–1345. doi: 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]; b) Giri R, Shi B-F, Engle KM, Maugel N, Yu J-Q. Transition Metal-Catalyzed C-H Activation Reactions: Diastereoselectivity and Enantioselectivity. Chem. Soc. Rev. 2009;38:3242–3272. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]
- (30).Wasa M, Worrell BT, Yu J-Q. Pd0/PR3-Catalyzed Arylation of Nicotinic and Isonicotinic Acid Derivatives. Angew. Chem., Int. Ed. 2010;49:1275–1277. doi: 10.1002/anie.200906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).a) Patai S, Rappoport ZE. The Chemistry of the Quinonoid Compounds. Wiley; New York: 1988. [Google Scholar]; b) Thomson RH. Naturally Occuring Quinones IV. Blackie Academic; London: 1997. [Google Scholar]
- (32).a) Fujiwara Y, Domingo V, Seiple IB, Gianatassio R, Del Bel M, Baran PS. Practical C-H Functionalization of Quinones with Boronic Acids. J. Am. Chem. Soc. 2011;133:3292–3295. doi: 10.1021/ja111152z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Molander GA, Colombel V, Braz VA. Direct Alkylation of Heteroaryls Using Potassium Alkyl- and Alkoxymethyltrifluoroborates. Org. Lett. 2011;13:1852–1855. doi: 10.1021/ol2003572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).a) Smart BE. Introduction: Fluorine Chemistry. Chem. Rev. 1996;96:1555–1555. doi: 10.1021/cr960075e. [DOI] [PubMed] [Google Scholar]; b) Welch JT. Advances in the Preparation of Biologically-Active Organofluorine Compounds. Tetrahedron. 1987;43:3123–3197. [Google Scholar]
- (34).a) Langlois BR, Billard T, Mulatier JC, Yezeguelian C. A New Preparation of Trifluoromethanesulfinate Salts. J. Fluorine Chem. 2007;128:851–856. [Google Scholar]; b) Langlois BR, Laurent E, Roidot N. Trifluoromethylation of Aromatic Compounds with Sodium Trifluoromethanesulfinate under Oxidative Conditions. Tetrahedron Lett. 1991;32:7525–7528. [Google Scholar]
- (35).Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond D, Baran PS. Innate C-H Trifluoromethylation of Heterocycles. Proc. Natl. Acad. Sci. U. S. A. 2011 doi: 10.1073/pnas.1109059108. 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. The Palladium-Catalyzed Trifluoromethylation of Aryl Chlorides. Science. 2010;328:1679–1681. doi: 10.1126/science.1190524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wang X, Truesdale L, Yu J-Q. Pd(II)-Catalyzed Ortho-Trifluoromethylation of Arenes Using TFA as a Promoter. J. Am. Chem. Soc. 2010;132:3648–3649. doi: 10.1021/ja909522s. [DOI] [PubMed] [Google Scholar]
- (38).Kino T, Nagase Y, Ohtsuka Y, Yamamoto K, Uraguchi D, Tokuhisa K, Yamakawa T. Trifluoromethylation of Various Aromatic Compounds by CF3I in the Presence of Fe(II) Compound, H2O2 and Dimethylsulfoxide. J. Fluorine Chem. 2010;131:98–105. [Google Scholar]
- (39).Gaich T, Baran PS. Aiming for the Ideal Synthesis. J. Org. Chem. 2010;75:4657–4673. doi: 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]
- (40).Newhouse T, Baran PS, Hoffmann RW. The Economies of Synthesis. Chem. Soc. Rev. 2009;38:3010–3021. doi: 10.1039/b821200g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vaillancourt V, Albizati KF. Synthesis and Absolute-Configuration of (+)-Hapalindole Q. J. Am. Chem. Soc. 1993;115:3499–3502. [Google Scholar]
- (42).Kobler C, Effenberger F. Chemo enzymatic synthesis of Rengyol and Isorengyol. Tetrahedron. 2006;62:4823–4828. [Google Scholar]
- (43).Breslow R, Yang J, Yan J. Biomimetic Hydroxylation of Saturated Carbons with Artificial Cytochrome P-450 Enzymes-Liberating Chemistry from the Tyranny of Functional Groups. Tetrahedron. 2002;58:653–659. [Google Scholar]