

Abstract
Objective
Recent studies have shown a role for Rac1 in regulating platelet functions, but how Rac1 is activated in platelets remains unclear. PIP3-dependent Rac exchanger 1 (P-Rex1) was originally identified in neutrophils that regulates phagocyte functions. We sought to examine whether P-Rex1 plays a role in platelet activation.
Methods and Results
Western blotting showed P-Rex1 expression in mouse and human platelets. Mice lacking P-Rex1 exhibited prolonged bleeding time and increased re-bleeding. When challenged with low doses of the G protein-coupled receptor (GPCR) agonists U46619 and thrombin, P-Rex1-/- platelets displayed significantly reduced secretion and aggregation compared to WT platelets. Increasing the concentration of these agonists could overcome the defect. Platelet aggregation induced by collagen, a non-GPCR agonist, was also compromised in the absence of P-Rex1. Along with these phenotypic changes were impaired Rac1 activation, reduced ATP secretion, decreased phosphorylation of Akt, JNK and p38 MAPK in P-Rex1-/- platelets upon agonist stimulation.
Conclusion
These results demonstrate for the first time the presence of P-Rex1 in platelets and its role in platelet secretion as well as aggregation induced by low-dose agonists for GPCR and by collagen.
Keywords: platelets, P-Rex1, Rac1, secretion, aggregation
Platelets play a critical role in physiological and pathological processes including hemostasis and thrombosis. At the site of blood vessel injury, platelets accumulate to the exposed subendothelial matrix and undergo an activation process that includes shape change, aggregation and granule secretion, leading to clot formation and initiating repair of the damaged vessel wall. A variety of agonists, including thrombin, thromboxane A2 (TXA2), adenosine diphosphate (ADP), and collagen, are involved in platelet activation and may work together to facilitate clot formation. Upon vessel wall injury, exposed subendothelial collagen plays an important role in the initiation of platelet activation. In addition, thrombin generated from the injury site, combined with TXA2 and ADP secreted from activated platelets, further induce platelet activation as well as formation of stable thrombi. Among these agonists, thrombin, TXA2 and ADP induce platelet activation via G protein-coupled receptors (GPCRs), whereas collagen engages multiple platelet receptors including the glycoprotein VI (GPVI)/Fc receptor γ, glycoprotein IV (GPIV) and the integrin α2β1 1. Thrombin receptors including the protease-activated receptors 1 and 4 (PAR1 and PAR4) in human and the TXA2 receptor are functionally coupled to the Gq and G12/13 signaling pathways 2-5. During platelet activation, the receptors for thrombin, TXA2 and collagen can mediate substantial activation of the small GTPase Rac, which is required for platelet aggregation and secretion.
Rac, a subfamily of the Rho small GTPases, contains three members including Rac1, Rac2 and Rac3. The Rac1 GTPase is ubiquitously expressed, whereas Rac2 is specifically expressed in hematopoietic cells of myeloid lineage 6. Rac3 GTPase is detected only in the brain during development 7. In platelets, Rac1 but not Rac2 is detected at the protein level 8. Rac1 deficient platelets display impaired aggregation and secretion, suggesting that the Rac1 GTPase is important to platelet functions 9. It is well known that activated Rac1 GTPase regulates lamellipodia formation through direct stimulation of p21-activated kinase (PAK), which in turn induces actin polymerization 9-11. A recent report showed that activated Rac GTPase also regulates the MAPK signaling pathways in platelets 12. Therefore, the role for the Rac GTPase is not confined to cytoskeletal rearrangement during platelet activation. Although the downstream effectors of activated Rac GTPase are well described, much less is known about the upstream mechanism for Rac GTPase activation in platelets.
Studies of Rac activation have identified a large number of guanine nucleotide exchange factors (GEFs), among which P-Rex1 is a Rac-specific GEF activated by the βγ subunits of G proteins and by phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which are produced through phosphoinositol 3-kinase (PI3K) activation 13, 14. Initially purified from neutrophils and found in high abundance both in leukocytes and in the brain 13, P-Rex1 is known to play important roles in the regulation of bactericidal functions of neutrophils. Based on studies of P-Rex1 deficient neutrophils, it was found that this GEF is required for optimal production of reactive oxygen species (ROS) and neutrophil chemotaxis through activation of the Rac2 GTPase 15, 16. During brain development, P-Rex1 is involved in neurotrophin-mediated neuronal migration 17. A more recent study showed that P-Rex1 is also involved in ErbB signaling in breast cancer cells 18, expanding its role beyond normal cells and physiological functions.
The expression and function of P-Rex1 in platelet has remained unknown. In the present study, we investigated whether P-Rex1 is important for platelet activation using the GPCR agonists thrombin and U46619, and a non-GPCR agonist collagen. Our study revealed that P-Rex1 is not only expressed in mouse platelets but also plays an important role in regulating platelet aggregation and dense granule secretion. Rac1 activation is attenuated in the absence of P-Rex1, and P-Rex1 deficient mice show an extended bleeding time as well as reduced activation of Akt, JNK and p38 MAPK. These results demonstrate that P-Rex1 is a major regulator of platelet activation by low-dose GPCR agonists and a potential target for therapeutic intervention.
Methods
Materials
The TXA2 analog U46619 and Rac inhibitor NSC23766 were purchased from Calbiochem (San Diego, CA). Thrombin, collagen and luciferase/luciferin reagent were purchased from Chrono-Log (Havertown, PA). Prostaglandin E1 and apyrase were obtained from Sigma-Aldrich (St Louis, MO). Mouse anti-P-Rex1 antibody 13, 15 was a kind gift from Dr. Marcus Thelen (Institute for Research in Biomedicine, Switzerland). A rabbit polyclonal antibody against phosphorylated Ser473 of Akt and antibodies against phosphorylated and non-phosphorylated forms of JNK (Thr183/Tyr185) were from Cell Signaling Technology (Boston, MA). The mouse monoclonal antibody against Rac1 were purchased from BD Biosciences (San Diego, CA).
Mice
P-Rex1 knockout mice were generated as described 16. The mice were backcrossed at least 7 generations to C57BL/6 background. Wide type C57BL/6 mice were purchased from Charles River Laboratories (Wilminton, MA). Mice 8 to 12 week of age were used in the study. All experiments involving the use of mice were conducted with protocols approved by the Institutional Animal Care and Use Committee at the University of Illinois at Chicago.
Platelet preparation
Male and female mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (5 mg/kg) in PBS. Whole blood was collected from the abdominal aorta using 100 μl volume of ACD (85 mM trisodium citrate, 83 mM dextrose, and 21 mM citric acid) as anticoagulant. For each experiment, blood was pooled from 5-6 mice. Platelets were isolated from platelet-rich plasma (PRP) by centrifugation of whole blood with 0.1 μg/ml prostaglandin E1 and 1 U/ml apyrase, washed twice with CGS buffer (0.12 M sodium chloride, 0.0129 M trisodium citrate, 0.03 M D-glucose, pH 6.5) with 1 U/ml apyrase and 5 mM EDTA, and resuspended in Tyrode's buffer (150 mM NaCl, 12 mM NaHCO3, 2.5 mM KCl, 10 mM HEPES, 5.5 mM D-Glucose, 1 mM CaCl2, 1 mM MgCl2 and 1 mg/ml BSA).
Platelet aggregation and secretion
Platelet aggregation was determined by the turbidometric method. Platelet dense granule secretion was evaluated by measuring the release of ATP using luciferin/luciferase reagent (Chrono-Lume). Washed platelets (3 × 108/ml) were mixed with luciferin/luciferase reagent within 1 min before stimulation. Platelet aggregation and secretion were recorded in a platelet aggregometer (Chrono-Log, Havertown, PA) at 37°C while stirring at 1,000 rpm. To detect the effect of Rac inhibition, washed platelets were pre-incubated with 1 μM NSC23766 for 1 min prior to stimulation with U46619. To investigate the effect of secretion on collagen-induced aggregation, platelets were pre-treated with 10 nM SQ29548 (Cayman Chemical) or 50 μM aspirin (Sigma) for 1 min. Platelet aggregation was then measured after collagen stimulation.
Western blotting assay
Washed platelets (400 μl, 1 × 108/ml) were stimulated with U46619 (60 nM) for different periods of time at 37°C while stirring at 1,000 rpm. The reaction was terminated by adding 4× loading buffer (NuPAGE, Invitrogen, Carlsbad, CA). Western blotting was performed as described previously 19. The lysate was separated on 10% SDS-PAGE. Phosphorylated and unphosphorylated Akt and MAPK were detected with specific antibodies against Akt (Ser473) and JNK (Thr183/Tyr185). The relative density of bands on gel blots was quantified using the ImageJ software (National Institutes of Health, Bethesda, MD). For detection of P-Rex1 in platelets, mouse and human platelets were lysed, the lysate was separated on 4-20% SDS-PAGE, and P-Rex1 was detected with the anti-P-Rex1 antibody. For positive control, HEK293T cells were transiently transfected with pRK5 plasmid vector containing human P-Rex1 cDNA, and the cell lysate was compared in Western blotting.
Analysis of Rac activation
Rac activation was determined using p21-activated kinase binding domain (PBD) as described previously 20. The PBD-GST fusion protein was expressed in E.coli strain HB101 and purified. Washed platelets (1 × 109/ml) were stimulated with 60 nM U46619 at different time points with stirring. The platelets were then lysed by addition of lysis buffer/wash buffer (6 mM Na2HPO4, 4 mM NaH2PO4, 1% Nonidet P-40, 150 mM NaCl, 30 mM MgCl2, 2 mM PMSF, Protease Inhibitor Cocktail I (Calbiochem), 0.1 mM Na3VO4, and 50 mM NaF). After centrifugation, the lysate was incubated with 20 μg of PAK1 PBD-GST recombinant protein at 4°C overnight, and then incubated with 30 μl of glutathione-Sepharose 4B beads (Little Chalfont, Buckinghamshire, UK) for another 1 h. Beads were washed five times with wash buffer and resuspended in a loading buffer. Aliquots of the total Rac protein and those bound to the beads (Rac-GTP) were analyzed by Western blotting.
Statistical analysis
For statistical analysis, a P-value < 0.05 was considered statistically significant. Student's t-test was performed. For bleeding time, difference between WT and P-Rex1 KO mice was evaluated with two-tailed Mann-Whitney tests.
Results
P-Rex1 deficient mice display impaired hemostasis
We first examined the expression of P-Rex1 in mouse and human platelets. Washed platelets isolated from P-Rex1-/- and WT mice (Figure 1A, upper panels) or from human blood (lower panels) were lysed and the expression of P-Rex1 was determined by Western blotting. An anti-P-Rex1 antibody detected a single species of expected size (∼190 kDa) in WT platelets. As a positive control, HEK293T cells transfected with human P-Rex1 cDNA-expression vector expressed a protein of similar size that was detected by the anti-P-Rex1 antibody (Figure 1A). This species was absent in platelets prepared from P-Rex1 knockout mice or in HEK293 cells transfected with an empty vector, confirming the identity of the detected protein as P-Rex1.
Figure 1. P-Rex1 deficient mice display unstable hemostasis in vivo.
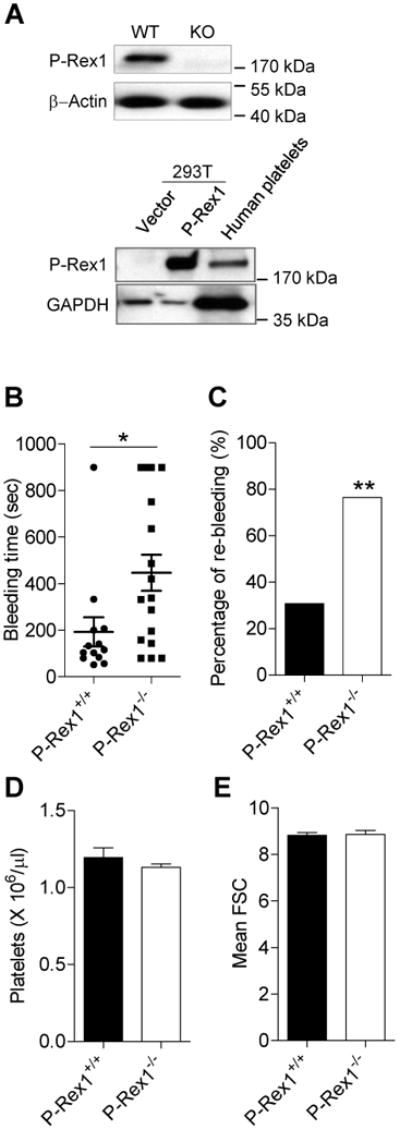
(A) P-Rex1 was expressed in the platelets. Platelets from WT and P-Rex1-/- (KO) mice were lysed and P-Rex1 protein was detected by Western blotting. β-actin was used as a loading control. HEK 293T cells were transiently transfected with expression vector (pRK5-human P-Rex1 as positive control. Lysate of 293T cells and washed human platelets were measured for P-Rex1 expression by using Western blotting. GAPDH was used as a loading control. (B) Bleeding time measurements in WT and P-Rex1-/- mice. Horizontal lines indicate the median values (with two-tailed Mann-Whitney tests, *, p<0.05), and the percentage of re-bleeding after 15 min (C) was also determined (with Chi-square test, **, p<0.01). Results in (B) and (C) were obtained from 13 WT and 17 KO mice. (D) The total number of platelets in peripheral blood of WT and KO were counted. Data are expressed as means ± SEM (n = 5). (E) The size of P-Rex1 platelet was estimated by flow cytometry using forward scatter (FSC). Data (FSC) are expressed as means ± SEM. D and E were based on data collected from 3 mice.
We next determined whether P-Rex1 is involved in hemostasis in vivo using a tail-bleeding assay. The bleeding time of P-Rex1-/- mice (446.8 ± 77.76 sec) was significantly longer than that of the WT controls (192.92 ± 62.61 sec) (Figure 1B; p<0.05). Of note, bleeding times longer than 900 sec were observed in nearly one quarter (23.5 %) of the P-Rex1-/- mice tested, compared to only 7.7 % of the WT controls. The percentage of mice showing re-bleeding, defined as recurrent bleeding within 15 min of the first occurrence, was more than 2.5 times higher in P-Rex1-/- mice than the WT controls (76.5% vs. 30.8%; Figure 1C). However, P-Rex1-/- mice did not display a significantly difference platelet count compared to the WT controls (1.19 ± 0.06 × 106 /μl in P-Rex1-/- mice vs. 1.13 ± 0.02 × 106 /μl in WT mice; Figure 1D), suggesting that P-Rex1 is not critical to the generation of platelets or its lifespan. Flow cytometry analysis showed that platelets with or without P-Rex1 were similar in size (Figure 1E). Platelet adhesion and spreading on fibrinogen was also unchanged in the absence of P-Rex1 (Supplemental Figure S1). Collectively, these data indicate that the observed difference in bleeding time between the WT and P-Rex1-/- mice might be attributed to functional changes in platelet activation, suggesting that P-Rex1 plays an important role in hemostasis.
P-Rex1 regulates platelet aggregation and dense granule secretion
Based on these findings, we next examined platelet aggregation and ATP release, which is a function of dense granule secretion. U46619 is a synthetic analog of TXA2 that induces platelet aggregation and secretion through the TXA2 receptor (TPR), primarily activating the Gαq-coupled pathways. Upon stimulation with a low concentration of U46619 (60 nM), platelet aggregation was markedly reduced in P-Rex1-/- mice (Figure 2A). Platelets isolated from WT mice showed a typical biphasic aggregation 21, of which the first phase was a direct result of U46619 activation through TPR, and the second phase presumably represented the combined effect of TPR activation and TPR-mediated ADP secretion through a paracrine or autocrine mechanism. A close examination of the P-Rex1-/- platelets found that it lacked the second phase (Figure 2A), suggesting that secretion-dependent aggregation might be compromised in P-Rex1 deficient platelets. Indeed, the phenotype of one-phase aggregation in P-Rex1-/- platelets was accompanied by a drastically altered pattern of ATP release. Whereas platelets from the WT mice showed two phases of ATP release upon U46619 stimulation, ATP secretion was completely absent in P-Rex1-/- platelets (Figure 2B). Increasing the concentration of U46619 to 100 nM partially restored aggregation of P-Rex1-/- platelets (Figure 2C). At 100 nM, U46619 induced a primary secretion in P-Rex1-/- platelets that was similar to ATP release from the WT platelets; however, secondary secretion in P-Rex1-/- platelets was markedly impaired (Figure 2D). At 300 nM, P-Rex1-/- and WT platelets showed similar aggregation and secretion (Figure 2E and F), suggesting that the effect of P-Rex1 deficiency can be overcome with high concentrations of the agonist (see supplemental Figure S3 for quantification). The restored platelet activation is largely the result of secretion-induced second phase activation. This notion was confirmed when ADP was added back to P-Rex1-/- platelets, which effectively restored second phage platelet aggregation (data not shown). Collectively, the results indicate that P-Rex1 plays an important role in platelet secretion.
Figure 2. P-Rex1 deficient platelets display impaired aggregation and ATP secretion upon stimulation with U46619.
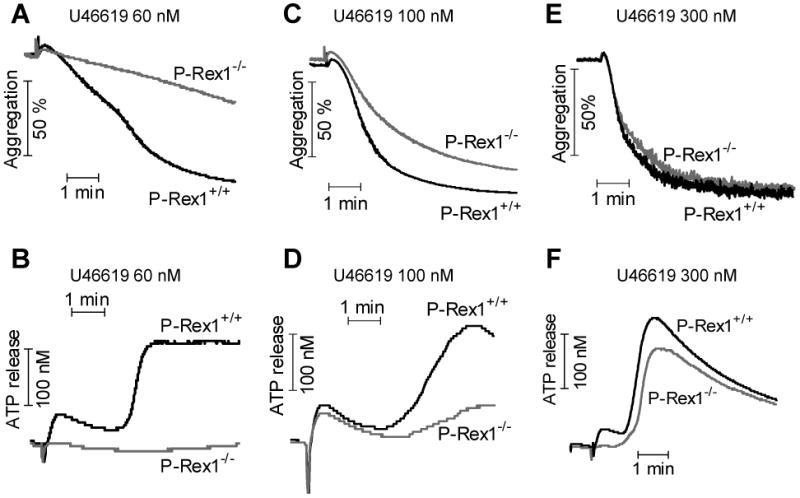
Washed platelets from WT and P-Rex1-/- mice were stimulated with U46619 at the concentrations of 60 nM (A and B), 100 nM (C and D), and 300 nM (E and F). Platelet aggregation (A, C, and E) was monitored as described in Methods. Platelet dense granule secretion (B, D, and F) was determined by measuring the release of ATP. The traces shown are representative of at least three independent experiments.
Thrombin is a potent agonist for platelet activation. A low concentration of thrombin (0.0125 U/ml) induced WT platelets to reversibly aggregate (Figure 3A) and to release ATP (Figure 3B), whereas both aggregation and secretion were absent in the P-Rex1-/- platelets. At a higher concentration (0.01875 U/ml), thrombin was able to induce platelet aggregation and ATP release without P-Rex1, although less than that in WT platelets (Figure 3C and 3D). Further increase of thrombin concentration to 0.025 U/ml completely overcame the defect of P-Rex1 in both aggregation and secretion assays (Figure 3E and 3F). Again, these results support an important role for P-Rex1 in platelet secretion by the GPCR agonists at low concentrations.
Figure 3. P-Rex1 deficient platelets displayed impaired aggregation and secretion upon stimulation with thrombin.
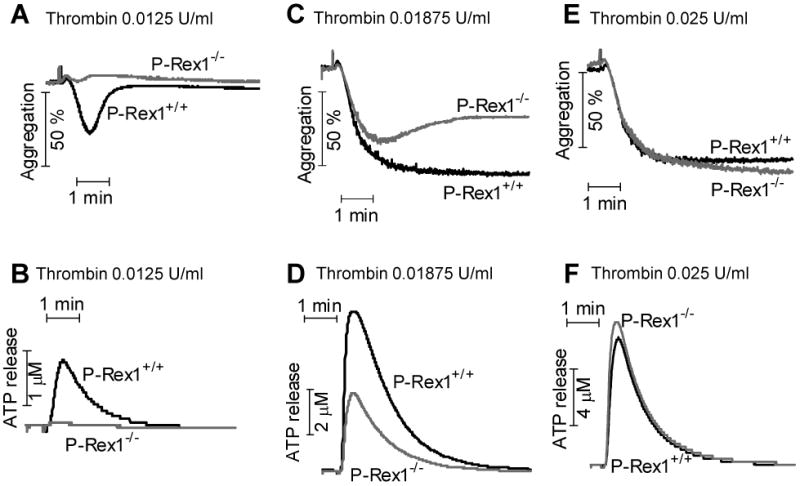
Washed platelets from WT and P-Rex1 KO mice were stimulated with thrombin at the indicated concentrations. Platelet aggregation (A, C and E) and secretion (B, D and F) was determined as described in Methods. The traces shown are representative of at least 3 independent experiments.
Collagen activates platelets through binding to several receptors including the glycoprotein VI (GPVI)/Fc receptor γ complex, integrin α2β1 and GPIV, none of them belonging to the 7-transmembrane domain GPCR family. A previous study showed that collagen stimulates activation of the small GTPase Rac in platelets 10. Therefore, we sought to determine whether P-Rex1 regulates collagen-mediated platelet activation. Upon stimulation with collagen (0.5 μg/ml and 1 μg/ml), platelets from P-Rex1-/- mice showed attenuated aggregation (Figure 4A, 4C) and secretion (Figure 4B, 4D) compared to WT platelets. To investigate whether collagen-induced aggregation is dependent on secretion and TPR feedback, we treated platelets with aspirin, an inhibitor for thromboxane synthase, and SQ29548, a highly selective thromboxane receptor antagonist. Both aspirin and SQ29548 markedly inhibited collagen-induced aggregation (Figure 4E and 4F). Therefore, collagen-induced aggregation is heavily dependent on the release of thromboxane. These results suggest that the attenuated response in P-Rex1 deficient platelets might be attributable to a defect in secretion and TPR feedback.
Figure 4. Impaired aggregation and secretion in P-Rex1-/- platelets upon collagen stimulation.
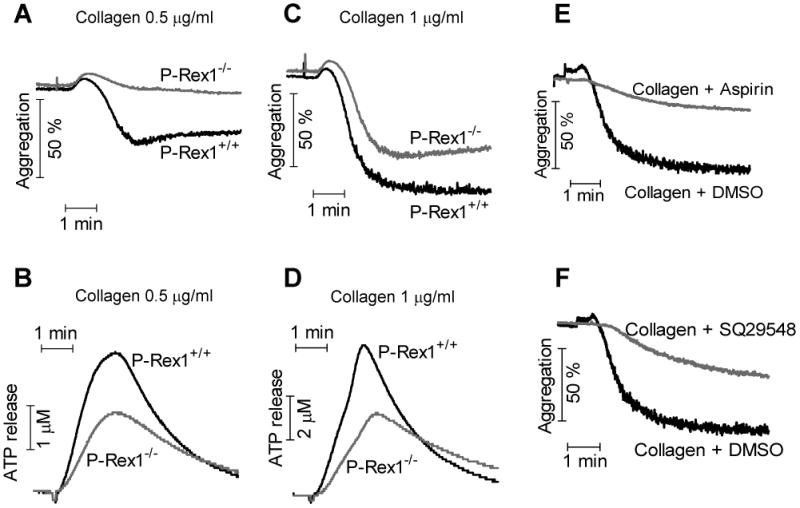
Washed platelets from WT and P-Rex1 KO mice were stimulated with collagen (equine tendon, Type I) at the concentration of 0.5 μg/ml (A and B) and 1 μg/ml (C and D). Platelet aggregation (A and C) and dense granule secretion (B and D) was described in Methods. The traces are representative of at least three independent experiments. Washed platelets from C57BL/6 mice were pretreated with 50 μM of aspirin (E) or 10 nM of SQ29548 (F) for 1 min. After stimulation of Collagen at 1 μg/ml, platelet aggregation was recorded by aggregometer. The traces are one of 3 independent experiments.
P-Rex1 regulates Rac activation in platelets
P-Rex1 is a GEF specific for Rac activation, as was documented in earlier studies using leukocytes 13. In platelets, Rac GTPase is rapidly activated after stimulation with the GPCR agonists U46619 and thrombin 22. Rac1-/- platelets exhibit impairment in aggregation and secretion 9. Since more than 70 GEFs for Rho GTPase are present, the GEFs that are primarily responsible for Rac activation in platelets remain undefined. To determine whether P-Rex1 regulates platelet functions through activation of Rac1, we conducted pull-down assay using the Rac-binding domain of p21-activated protein kinase (PBD) 20. A rapid activation of Rac1 was detected in WT platelets following stimulation with U46619 (60 nM), and the results were reproducible. Rac1 activation peaked at ∼15 sec and subsided by 60 sec. In P-Rex1-/- platelets, U46619 induced less that 50% of Rac1 activation at 15 sec compared to WT platelets (Figure 5A, 5B). To further verify that P-Rex1 regulates platelet activation through activating Rac GTPase, we examined whether WT platelets treated with the Rac inhibitor NSC23766 23 could simulate the phenotype of P-Rex1-/- platelets. In platelets receiving NSC23766 (1 μM), U46619 (60 nM)-induced aggregation was markedly attenuated (Figure 5C). Similar to the P-Rex1-/- platelets, NSC23776-treated platelets lacked the second phase of aggregation upon U46619 stimulation. The U46619-induced ATP release was totally abolished after treatment with NSC23766 (Figure 5D). Along with the inhibition of platelet activation, NSC23766 blocked Rac1 activation (Figure 5E). Interestingly, increasing U46619 concentration to 300 nM and 600 nM did not overcome the inhibitory effect of NSC23766 on platelet aggregation and dense granule secretion (supplemental Figure S2). In U46619-stimulated platelets, granule release exhibits two phases. NSC23766 is highly potent in blocking the second-phase secretion, but only slightly reduced the first-phase secretion in WT platelets stimulated with 300 nM of U46619 (Supplemental Figure S2F). In comparison, P-Rex1 deficiency reduced first-phase secretion but had little effect on the second-phase secretion when the platelets were exposed to the same concentration of U46619. In P-Rex1-deficient platelets, NSC23766 did not exert additional inhibition of either aggregation (Supplemental Figure S2E) or first-phase secretion, although it blocked the second-phase secretion (Supplementa Figure S2F). These results suggest that Rac1 is required for platelet dense granule secretion, and P-Rex1 is important for maximal platelet aggregation and secretion by serving as an important Rac1 activator, especially at low concentrations of GPCR agonists.
Figure 5. P-Rex1 deficient platelets display impaired Rac activation.
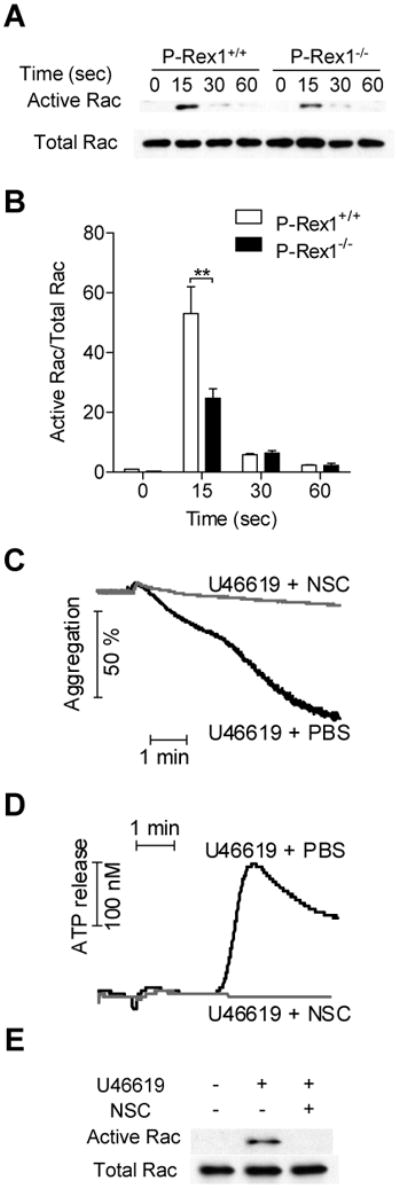
(A) Activation of Rac1 was down-regulated in P-Rex1-/- platelets upon stimulation with U46619. Platelets from WT and P-Rex1-/- mice were stimulated with 60 nM of U46619 for 0, 15, 30 and 60 sec. PBD pull-down assay were performed as described in Materials and Methods and the activated Rac1 was detected with a Rac1-specific antibody. Rac1 from total lysis was detected as input control. (B) Densitometry analysis was conducted to determine the relative level of Rac1 activation (means ± SEM based on 3 experiments; **, p<0.01). (C and D) The Rac inhibitor NSC23766 was used for treatment of WT platelets (NSC, 1 μM, 1 min). The platelets were then stimulated with U46619 (60 nM), and platelet aggregation (C) and ATP release (D) were measured. The effect of NSC23766 on Rac activation was detected (E). Data shown are representative of 3 independent experiments.
P-Rex1 is required for optimal signaling through Akt and MAPK
The PI3K - Akt signaling pathway is involved in platelet aggregation and secretion under various stimulation conditions 24-27. Rac has been implicated in Akt activation. To determine whether P-Rex1 regulates Akt activation, we stimulated platelets from WT and P-Rex1-/- mice with 60 nM of U46619. Peak Akt-Ser473 phosphorylation was observed at 3 min (Figure 6A). Densitometry analysis showed that the peak level of phosphorylation at Ser473 is lower in P-Rex1-/- platelets than in WT platelets, suggesting that P-Rex1 is required for optimal activation of Akt in platelets. Similar to results obtained in aggregation and secretion assays, Akt phosphorylation was identical between WT and P-Rex1-/- platelets when high concentrations of agonists (e.g. 300 nM of U46619) were used (Supplemental Figure S4).
Figure 6. Akt and JNK phosphorylation is attenuated in P-Rex1 deficient platelets.
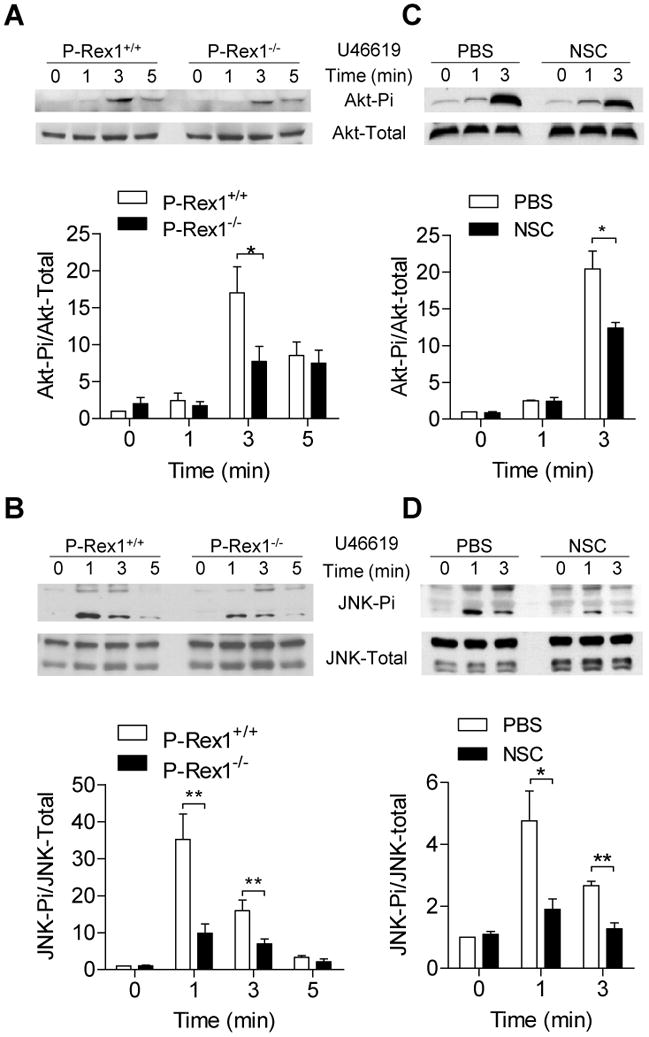
Washed platelets from WT and P-Rex1 KO mice were stirred and stimulated with 60 nM U46619 for 0, 1, 3 and 5 min at 37°C in the aggregometer (A, B). In (C, D) the platelets were treated with either PBS or NSC23766 (NSC, 1 μM, 1 min) before U46619 stimulation. The reaction was terminated with the addition of 4 × loading buffer. Western blotting was performed using platelet lysate and an anti-phospho-Akt antibody (Ser437) (A and C) or an anti-phospho-JNK antibody (Thr183/Tyr185) (B and D). The total protein content of these kinases were also detected and shown as loading controls. Phosphorylation was performed by anti-Akt Ser 437 antibody. The level of phosphorylation was determined using densitometry and relative phosphorylation was shown in bar charts (means ± SEM based on 3 experiments; *, p<0.05 and **, p<0.01).
It has been previously reported that the MAPK signaling pathways (JNK, p38 MAPK and ERK) can be quickly activated by the GPCR agonist U46619 and contributes to platelet activation 28, 29. Since Rac activation is also required for MAPK activation, we postulated that activation of MAPKs might be a downstream event of the P-Rex1-Rac axis. After stimulation with U46619, the phosphorylation of JNK (Figure 6B) was markedly elevated in WT platelets, but the induced phosphorylation was much less in P-Rex1-/- platelets (Figure 6B). Similar results were obtained for ERK and p38 MAPK (data not shown). These findings suggest that P-Rex1 regulates the activation of MAP kinases. The relationship between P-Rex1 and induced phosphorylation of Akt and MAPKs was next examined. Platelets treated with NSC23766 displayed a significantly reduced phosphorylation of Akt (Figure 6C) and JNK (Figure 6D). These results suggest that Rac is upstream of Akt and JNK for their phosphorylation, and P-Rex1 mediated phosphorylation of these kinases was accomplished at least in part through its role as a Rac GEF.
Discussion
Platelets express a large number of GPCRs including proteinase-activated receptors for thrombin, P2Y purinergic receptors, and thromboxane A2 receptor. Although GPCR-mediated Rac activation in platelets has been reported previously 8, 9, 11, 30, the GEFs responsible for this action in platelets remain unidentified. Results from the present study demonstrate that P-Rex1, one of the Rac GEFs activated by PIP3 and Gβγ 13, is expressed in platelets. Moreover, we have shown that P-Rex1 plays an important role in platelet activation upon stimulation with low concentrations of the GPCR agonists U46619 and thrombin. Mice lacking P-Rex1 exhibit prolonged bleeding time and increased re-bleeding, suggesting a deficiency in platelet functions. Since the gene deletion procedure abrogates P-Rex1 expression in all tissues including blood vessels, we further investigated platelet functions ex vivo. P-Rex1 gene deletion does not appear to affect platelet development, as the number and size of platelets are indistinguishable between WT and P-Rex1-/- mice. Furthermore, maximal secretion in WT and P-Rex1 deficient platelets, induced by a high dose of thrombin, was also indistinguishable suggesting that the granule contents of these platelets are similar. It was the platelet aggregation measured after stimulation with low concentrations of the GPCR agonists U46619 and thrombin that was significantly changed in the absence of P-Rex1. Under these experimental conditions, defective secretion was also observed. However, increasing the concentrations of the GPCR agonists overcomes the defective aggregation of P-Rex1-/- platelets.
The observation that compromised functions in P-Rex1-/- platelets could be overcome with high dose of GPCR agonists suggests that, under these experimental conditions, another signaling pathway triggered by the GPCR agonists apparently can replace P-Rex1. It is likely that other Rac GEFs are activated by high concentrations of the GPCR agonists. P-Rex1 is one of the multi-domain Rho-GEF of the Dbl family with high specificity for Rac 13. More than 70 members of the Dbl family Rho GEFs have been identified to date, which far exceeds the number of available Rho GTPases and suggests that GEFs confer tissue specificity 31. Extensive studies have been conducted to investigate the diverse functions of Rho GEFs in a variety of tissues and cells, but very little is known about the presence and functions of Rho GEFs in platelets. Among the few published studies conducted on Rac GEFs in platelets, the Vav subfamily member Vav1 and Vav3 have been found to mediate collagen-induced platelet aggregation 32. The Vav proteins are activated downstream of the ITAM-coupled collagen receptor, GPVI, and mediate PLCγ2 activation which is necessary for the secretion of platelet granules upon collagen stimulation. In a recent study, Vav proteins have been shown to work together with P-Rex1 to regulate leukocyte functions 33. Therefore it is likely that Vav proteins are alternative to P-Rex1 when platelets are stimulated with higher concentrations of GPCR agonists. Interestingly, in neutrophils the two GEFs play different roles in the regulation of LPS priming for oxidant production (requiring P-Rex1 and Vav), particle-induced oxidant production (Vav) and cell spreading (Vav) 33. Our finding that P-Rex1 is not involved in platelet spreading is consistent with these results from neutrophils. Although a full comparison of different Rac GEFs is a subject beyond this work, our preliminary data indeed suggest the presence of such regulatory mechanisms. We have shown that pharmacological inhibition of Rac produced an effect similar to that of P-Rex1 gene deletion in U46619-stimulated platelets. The reduction in platelet aggregation and ATP release was accompanied by a ∼50% decrease in Rac1 activation following U46619 stimulation, supporting a role for the activated Rac in platelet aggregation. The partial loss of Rac1 activation also indicates that P-Rex1 is not the only GEF responsible for Rac1 activation in platelets 22.
The functional involvement of Rac in platelet activation remains an unsettled subject. Our findings corroborate those reported by Zheng and colleagues, who combined pharmacological and genetic approaches to demonstrate an essential role for Rac1 activation in platelet secretion and aggregation 9. Other published reports showed that Rac1 is required for the stability of platelet aggregation under flow 8 and for GPVI- and CLEC-2-dependent platelet aggregation through regulation of PLC-γ2 activation 34. Interestingly, it was not reported that Rac1 deficiency alter GPCR agonist-induced platelet aggregation in these studies. We believe that the concentrations of the GPCR agonists used in these studies might be too high to detect a role for Rac in platelet activation. In the report by McCarty et al 8, thrombin was used at 0.1 U/ml and no difference in platelet aggregation was observed between WT and Rac1 / Rac2 double-KO platelets. In our study, we found that thrombin at 0.0125 U/ml failed to induce the second-phase aggregation in P-Rex1-/- platelets, but at a concentration of 0.01875 U/ml platelet aggregation was markedly improved. At 0.025 U/ml of thrombin, the difference between WT and P-Rex1-/- platelets was completely lost. Likewise, aggregation was markedly different between WT and P-Rex1-/- platelets when stimulated with 60 nM of U46619, but the difference was much smaller at 100 nM of U46619. Pleines et al observed identical platelet aggregation in WT and Rac1-/- platelets when U46619 was used at 300 nM and 3 μM 34. These observations are consistent with those using the Rac inhibitor NSC23766, which inhibits both platelet secretion and aggregation but the inhibitory effect was reversed with increased agonist concentration 9. Taken together, P-Rex1 and Rac1 are required for platelet aggregation at low concentrations of both GPCR agonists and collagen, but as the agonist concentration increases, other GEFs may replace P-Rex1 for Rac1 activation. Rac1 plays a non-redundant role in platelet secretion, and high concentrations of the GPCR agonist U46619 (e.g., 300 nM) cannot overcome the inhibitory effect of NSC23766 (Supplemental Figure S2). Therefore, P-Rex1 regulates both first phase release and second phase release when platelets are exposed to a GPCR agonist at low dose. P-Rex1 gene deletion becomes less critical to platelet activation when the agonist concentration increases. In comparison, NSC23766 potently blocks the second phase release even at high concentrations of a GPCR agonist. These observations suggest that a GEF-specific inhibition by NSC23766 exists when platelets are exposed to low agonist concentration, but as agonist concentration increases, additional GEFs may be activated and NSC23766 becomes less specific for Rac activation by a particular GEF.
Dense granule secretion plays a less important role in platelet aggregation when stimulated with high concentrations of the GPCR agonists. Consistent with these findings, ATP release from the P-Rex1-/- platelets was also defective at low agonist concentrations (60 nM of U46619 and 0.0125 U/ml of thrombin). When the agonist concentration was increased to 100 nM of U46619 and 0.01857 U/ml of thrombin, the secretion of ATP from P-Rex1-/- platelets was partially restored but still remained markedly attenuated compared to WT platelets. Based on the time course of platelet aggregation and secretion, there was a close correlation between defective ATP release and reduced aggregation in P-Rex1-/- platelets, suggesting that compromised dense granule secretion substantially contributes to the observed reduction in platelet aggregation. Data obtained from this study support those published previously and together suggest an important role for Rac1 and the P-Rex1 – Rac1 axis to regulate platelet dense granule secretion.
In addition to the GPCR agonists, collagen was used at two different concentrations to stimulate WT and P-Rex1-/- platelets. We found that platelet aggregation induced by 0.5 μg/ml collagen was significantly reduced in the absence of P-Rex1; however, raising the concentration of collagen to 1 μg/ml markedly improved platelet aggregation. Although none of the collagen receptors are known GPCRs, collagen-stimulates TXA2 production. As a result, collagen-induced secretion contributes to platelet aggregation through a paracrine mechanism involving TXA2, especially when collagen is used at low concentrations. Consistent with this explanation, collagen induced ATP release was compromised in P-Rex1-/- platelets, much like those stimulated with the GPCR ligands. ATP along with other secreted products such as TXA2 serve to stimulate the second phase of platelet aggregation. Consistent with this notion, we observed that aspirin and the TP receptor blocker SQ29548 effectively reduced platelet aggregation (Figure 4E, 4F). In addition, since activation of the collagen receptor GPVI leads to PI3K activation, the resulting accumulation of PIP3 may contribute to P-Rex1 activation. Indeed, inhibition of PI3K negatively impacts collagen-induced platelet secretion and aggregation 35-37, although the isoform of Class I PI3K involved in collagen signaling might be different from the one downstream of GPCRs 36. Taken together, P-Rex1 appears to play an important role downstream of several platelet receptors, including both GPCRs and non-GPCRs, that lead to platelet secretion and aggregation.
Multiple signaling pathways are activated upon stimulation of platelets with GPCR agonists and collagen. In the present study, we assessed the impact of P-Rex1 deficiency on selected signaling pathways. The Ser/Thr kinase Akt is a downstream effector of PI3K, as its activation requires PIP3 production. Both the Akt and MAPK signal pathways are required for platelet activation. Platelets from Akt1-/- or Akt2-/- mice display impaired aggregation similar to platelets treated with the Akt inhibitor SH-6, suggesting that Akt-mediated signal pathway is required for platelet activation 38. The results from experiment using the JNK1-/- mice showed that JNK1 is involved in regulation of platelet functions 29. JNK1 deficient mice display a prolonged bleeding time and impaired thrombus formation, which is associated with deficiencies in platelet aggregation and secretion. In addition, p38 MAPK and ERK are required for platelet activation based on the use of small molecular inhibitors SB203580 and PD98059 12, 28. In P-Rex1-/- platelets, both Akt and MAPK signaling pathways downstream of the TXA2 receptor was defective, suggesting that these signal pathways are modulated by P-Rex1. However, the mechanism for P-Rex1 regulation of Akt and MAPK signal pathways remained unknown. One interpretation is that P-Rex1 modulates the activation of Akt and MAPK through Rac activation. In cardiomyocytes, Rac1-dependent Akt and JNK signaling is required for interleukin-18 expression 39. In human lung epithelial cells, thrombin-induced Akt activation is also dependent on Rac1 activation 40. Rac1-deficient platelets exhibit decreased p38 MAPK and ERK activation 12. Our data show that the Rac1 inhibitor used can alleviate U46619-induced JNK and Akt activation. Because Rac1 was downstream of P-Rex1, we concluded that the P-Rex1-Rac1 axis plays a role in the activation of Akt and JNK, which contributes to platelet activation. However since P-Rex1 is a large protein with several motifs of defined functions, it can possibly serve as a scaffold for protein-protein interaction that brings together signaling molecules and facilitate platelet aggregation through mechanisms other than Rac1 activation.
In summary, results from this study demonstrate an expanded role for P-Rex1 in cells other than neutrophils and neurons, in which P-Rex1 was originally identified. The findings provide evidence that GEFs dictate the specificity of the small GTPase Rac in different tissues and cell types, where they are expressed. In platelets, P-Rex1 appears to be important for the secretion functions, especially when stimulated with low doses of the GPCR ligands U46619 and thrombin. However, cautions should be taken to interpret the data obtained so far, because the studies were conducted using mouse platelets and we have not yet obtained a P-Rex1 inhibitor for examination of human platelets. The identification of P-Rex1 expression in human platelets as well as the conservation between these proteins across mammals suggests that P-Rex1 play an important role in human platelets. Since secretion and the resulting paracrine signaling amplify platelet activation, P-Rex1 may be a target for therapeutic intervention of atherothrombosis given that it is not only used by GPCRs but also by the collagen receptor(s).
Supplementary Material
Acknowledgments
Source of Funding: This study was supported in part by grants AI033503 and HL077806 (R.D.Y.), HL24530 (G.C.L.), HL080706 and HL070694 (D.W.) from the U.S. Public Health Services.
Footnotes
Disclosure: The authors declare no competing financial interests.
References
- 1.Herr AB, Farndale RW. Structural insights into the interactions between platelet receptors and fibrillar collagen. J Biol Chem. 2009;284:19781–19785. doi: 10.1074/jbc.R109.013219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knezevic I, Borg C, Le Breton GC. Identification of Gq as one of the G-proteins which copurify with human platelet thromboxane A2/prostaglandin H2 receptors. J Biol Chem. 1993;268:26011–26017. [PubMed] [Google Scholar]
- 3.Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997;389:183–186. doi: 10.1038/38284. [DOI] [PubMed] [Google Scholar]
- 4.Djellas Y, Manganello JM, Antonakis K, Le Breton GC. Identification of Galpha13 as one of the G-proteins that couple to human platelet thromboxane A2 receptors. J Biol Chem. 1999;274:14325–14330. doi: 10.1074/jbc.274.20.14325. [DOI] [PubMed] [Google Scholar]
- 5.Sambrano GR, Weiss EJ, Zheng YW, Huang W, Coughlin SR. Role of thrombin signalling in platelets in haemostasis and thrombosis. Nature. 2001;413:74–78. doi: 10.1038/35092573. [DOI] [PubMed] [Google Scholar]
- 6.Didsbury J, Weber RF, Bokoch GM, Evans T, Snyderman R. rac, a novel ras-related family of proteins that are botulinum toxin substrates. J Biol Chem. 1989;264:16378–16382. [PubMed] [Google Scholar]
- 7.Bolis A, Corbetta S, Cioce A, de Curtis I. Differential distribution of Rac1 and Rac3 GTPases in the developing mouse brain: implications for a role of Rac3 in Purkinje cell differentiation. Eur J Neurosci. 2003;18:2417–2424. doi: 10.1046/j.1460-9568.2003.02938.x. [DOI] [PubMed] [Google Scholar]
- 8.McCarty OJ, Larson MK, Auger JM, Kalia N, Atkinson BT, Pearce AC, Ruf S, Henderson RB, Tybulewicz VL, Machesky LM, Watson SP. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem. 2005;280:39474–39484. doi: 10.1074/jbc.M504672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akbar H, Kim J, Funk K, Cancelas JA, Shang X, Chen L, Johnson JF, Williams DA, Zheng Y. Genetic and pharmacologic evidence that Rac1 GTPase is involved in regulation of platelet secretion and aggregation. J Thromb Haemost. 2007;5:1747–1755. doi: 10.1111/j.1538-7836.2007.02646.x. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki-Inoue K, Yatomi Y, Asazuma N, Kainoh M, Tanaka T, Satoh K, Ozaki Y. Rac, a small guanosine triphosphate-binding protein, and p21-activated kinase are activated during platelet spreading on collagen-coated surfaces: roles of integrin alpha(2)beta(1) Blood. 2001;98:3708–3716. doi: 10.1182/blood.v98.13.3708. [DOI] [PubMed] [Google Scholar]
- 11.Vidal C, Geny B, Melle J, Jandrot-Perrus M, Fontenay-Roupie M. Cdc42/Rac1-dependent activation of the p21-activated kinase (PAK) regulates human platelet lamellipodia spreading: implication of the cortical-actin binding protein cortactin. Blood. 2002;100:4462–4469. doi: 10.1182/blood.V100.13.4462. [DOI] [PubMed] [Google Scholar]
- 12.Flevaris P, Li Z, Zhang G, Zheng Y, Liu J, Du X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood. 2009;113:893–901. doi: 10.1182/blood-2008-05-155978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welch HC, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, Tempst P, Hawkins PT, Stephens LR. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–821. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- 14.Mayeenuddin LH, McIntire WE, Garrison JC. Differential sensitivity of P-Rex1 to isoforms of G protein betagamma dimers. J Biol Chem. 2006;281:1913–1920. doi: 10.1074/jbc.M506034200. [DOI] [PubMed] [Google Scholar]
- 15.Welch HC, Condliffe AM, Milne LJ, Ferguson GJ, Hill K, Webb LM, Okkenhaug K, Coadwell WJ, Andrews SR, Thelen M, Jones GE, Hawkins PT, Stephens LR. P-Rex1 regulates neutrophil function. Curr Biol. 2005;15:1867–1873. doi: 10.1016/j.cub.2005.09.050. [DOI] [PubMed] [Google Scholar]
- 16.Dong X, Mo Z, Bokoch G, Guo C, Li Z, Wu D. P-Rex1 is a primary Rac2 guanine nucleotide exchange factor in mouse neutrophils. Curr Biol. 2005;15:1874–1879. doi: 10.1016/j.cub.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 17.Yoshizawa M, Kawauchi T, Sone M, Nishimura YV, Terao M, Chihama K, Nabeshima Y, Hoshino M. Involvement of a Rac activator,P-Rex1, in neurotrophin-derived signaling and neuronal migration. J Neurosci. 2005;25:4406–4419. doi: 10.1523/JNEUROSCI.4955-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sosa MS, Lopez-Haber C, Yang C, Wang H, Lemmon MA, Busillo JM, Luo J, Benovic JL, Klein-Szanto A, Yagi H, Gutkind JS, Parsons RE, Kazanietz MG. Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol Cell. 2010;40:877–892. doi: 10.1016/j.molcel.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian F, Deng J, Cheng N, Welch EJ, Zhang Y, Malik AB, Flavell RA, Dong C, Ye RD. A non-redundant role for MKP5 in limiting ROS production and preventing LPS-induced vascular injury. EMBO J. 2009;28:2896–2907. doi: 10.1038/emboj.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benard V, Bohl BP, Bokoch GM. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J Biol Chem. 1999;274:13198–13204. doi: 10.1074/jbc.274.19.13198. [DOI] [PubMed] [Google Scholar]
- 21.Li Z, Zhang G, Le Breton GC, Gao X, Malik AB, Du X. Two waves of platelet secretion induced by thromboxane A2 receptor and a critical role for phosphoinositide 3-kinases. J Biol Chem. 2003;278:30725–30731. doi: 10.1074/jbc.M301838200. [DOI] [PubMed] [Google Scholar]
- 22.Gratacap MP, Payrastre B, Nieswandt B, Offermanns S. Differential regulation of Rho and Rac through heterotrimeric G-proteins and cyclic nucleotides. J Biol Chem. 2001;276:47906–47913. doi: 10.1074/jbc.M104442200. [DOI] [PubMed] [Google Scholar]
- 23.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, De S, Damron DS, Chen WS, Hay N, Byzova TV. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104:1703–1710. doi: 10.1182/blood-2003-10-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho MJ, Pestina TI, Steward SA, Lowell CA, Jackson CW, Gartner TK. Role of the Src family kinase Lyn in TxA2 production, adenosine diphosphate secretion, Akt phosphorylation, and irreversible aggregation in platelets stimulated with gamma-thrombin. Blood. 2002;99:2442–2447. doi: 10.1182/blood.v99.7.2442. [DOI] [PubMed] [Google Scholar]
- 26.Holinstat M, Preininger AM, Milne SB, Hudson WJ, Brown HA, Hamm HE. Irreversible platelet activation requires protease-activated receptor 1-mediated signaling to phosphatidylinositol phosphates. Mol Pharmacol. 2009;76:301–313. doi: 10.1124/mol.109.056622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson SP, Schoenwaelder SM. PI 3-Kinase p110beta regulation of platelet integrin alpha(IIb)beta3. Curr Top Microbiol Immunol. 2010;346:203–224. doi: 10.1007/82_2010_61. [DOI] [PubMed] [Google Scholar]
- 28.Li Z, Zhang G, Feil R, Han J, Du X. Sequential activation of p38 and ERK pathways by cGMP-dependent protein kinase leading to activation of the platelet integrin alphaIIb beta3. Blood. 2006;107:965–972. doi: 10.1182/blood-2005-03-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adam F, Kauskot A, Nurden P, Sulpice E, Hoylaerts MF, Davis RJ, Rosa JP, Bryckaert M. Platelet JNK1 is involved in secretion and thrombus formation. Blood. 2009;115:4083–4092. doi: 10.1182/blood-2009-07-233932. [DOI] [PubMed] [Google Scholar]
- 30.Soulet C, Gendreau S, Missy K, Benard V, Plantavid M, Payrastre B. Characterisation of Rac activation in thrombin- and collagen-stimulated human blood platelets. FEBS Lett. 2001;507:253–258. doi: 10.1016/s0014-5793(01)02984-2. [DOI] [PubMed] [Google Scholar]
- 31.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 32.Pearce AC, Senis YA, Billadeau DD, Turner M, Watson SP, Vigorito E. Vav1 and vav3 have critical but redundant roles in mediating platelet activation by collagen. J Biol Chem. 2004;279:53955–53962. doi: 10.1074/jbc.M410355200. [DOI] [PubMed] [Google Scholar]
- 33.Lawson CD, Donald S, Anderson KE, Patton DT, Welch HC. P-Rex1 and Vav1 cooperate in the regulation of formyl-methionyl-leucyl-phenylalanine-dependent neutrophil responses. J Immunol. 2011;186:1467–1476. doi: 10.4049/jimmunol.1002738. [DOI] [PubMed] [Google Scholar]
- 34.Pleines I, Elvers M, Strehl A, Pozgajova M, Varga-Szabo D, May F, Chrostek-Grashoff A, Brakebusch C, Nieswandt B. Rac1 is essential for phospholipase C-gamma2 activation in platelets. Pflugers Arch. 2009;457:1173–1185. doi: 10.1007/s00424-008-0573-7. [DOI] [PubMed] [Google Scholar]
- 35.Gilio K, Munnix IC, Mangin P, Cosemans JM, Feijge MA, van der Meijden PE, Olieslagers S, Chrzanowska-Wodnicka MB, Lillian R, Schoenwaelder S, Koyasu S, Sage SO, Jackson SP, Heemskerk JW. Non-redundant roles of phosphoinositide 3-kinase isoforms alpha and beta in glycoprotein VI-induced platelet signaling and thrombus formation. J Biol Chem. 2009;284:33750–33762. doi: 10.1074/jbc.M109.048439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim S, Mangin P, Dangelmaier C, Lillian R, Jackson SP, Daniel JL, Kunapuli SP. Role of phosphoinositide 3-kinase beta in glycoprotein VI-mediated Akt activation in platelets. J Biol Chem. 2009;284:33763–33772. doi: 10.1074/jbc.M109.048553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weng Z, Li D, Zhang L, Chen J, Ruan C, Chen G, Gartner TK, Liu J. PTEN regulates collagen-induced platelet activation. Blood. 2010;116:2579–2581. doi: 10.1182/blood-2010-03-277236. [DOI] [PubMed] [Google Scholar]
- 38.Yin H, Stojanovic A, Hay N, Du X. The role of Akt in the signaling pathway of the glycoprotein Ib-IX induced platelet activation. Blood. 2008;111:658–665. doi: 10.1182/blood-2007-04-085514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Venkatesan B, Valente AJ, Prabhu SD, Shanmugam P, Delafontaine P, Chandrasekar B. EMMPRIN activates multiple transcription factors in cardiomyocytes, and induces interleukin-18 expression via Rac1-dependent PI3K/Akt/IKK/NF-kappaB andMKK7/JNK/AP-1 signaling. J Mol Cell Cardiol. 2010;49:655–663. doi: 10.1016/j.yjmcc.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin CH, Cheng HW, Ma HP, Wu CH, Hong CY, Chen BC. Thrombin Induces NF-{kappa}B Activation and IL-8/CXCL8 Expression in Lung Epithelial Cells by a Rac1-dependent PI3K/Akt Pathway. J Biol Chem. 2011;286:10483–10494. doi: 10.1074/jbc.M110.112433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.