

SUMMARY
A murine low-dose (LD) aerosol model is commonly used to test tuberculosis vaccines. Doses of 50-400 CFU (24-hour lung CFU) infect 100% of exposed mice. The LD model measures progression from infection to disease based on organ CFU at defined time points. To mimic natural exposure, we exposed mice to an ultra-low dose (ULD) aerosol. We estimated the presented dose by sampling the aerosol. Female C57BL/6 mice were exposed to Mycobacterium tuberculosis H37Rv aerosol at 1.0, 1.1, 1.6, 5.4, and 11 CFU presented dose, infecting 27%, 36%, 36%, 100%, and 95% of mice, respectively. These data are compatible with a stochastic infection event (Poisson distribution, weighted R2= 0.97) or with a dose-response relationship (sigmoid distribution, weighted R2= 0.97). Based on the later assumption, the ID50 was 1.6 CFU presented dose (95% confidence interval, 1.2 to 2.1). We compared organ CFU after ULD and LD aerosols (5.4 vs. 395 CFU presented dose). Lung burden was 30-fold lower in the ULD model at 4 weeks (3.4 vs. 4.8 logs, p<0.001) and 18 weeks (≤3.6 vs. 5.0 logs, p=0.01). Mice exposed to ULD aerosols as compared to LD aerosols had greater within-group CFU variability. Exposure to ULD aerosols leads to infection in a subset of mice, and to persistently low organ CFU. The ULD aerosol model may resemble human pulmonary tuberculosis more closely than the standard LD model, and may be used to identify host or bacterial factors that modulate the initial infection event.
Keywords: Aerosol, animal model, median infectious dose, Mycobacterium tuberculosis
INTRODUCTION
Human infection with Mycobacterium tuberculosis (MTB) occurs after inhalation of aerosolized bacteria from an infected person. When contacts of an infected source patient are evaluated only a subset of the exposed humans are infected based on tuberculin skin test conversion. The infection rate varies based on the infectivity of the source and the proximity and duration of the exposure. Infection rates after the identification of single source patients averaged 5% in Arkansas prisons, and 12% in Arkansas nursing homes (19). An exposure in the confined environment of a submarine infected 80% of humans sleeping in the same compartment as the source (12).
The natural event of human tuberculosis exposure is assumed to be stochastic (12). Under the stochastic assumption, the inhalation of an infectious quantum results in infection, and the relationship between the aerosol dose and the infection rate is modeled by the Poisson distribution. An alternative model is the biological dose-response relationship represented by a sigmoid curve.
In a classic study demonstrating that MTB is spread through the aerosol route, guinea pigs were placed in the exhaust air ducts of a tuberculosis ward, resulting in MTB infection in 19% of the animals (16). Subsequent animal models have used nebulizers to generate aerosol MTB doses at high multiples of the infectious dose. These models generate uniform infection of all animals. These models have been useful for natural history studies, and for testing vaccines and therapeutics. However these models do not mimic the lower doses associated with natural human infections that lead to only a portion of the population acquiring infection.
A standard low-dose (LD) murine model has been widely used to test vaccines (2, 3, 9, 20). Groups of vaccinated and unvaccinated mice are simultaneously challenged with an aerosol dose between 50 and 400 CFU based on the retained dose, as measured by lung necropsy at 24 hours after exposure (13, 15). At these doses, all mice in the aerosol challenge are infected. The lung bacterial burden increases rapidly during the first four weeks of infection and then plateaus at around 105 to 107 CFU. In the early phase of infection, the MTB also disseminates from the lungs to other organs such as the liver and spleen (4). The infection leads to chronic disease with progressive tissue destruction in lung, liver, and spleen, eventually leading to the death of the mouse. Vaccine efficacy is identified by reduced organ CFU or by increased survival time.
To establish a partial infection model in which only a subset of animals are infected, we exposed mice to an ultra-low-dose (ULD) aerosol. At these low doses, sentinel necropsy does not provide a reliable estimate of the retained dose. Instead, we measured the presented dose using Guyton’s formula to estimate the minute ventilatory volume of the mice and a BioSampler to measure the viable aerosol concentration (18).
The goals of the current study were (1) determine the presented dose required to produce a partial infection in mice, (2) determine if the relationship between presented dose and infection rate is compatible with a stochastic model, and (3) estimate MTB median infectious dose (ID50) in C57BL/6 mice.
MATERIALS AND METHODS
Bacterial strains and growth conditions
MTB H37Rv was used in all aerosol experiments. Frozen stock of 0.72 optical density was used to prepare the inoculum. After thawing the frozen stock a calculated volume of stock was diluted in 2 mL solution of phosphate-buffered saline (PBS) containing 0.05% tyloxapol. The solution was then sonicated twice for 15 seconds with a two-minute interval using an ultrasonicator (Misonix S-4000, Qsonica, LLC). It was then diluted in PBS-tyloxapol to bring the final volume of the starting inoculum to 15 mL. Antifoam Y-30 (A5758, Sigma-Aldrich Co.) at a concentration of 0.002% was added to the inoculum prior to the aerosol exposure. Bacterial titers were performed to determine the viable concentration of the inoculum before and after aerosolization, and to determine the BioSampler concentration.
Animals
The animals were handled in the Regional Biocontainment Laboratory at Duke according to an approved protocol by the Institutional Animal Care and Use Committee. 6-8 week old C57BL/6 mice from Jackson Labs (Catalog No. 000664) were used for the aerosol exposures. One week prior to the aerosol challenge animals were acclimatized in the Biosafety Level 3 animal holding area. The area is set to a 12-hour on/off light cycle and is environmentally controlled at 21°C room temperature and 50% relative humidity.
Aerosol exposures
A whole-body Madison exposure chamber connected to a Class III biological safety cabinet was used for aerosol challenges (18). Aerosol was generated using a 6-jet Collision nebulizer (CN25, BGI Inc.). The nebulizer was operated at 19±1 liters per minute (Lpm) and 35±1 pounds per square inch (1). The aerosol exposure was controlled by a turnkey system, Aerosol Management platform (AeroMP, Biaera Tech., LLC) (8). The AeroMP system controls, monitors, and records the aerobiology parameters of nebulizer, dilution, and sampler air flow rates. The total airflow through the Madison chamber was set to 50 Lpm by the AeroMP. The animals were loaded into the chamber with the air flow running through the system. Time duration of aerosol exposures was 20 minutes, initiated and terminated by the AeroMP. A BioSampler (SKC Inc.) with a flow rate of 12.5 Lpm was used to sample the aerosol through a port on the door of the Madison chamber.
Madison chamber has been designed to trim large particles on to a baffle and allow small particles to enter the mixing section of the chamber. The aerosol sampling from the mixing chamber is anisokinetic. The chamber has been validated to disperse particles of consistent size and concentration. We have previously demonstrated reproducible pulmonary delivery among mice using this chamber (18).
Determination of presented dose and retained dose
The presented dose represents the number of viable bacterial CFU inhaled by the mouse during the exposure. Presented dose was calculated using exposure volume and viable aerosol concentration in the exposure chamber. Guyton’s formula was used to estimate the minute ventilatory volume: VM= 2.1 × (BW) 0.75, where BW is the body weight of mouse (8, 17). VE is exposure volume VE = VM × t, where t is exposure time. Dose presented: DP= VE × Ca, where Ca is the viable aerosol concentration. Ca was calculated using the BioSampler. Ca= (CSAMPLE × (VSAMPLER-(EC × t)))/(QSAMPLER × t), where CSAMPLE is concentration in BioSampler, VSAMPLER is sampler initial volume, EC is evaporation rate, t is exposure time duration, and QSAMPLER is sampler airflow rate.
The aerosol concentration was varied by changing the inoculum concentrations. The retained dose was determined by lung necropsy in four mice sacrificed 24 hours after exposure. Lungs removed from the mice were placed in a WhirlPak bag containing 2 mL of PBS and homogenized by manually rolling a pipette on the bag. Defined volumes of the neat homogenate and serial dilutions were plated on 7H10 agar plates (#221174, BD Diagnostic Systems). The plates were incubated for 4 weeks at 37° C. The limit of detection (LOD) for each group was determined based on the volume and dilutions plated. The volume and dilutions were modified for different organs and time points based on predicted organ CFUs. The upper limit for colony counting was 400 colonies per plate. When the most dilute sample from an organ generated over 400 colonies per plate, the upper limit of quantitation (LOQ) was used in place of the exact CFU.
Analysis
The primary and secondary endpoints were the proportion of mice infected and the organ CFU counts, respectively. Log-transformed organ CFUs were compared by t-test. The correlation between the log-transformed presented dose and the log-transformed organ CFU was assessed by linear regression.
The ID50 was estimated by plotting the proportion of mice infected as a function of the presented dose, then fitting curves for a Poisson distribution and sigmoid dose-response. The Poisson curve was represented by P = 1 − eˆ(-Dp/DI), where P is the proportion of animals infected, Dp is the presented dose, and the constant DI represents the presented dose corresponding to an infection event (infectious quantum). This formula was derived from the general Poisson’s formula, P(κ; λ) = (λˆκ × eˆ-λ)/κ!, expressing the probability of exactly κ occurrences, where λ is the expected number of occurrences. This formula was solved for κ=0, representing the probability of no infection, eˆ-λ. This was subtracted from 1 to yield the probability of infection, 1 - eˆ-λ. Lastly, the expected number of occurrences, λ, was replaced by the ratio Dp/DI. The constant DI was then varied to produce the best fit to the data, based on least squares, weighted by group size.
The sigmoid curve was represented by P = K/ (1 + eˆ(a + (b × log Dp))), where P is the proportion of animals infected, and Dp is the presented dose. The constant K represents the maximal response, while constants a and b together generate the slope and intercept. GraphPad Prism version 5.01 was used to determine the best-fit curve and the ID50. Lastly, the ID50 was calculated using the traditional method of Reed and Muench (14).
RESULTS
In the first set of experiments, four groups of mice were exposed to varying concentrations of MTB aerosol, and then euthanized at multiple time points to determine the proportion of infected mice and the organ CFU (Table 1, Fig. 1). Groups A through D received presented doses ranging from 1 to 395 CFU/ mouse, corresponding to a retained dose of <2 to 53. Presented dose for groups A, B, C, and D were based on the mouse weights of 20.6, 16.5, 18.7, and 18.7 grams. We refer to group D as an example of LD aerosol exposure and to all other groups as ULD exposures.
Table 1.
Results from four aerosol exposures with varied doses of MTB*
| Aerosol Exposure Group | Starting inoculum OD × 10-4 | Pre-nebulizer (CFU/mL) × 104 | Post-nebulizer (CFU/mL) × 104 | Dose (CFU/mouse) | Infected/Total Mice | Combined Infected/Total Mice (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| Presented | Retained ± SD (24 h) | 4 week | 10 week | 18 week | |||||
| A | 3.3 | 6 | 0.6 | 1.0 | <0.5+ | 1/4 | 1/4 | 1/3 | 03/11 (27) |
| B | 33 | 68 | 4.2 | 5.4 | 7.5 ±3.42 | 4/4 | ND | 3/3 | 7/7 (100) |
| C | 330 | 280 | 98 | 106 | 17.5 ±6.61 | 4/4 | 4/4 | 3/3 | 11/11 (100) |
| D | 3300 | 8000 | 251 | 395 | 53.0 ±7.02 | 4/4 | 4/4 | 3/3 | 11/11 (100) |
Groups of female C57BL/6 mice were exposed to various concentrations of MTB aerosol in the whole-body Madison chamber for 20 minutes. Inoculae were prepared at defined optical densities by dilution of a frozen stock. Bacterial titers were determined from the inoculum before nebulization (pre-nebulizer) and from the material remaining in the Collison nebulizer after nebulization (post-nebulizer). A BioSampler was used to determine the viable aerosol concentration in the chamber, and to estimate the presented dose of MTB to the mice. The dose retained in the lungs was measured by culturing lung homogenates from four mice 24 hours after exposure. The proportion of mice infected was based on cultures of lung, liver and spleen at 4 weeks, 10 weeks, and 18 weeks after exposure. CFU data from these exposures are displayed in Figure 1. Abbreviations: OD, optical density; CFU, colony-forming units, ND, not done.
No bacteria were detected lungs from any of four mice assayed. The group limit of detection was 0.5 CFU.
Figure 1. MTB CFU at varied time points after infection.
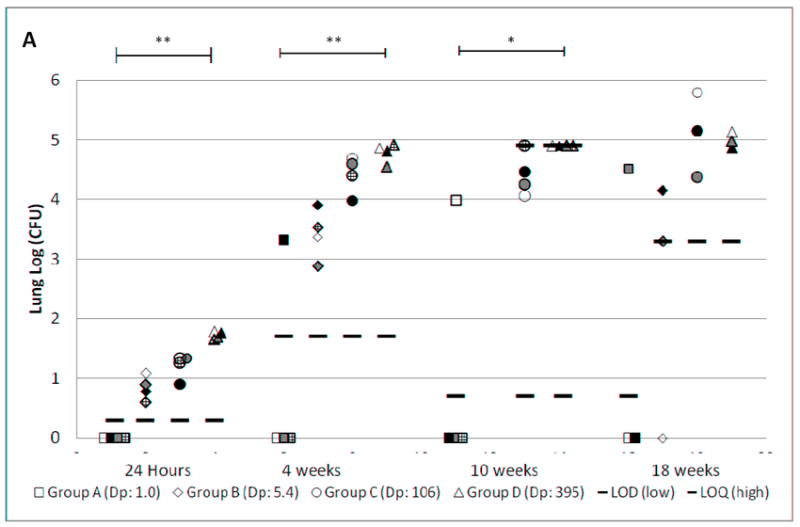
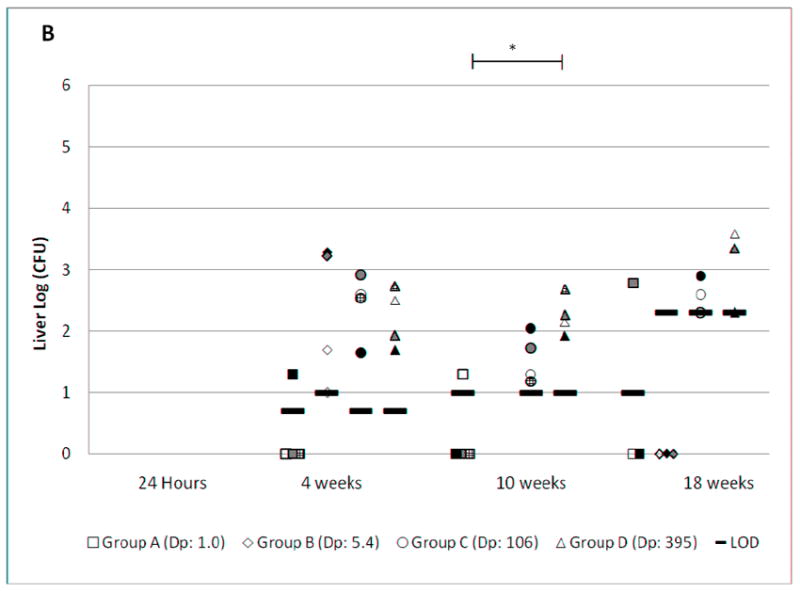
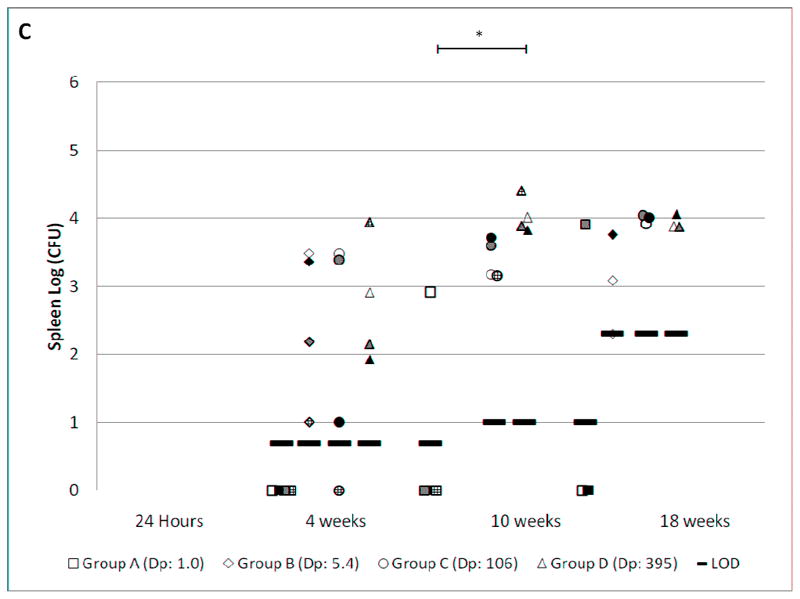
Female C57BL/6 mice were exposed to various concentrations of MTB aerosol (Table 1), resulting in the presented doses (Dp) indicated in the legend. CFU from lung (Fig. 1A), liver (Fig. 1B) and spleen (Fig. 1C) were determined by necropsy at the time points shown. The limit of detection (LOD) is shown for each group at each time point. The upper limit of quantitation (LOQ) is also shown when applicable. Groups A, B, C, and D are represented by square, diamond, circle, and triangle shapes, respectively. Individual mice within each exposure group have the same pattern in the lung, liver, and spleen charts. When no bacteria were grown, the results are plotted on the X-axis. Mean log CFU±SD for infected mice in each exposure group are shown in the box. The brackets show statistically significant linear correlations between the log-transformed Dp and log-transformed organ CFU across the four exposure groups (**, p<0.001. *, p<0.05).
All mice in groups B, C, and D were infected as shown by the presence of MTB in lung, liver, and spleen at 4, 10, and 18 weeks post-exposure. At the lowest dose (group A), infection was detected in only one mouse at each time point. As expected, there was a positive dose-response relationship between the presented dose and the lung CFU at each time point. Based on linear regression between log-transformed presented dose and log-transformed lung CFU, the p-values for the correlation were <0.001, <0.001, 0.021, and 0.078 at 24 hours, 4 weeks, 10 weeks, and 18 weeks, respectively.
The lung burden of mice in all four groups increased rapidly during the first 4 weeks, then slowed between weeks 4 and 18. This pattern has been observed in previous reports using the LD aerosol infection model. However, the ULD exposure led to a persistently lower lung CFU than the LD exposure. The lung CFU was lower in group B compared to D at 24 hours, 4 weeks, and 18 weeks by 0.8, 1.4, and 1.4 logs respectively (p = <0.001, <0.001, and 0.01). Dissemination to liver and spleen was observed at 4, 10, and 18 weeks. There was greater variation in CFU at these sites than in the lungs. The dose-response relationship was statistically significant in both liver and spleen at 10 weeks (p <0.05), but not at 4 and 18 weeks.
In a second set of experiments, larger groups of mice were exposed to ULD aerosols to confirm the reproducibility of the establishment of a partial infection, and to estimate the ID50. Groups E, F, and G received presented doses of 1.1, 1.6, and 11 CFU/ mouse, resulting in infection of 36%, 36%, and 95% of mice, respectively (Table 2). We did not measure the retained dose in these groups, as it was predicted to be too low for accurate quantitation. Presented doses for groups E, F, and G were based on a theoretical mouse weight of 18.9 grams. Presented dose correlated well with the proportion of infected mice (Tables 1 and 2).
Table 2.
Results from three aerosol exposures with ultra-low doses of MTB*
| Aerosol Exposure Group | Starting inoculum OD × 10-4 | Pre-nebulizer (CFU/mL) × 104 | Post-nebulizer (CFU/mL) × 104 | Presented Dose (CFU/ mouse) | Time of necropsy (weeks) | Infected/Total Mice (%) |
|---|---|---|---|---|---|---|
| E | 2.5 | 3.05 | 0.10 | 1.1 | 5 | 10/28 (36%) |
| F | 0.8 | 0.35 | 0.30 | 1.6 | 3 | 5/14 (36%) |
| G | 10 | 33 | 10.6 | 11.0 | 4 | 19/20 (95%) |
Figure 2 shows the lung CFU in these larger groups of mice exposed to ULD aerosol. The SD in lung CFU in groups E, F, and G was 0.51, 1.85, and 0.39 logs respectively. This is higher than the SD typically observed in the standard LD aerosol model of around 0.2 logs at the 4-week time point. As an example of LD aerosol exposure, the SD in group D was 0.17 logs. This increased SD in the ULD as compared to the LD exposures may be explained by the greater stochastic variation in the actual dose delivered to each mouse.
Figure 2. MTB CFU in lungs after ULD exposure.
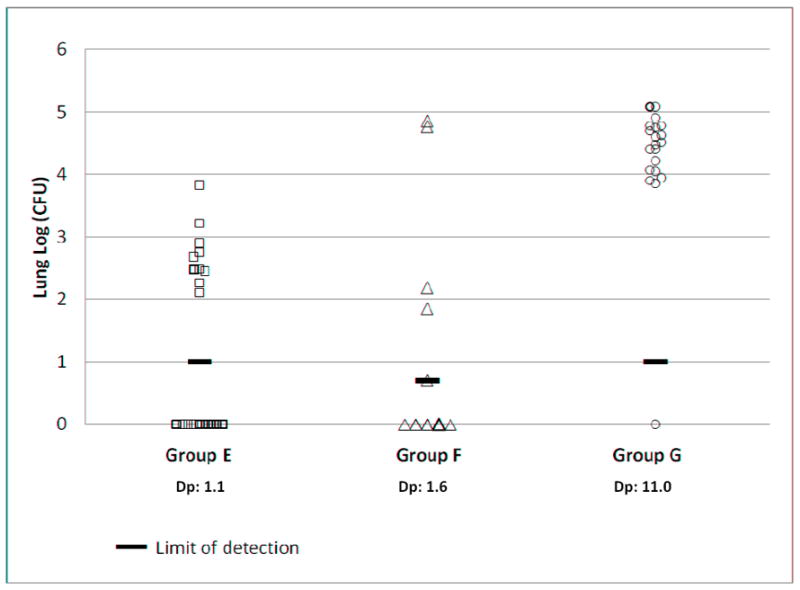
Aerosol exposure groups are summarized in Table 2. CFU was determined by lung necropsy of groups E, F, and G at 5, 3, and 4 weeks, respectively post aerosol exposure. The limit of detection (LOD) is shown for each group. When no bacteria were grown, the results are plotted on the X-axis. Dp: presented dose (CFU/mouse).
Necropsies in Groups E and G were carried out at 5 weeks and 4 weeks after infection, respectively. There was a clear separation between infected and uninfected mice in these groups with the lowest lung CFU at least one log over the limit of detection (Figure 2). Necropsy in Group F was carried out earlier, at 3 weeks after infection. The separation between infected and uninfected mice is less clear in this group as several infected mice had a lung CFU near the limit of detection. The aerosol concentration in the exposure chamber for the ULD groups A, E, F, and G, was 2.67 × 10-3, 3.13 × 10-3, 4.32 × 10-3, and 2.90 × 10-2 CFU/mL, respectively. The aerosol concentration for groups B, C, D that led to 100% infection was 1.56 × 10-2, 2.76 × 10-1, and 1.03 × 100 CFU/mL, respectively.
Figure 3 shows the proportion of infected mice as a function of the log presented dose for all seven exposure groups. Best fit curves are shown based on a Poisson distribution and sigmoid dose-response relationship. The Poisson curve corresponds to the stochastic (all-or-none) model of infection, while the sigmoid curve corresponds to a biological dose-response. The Poisson curve provided a good fit (weighted R2 = 0.973) with an ID50 estimate of 1.9 CFU presented dose, and an estimate of 2.8 CFU as the infectious quantum, the presented dose corresponding to an infection event. The sigmoid curve also provided a good fit to the data (weighted R2 = 0.970) with an ID50 estimate of 1.6 CFU presented dose (95% confidence interval, 1.2 to 2.1), and an estimated maximal response of 100% infected. The traditional method of Reed and Muench provided an ID50 estimate of 1.4 CFU presented dose.
Figure 3. Proportion of mice infected based on presented dose.
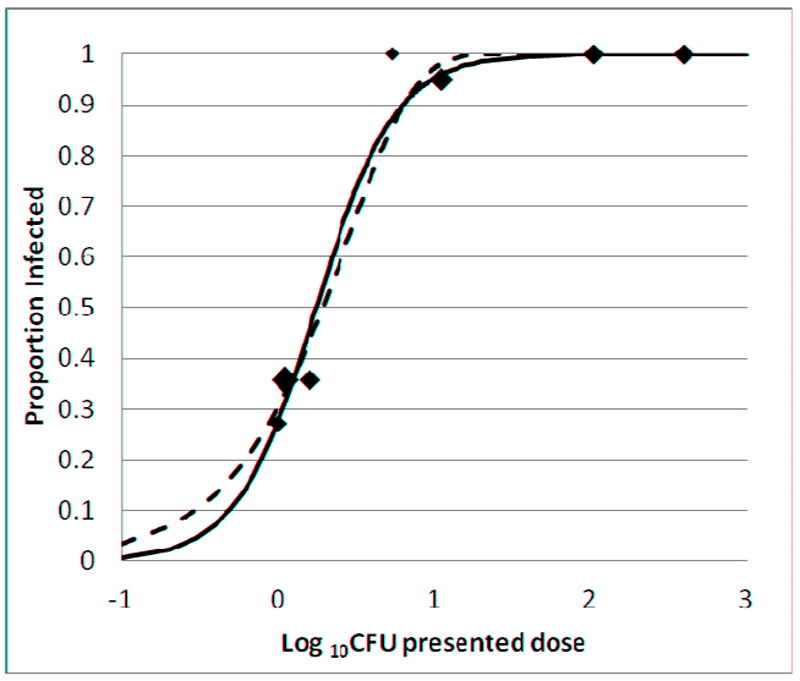
Aerosol exposure groups A-G are described in Tables 1 and 2. The area of each data point is proportional to the total number of mice in the group. Curves were plotted based on a sigmoid dose-response (solid line) and a Poisson distribution (dashed line) as described in Methods. The best-fit curves were determined by weighted least squares.
DISCUSSION
We delivered ULD MTB aerosol to mice, leading to infection in a portion of mice in four independent experiments. The murine infection curve was consistent with both the Poisson and sigmoid distribution. The stochastic (Poisson) assumption is supported in this study by low infectious dose, the steep slope of the dose response curve, and the maximal response of 100%. Using the stochastic assumption, the ID50 of 1.9 CFU correlates with an infectious quantum of 2.8 CFU presented dose. Since only a portion of the presented dose is delivered to the lungs (18), these data are consistent with an infectious quantum of a single CFU. Mycobacteria aggregate in growth media, so one MTB CFU in our aerosol may correspond to multiple bacteria. We previously reported a mass mean aerodynamic diameter in our MTB aerosols of 1.98 microns, larger than the predicted aerodynamic diameter of one micron for a single bacterium (18). Aggregation in the starting inocula may have contributed to the lack of linear correlation between the inoculum OD and CFU titer in Tables 1 and 2.
The data gathered so far are compatible with either a stochastic or dose-response model. As shown in Fig. 3, it is possible to distinguish stochastic and dose-response curves at very low doses. The stochastic model predicts a near-linear relationship between event rate and dose at very low doses, while the dose-response model predicts a threshold below which the event rate is infinitesimal. For example, at a presented MTB aerosol dose of 0.2 CFU, the stochastic model predicts an infection rate of 7.4% compared to the dose-response prediction of 2.1%. At a presented dose of 0.1 CFU, the predictions are 3.8% and 0.6% respectively. A murine exposure study could be carried out to distinguish between these two predictions, though a very large number of mice would be required.
Infection by a single bacterium was hypothesized by Dannenberg and the results of our study support his hypothesis (5). Dannenberg had postulated that upon entry of a single bacterium, if it successfully evades the host defense mechanism, infects the macrophages and multiplies to cause infection (5). Our results also validate the concept that infection is an all-or-none event.
Several limitations of the ULD model should be noted. The primary outcome in the ULD model is the proportion of mice infected. Larger group sizes are typically necessary for pairwise comparisons using a dichotomous variable such as the proportion infected, as compared to a continuous variable such as organ CFU. Though CFUs are measured in the ULD model, we observed greater within-group variation (Figure 2) as compared to typical LD results. Lastly, it is difficult to accurately measure the retained dose in the ULD model, since the number of bacteria delivered is so low. This limitation could be overcome by plating complete lung homogenates from a large number of mice. We have instead used the presented dose as the standard dose measurement for these studies. If the ULD model is used to assess the efficacy of a vaccine or therapeutic against the MTB infection event, then it will be essential to expose control and experimental groups simultaneously to identical aerosols. The capacity of the Madison chamber (72 mice) makes this feasible. Experiments requiring larger numbers of mice may be accomplished with multiple exposures if the experimental groups are stratified across exposures.
Previous studies in other vertebrate species have demonstrated a low infectious dose for mycobacteria. Aerosol delivery of 3-5 CFU of MTB caused infection in guinea pigs as demonstrated by granuloma formation, cytokine production, and bacterial burden at 3 to 4 weeks post infection (10, 11). A low dose of 5 CFU of M. marinum in adult zebra fish delivered by intraperitoneal injection caused mortality (21). Intra-bronchial delivery of 10 MTB CFU in Rhesus monkeys caused asymptomatic tuberculosis in all the challenged monkeys (7).
We are aware of two studies using low enough doses of mycobacteria to cause partial infection in experimental animals. The first was the classic report by Riley and colleagues demonstrating that MTB can be transmitted by the aerosol route (16). Guinea pigs were placed in the exhaust duct of a tuberculosis patient ward over a period of two years. MTB infection was demonstrated in 71 out of 373 guinea pigs. The viable MTB aerosol concentration was not measured in this study. More recently, Dean and colleagues delivered low doses of M. bovis to calves by the intra-tracheal route (6). Infection was identified by sequential measurement of immune responses and by post-mortem examination. Intra-tracheal delivery of 1, 10, 100, and 1000 CFU led to infection in 50%, 83%, 75%, and 75% of calves, respectively. The authors estimated that one CFU in their intra-tracheal inoculum represented eight bacteria (6). A sigmoid dose response curve fits these results well (R2 = 0.92), yielding an ID50 estimate of 1 CFU, and an estimated maximal response of 79% infected. Since even high doses did not lead to 100% infection, these data does not fit a Poisson curve (R2 = -0.02), and are not consistent with an all-or-none infection event.
We chose inbred mice for this study to minimize within-group variation among the animals. This relatively clean system generated a reasonably precise estimate of the ID50, and generated data that was consistent with the stochastic all-or-none model. Many variables present in human populations are not captured in this model, and are likely to influence the infection event. These factors may include varied genetic backgrounds, prior mycobacterial exposure, immune suppression, and lung damage related to non-infectious causes. These factors may influence the ID50, the slope of the dose-response curve, and the peak response. Variations on this murine model may allow the effect of some of these factors on the infection rate to be assessed in a controlled laboratory environment.
The ULD model has some features that may mimic human tuberculosis infection more closely than the standard LD model. A typical human exposure to infectious aerosol leads to infection in only a small proportion of the exposed population. Human pulmonary tuberculosis typically begins at a single pulmonary site, compatible with a single infectious event. ULD model mimics the natural event, route, and dose of exposure.
The ULD model can be used to identify host or bacterial factors that modulate the initial infection event. These factors may be different from those that influence progression from the initial infection to disease. The ULD may be used to assess the effects of vaccination on the infection event by comparing infection rates in control and vaccinated groups exposed to the same aerosol.
Acknowledgments
The authors thank T. Scott Alderman for his assistance in providing biosafety training. This work was supported by National Institute of Health grants U54 AI057157 (Southeast Regional Center of Excellence for Emerging Infections and Biodefense), P30 AI051445 (Duke Center for Translational Research), UC6 AI058607 (Regional Biocontainment Laboratory at Duke), and by National Science Foundation Graduate Research Fellowship 2011083676 (C. C. P.).”
ABBREVIATIONS
- AeroMP
Aerosol Management Platform (Biaera Tech., LLC)
- DI
presented dose corresponding to an infection event
- DP
presented dose
- ID50
median infectious dose
- PBS
phosphate-buffered saline
- LD
low-dose
- Lpm
liters per minute
- MTB
Mycobacterium tuberculosis
- SD
Standard deviation
- ULD
Ultra-low dose
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.The Baker Company. Baker Manual. Operating and Maintenance Manual for IsoGARD Isolator S/N 89357. Stanford; 2006. p. 53. [Google Scholar]
- 2.Beamer Gl, Turner J. Murine models of susceptibility to tuberculosis. Arch Immunol Ther Exp. 2005;53:469–483. [PubMed] [Google Scholar]
- 3.Cardona PJ, Gordillo S, Diaz J, Tapia G, Amat I, Pallares A, Vilaplana C, Ariza A, Ausina V. Widespread bronchogenic dissemination makes DBA/2 mice more susceptible than C57BL/6 mice to experimental aerosol infection with Mycobacterium tuberculosis. Infect Immun. 2004;72:1065–1071. doi: 10.1128/IAI.71.10.5845-5854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun. 2002;70:4501–4509. doi: 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dannenberg AM. Pathogenesis of pulmonary Mycobacterium bovis infection: basic principles established by the rabbit model. Tuberculosis. 2001;81:87–96. doi: 10.1054/tube.2000.0260. [DOI] [PubMed] [Google Scholar]
- 6.Dean GS, Rhodes SG, Coad M, Whelan AO, Cockle PJ, Clifford DJ, Hewinson RG, Vordermeier HM. Minimum Infective Dose of Mycobacterium bovis in Cattle. Infect Immun. 2005;73:6467–6471. doi: 10.1128/IAI.73.10.6467-6471.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gormus BJ, Blanchard JL, Alvarez XH, Didier PJ. Evidence for a rhesus monkey model of asymptomatic tuberculosis. J Med Primatol. 2004;33:134–145. doi: 10.1111/j.1600-0684.2004.00062.x. [DOI] [PubMed] [Google Scholar]
- 8.Hartings JM, Roy CJ. The automated bioaerosol exposure system: Preclinical platform development and a respiratory dosimetry application with nonhuman primates. J Pharmacol Toxicol Meth. 2004;49:39–55. doi: 10.1016/j.vascn.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Hovav AH, Mullerad J, Maly A, Davidovitch L, Fishman Y, Bercovier H. Aggravated infection in mice co-administered with Mycobacterium tuberculosis and the 27-kDa lipoprotein. Microbes Infect. 2006;8:1750–1757. doi: 10.1016/j.micinf.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 10.Ly L, Russell M, McMurray D. Cytokine profiles in primary and secondary pulmonary granulomas of Guinea pigs. Am J Respir Cell Mol Biol. 2008;38:455–462. doi: 10.1165/rcmb.2007-0326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMurray D. Hematogenous reseeding of the lung in low-dose, aerosol-infected guinea pigs. Tuberculosis (Edinb) 2003;83:131–134. doi: 10.1016/s1472-9792(02)00079-3. [DOI] [PubMed] [Google Scholar]
- 12.Nardell EA, Piessens WF. Infectious Disease Aerobiology: Aerosol Challange Methods. In: Raviglione MC, editor. Transmission of Tuberculosis Tuberculosis: A Comprehensive, International Approach. New York: Marcel Dekker; 2000. pp. 215–240. [Google Scholar]
- 13.Orme I, Gonzalez-Juarrero M. Animal models of M. tuberculosis Infection. In: Coico R, Kowalik T, Quarles J, Stevenson B, Taylor R, editors. Curr Protoc Microbiol. Vol. 7. Wiley Online Library; 2007. pp. 10A.15.11–10A.15.29. [DOI] [PubMed] [Google Scholar]
- 14.Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Am J Epidemiol. 1938;27:493–497. [Google Scholar]
- 15.Rhoades ER, Frank AA, Orme IM. Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tuber Lung Dis. 1997;78:57–66. doi: 10.1016/s0962-8479(97)90016-2. [DOI] [PubMed] [Google Scholar]
- 16.Riley RL, Mills CC, Nyka W, Weinstock N, Storey PB, Sultan LU, Riley MC, Wells WF. Aerial dissemination of pulmonary tuberculosis. A two-year study of contagion in a tuberculosis ward. Am J Epidemiol. 1959;70:185–196. doi: 10.1093/oxfordjournals.aje.a117542. [DOI] [PubMed] [Google Scholar]
- 17.Roy JC, Pitt ML. Infectious Disease Aerobiology: Aerosol Challenge Methods. In: Swearengen JR, editor. Biodefense: Research Methodology and Animal Models. Boca Raton: CRC Press; 2005. pp. 61–76. [Google Scholar]
- 18.Saini D, Hopkins GW, Chen CJ, Seay SA, Click EM, Lee S, Hartings JM, Frothingham R. Sampling port for real-time analysis of bioaerosol in whole body exposure system. J Pharmacol Toxicol Meth. 2011;63:143–149. doi: 10.1016/j.vascn.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stead WW, Senner JW, Reddick WT, Lofgren JP. Racial differences in susceptibility to infection by Mycobacterium tuberculosis. N Engl J Med. 1990;15:422–427. doi: 10.1056/NEJM199002153220702. [DOI] [PubMed] [Google Scholar]
- 20.Steenwinkel JD, Knegt GJD, Kate MTT, Belkum AV, Verbrugh HA, Hernandez-Pando R, Soolingen DV, Bakker-Woudenberg IAJM. Immunological parameters to define infection progression and therapy response in a well-defined tuberculosis model in mice. Int J Immunopathol Pharmacol. 2009;22:723–724. doi: 10.1177/039463200902200318. [DOI] [PubMed] [Google Scholar]
- 21.Swaim LE, Connolly LE, Volkman HE, Humbert O, Born DE, Ramakrishnan L. Mycobacterium marinum infection of adult zebrafish causes caseating granulomatous tuberculosis and is moderated by adaptive immunity. Infect Immun. 2006;74:6108–6117. doi: 10.1128/IAI.00887-06. [DOI] [PMC free article] [PubMed] [Google Scholar]