

Abstract
Pax6 is an essential transcription factor for lens, lacrimal gland and pancreas development. Previous transgenic analyses have identified several Pax6 regulatory elements, but their functional significance and binding factors remain largely unknown. In this study, we generated two genomic truncations to delete three elements that were previously shown to bind to the Meis/Prep family homeoproteins. One 3.1 kb deletion (Pax6 ΔDP/ΔDP) removed two putative pancreatic enhancers and a previously identified ectodermal enhancer, while a 450 bp sub-deletion (Pax6ΔPE/ΔPE) eliminated only the promoter-proximal pancreatic enhancer. Immunohistochemistry and quantitative RT-PCR showed that the Pax6ΔPE/ΔPE pancreata had a significant decrease in Pax6, glucagon, and insulin expression, while no further reductions were observed in the Pax6ΔDP/ΔDP mice, indicating that only the 450 bp region is required for pancreatic development. In contrast, Pax6ΔDP/ΔDP, but not Pax6ΔPE/ΔPE mice, developed stunted lacrimal gland and lens hypoplasia which was significantly more severe than that reported when only the ectodermal enhancer was deleted. This result suggested that the ectodermal enhancer must cooperate with its neighboring sequences to regulate the Pax6 ectodermal expression. Finally, we generated conditional knockouts of Prep1 in lens and pancreas, but surprisingly, did not observe any developmental defects. Together, these results provide functional evidence for the independent and synergistic roles of the Pax6 upstream enhancers, and they suggest the potential redundancy of Meis/Prep protein in Pax6 regulation.
Keywords: Pax6, Prep1, pknox1, Meis, lens, pancreas, lacrimal gland
INTRODUCTION
Pax6 is an evolutionarily conserved transcription factor critical for embryogenesis. By E9.5 during murine eye development, Pax6 is expressed in both the optic cup, which will form the neural retina, and a portion of the head surface ectoderm, which will eventually invaginate to form the lens. Pax6-null mice cannot form these eye structures, nor the proper nasal structures to sustain breathing, and die at birth (Grindley et al., 1995). Furthermore, humans heterozygous for PAX6 develop blindness, aniridia (iris hypoplasia), Peter’s anomaly (lens-cornea attachment), colobomas, glaucoma and cataracts, while reduced expression of Pax6 in mice lead to microphthalmia and numerous defects in the anterior chamber of the eye (Glaser et al., 1994; Hill et al., 1991; Hogan et al., 1986; Kokotas and Petersen, 2010). These evidences demonstrate that eye development requires a precise level of Pax6 expressed in an exact spatiotemporal manner.
Pax6 is also expressed as early as E8.5 in the early murine pancreatic bud, but later becomes restricted to the four islet endocrine cell types (α, β, δ and ε) that make up the Islets of Langerhans which secrete insulin and glucagon. The promoter regions of the genes encoding glucagon, insulin and somatostatin all contain Pax6 binding sites, and Pax6 has been shown to actively regulate these genes in cell culture (Andersen et al., 1999; Ritz-Laser et al., 1999; Sander et al., 1997). Consistent with this, the Pax6-null mutants lack the glucagon-producing alpha cells, and the remaining endocrine cells form disorganized islets intermixed with exocrine cells (St-Onge et al., 1997). In addition, conditional inactivation of Pax6 in islet cells causes mutants to die several days after birth as a result of an overt diabetic phenotype, indicating a crucial role for Pax6 in β cell function (Ashery-Padan et al., 2004). In humans, glucose intolerance and diabetes have also been observed in patients with PAX6 heterozygous mutations, further underscoring the exquisite sensitivity of human physiology to PAX6 dosage (Yasuda et al., 2002).
The intricate spatiotemporal expression pattern of Pax6 is controlled by a complex array of regulatory enhancer regions. A well characterized cis-regulatory region located at 3.9 kb upstream to the murine Pax6 P0 promoter, termed the ectodermal enhancer (EE), has been shown to direct lens-specific expression in transgenic mouse (Kammandel et al., 1999; Williams et al., 1998; Zhang et al., 2002). The functional significance of this 341 bp enhancer element was further demonstrated by target deletion in mouse genome, which led to diminished Pax6 expression and lens hypoplasia (Dimanlig et al., 2001). Nevertheless, significant Pax6 expression persisted in this mutant to allow lens formation, indicating the existence of additional lens specific enhancer(s). We have also previously identified at 1.9 kb upstream of the Pax6 P0 promoter a 450 bp sequence that is conserved from human, mouse to fugu fish (Zhang et al., 2003; Zhang et al., 2006). This more promoter proximal enhancer, hereafter referred as PE, can drive islet-specific expression as shown by transient transgenic analysis. However, Kammandel and colleagues have instead localized the Pax6 pancreatic enhancer in a 1.1 kb distal element (hereafter referred as DE) at 4.6 kb upstream from the Pax6 P0 promoter (Kammandel et al., 1999). In further confusion, the Pax6 pancreatic expression was reported to be replicated by a 160 kb murine BAC-GFP transgene carrying the two putative Pax6 pancreatic enhancers (PE and DE), but not by a 420 kb YAC-GFP reporter that carries the homologous human sequences (Kim and Lauderdale, 2006; Kleinjan et al., 2006). Therefore, extensive transgenic studies have thus far failed to resolve the identity of the mammalian Pax6 pancreatic enhancer(s).
Little is known about the upstream transcription factors that directly control the Pax6 expression. Combining biochemical and transgenic analysis, we have first showed that the closely related Meis1 and Meis2 can bind to the Pax6 EE sequence to regulate lens expression (Zhang et al., 2002). Extending this finding further, we next observed that the Pax6 PE sequence contains a composite site for Meis and Pbx homeoprotein binding, which is necessary for the Pax6 pancreatic enhancer activity in mouse (Zhang et al., 2006). Interestingly, biochemical studies showed that this site binds relatively weakly to Meis1 and Meis2, but more strongly to the more distantly related Prep1 (pknox1) and Prep2 (pknox2), which belongs to the same TALE class homeodomain transcription factors. This has been confirmed in studies of the zebrafish pax6b gene, where the corresponding PE and DE sequences were also found to bind the Prep/Pbx complex in vitro (Delporte et al., 2008). More recently, Rowan et al showed that the systemic inactivation of Prep1 disrupts Pax6 expression and lens formation (Rowan et al., 2010). Taken together, these studies suggest that Prep1 binds to multiple upstream enhancers to regulate Pax6 expression in both lens and pancreatic development.
In our present study, we first tested this model of conserved mechanism of Pax6 regulation by Meis/Prep by generating a series of Pax6 enhancer knockout mice. We showed that deletion of the 450 bp Pax6 PE sequence significantly reduced pancreatic Pax6 expression and the number of endocrine cells. In contrast, a larger 3.1 kb deletion encompassing the Pax6 PE, DE and EE elements did not further diminish Pax6 expression in pancreas, but led to more severe lens defects than previously observed by deleting the EE enhancer alone. This suggests that the Pax6 PE sequence plays a critical role in promoting the Pax6 pancreatic expression, while the Pax6 lens expression is control by both the EE element and its surrounding sequences. Finally, we generated a conditional knockout of Prep1 in the lens and the pancreas, but the resulting mutant did not exhibit any Pax6 expression changes or developmental defects. These results thus reveal the complexity and redundancy of Pax6 transcriptional regulation.
MATERIALS AND METHODS
Generation of the Pax6 enhancer knockout mice
The Pax6 enhancer targeting vector was generated using the recombineering method (Fig. 1A) (Liu et al., 2003). In brief, a 12.3 kb Pax6 genomic sequence containing the 1.1 kb distal enhancer element (DE), the ectodermal enhancer element (EE) and the 450 bp proximal enhancer element (PE) was first cloned from a 129S6/SvEvTac BAC clone (BACPAC Resources Center at Children’s Hospital Oakland Research Institute, catalogue number RP22-55A14) by gap repair into pPL253, a MC1TK-containing plasmid. Through homologous recombination, a loxP site and a Sph I site was inserted next to the Spe I site upstream to DE sequence, whereas the 450 bp PE sequence was replaced by an frt-flanked Neo selection cassette. The resulting Pax6 enhancer targeting vector was verified by direct sequencing and linearized to transfect ES cells (129S6/SvEvTac) by electroporation. After drug selection, the positive ES cell clones were further screened by Southern blot using both 5′ (Sph I) and 3′ (Spe I) external probes and injected into C57BL/6 mouse blastocysts to generate Pax6Neo mice. Tail biopsies were then collected and genotype-PCR performed to confirm the correct targeting (primers for Neo insert: Pax6Neo F: 5′-GAAGGGACTGGCTGC TATTG-3′ and Pax6Neo R: AATATCACGGGTAGCCAACG-3′; primers for the wild type allele: Pax6+ F: 5′-CGCCGAATTCAGTGTGGCCTAGAGACGCTG-3′ and Pax6+ R: CCAGCTTATCTATCTGTCTGTCAATAAAGGGC-3′). The Pax6Neo/+ mice were next crossed to the FLP-recombinase mice (stock number 009086, Jackson Laboratory, Bar Harbor, ME) to remove the frt-flanked Neo cassette in the germline. The resulting Pax6ΔPE/+ mice were then confirmed by genotype-PCR (primers: Pax6ΔPE F: 5′-CAAAAGCTTGGAAAGGACGCTCCAGCA TCCCAG-3′ and Pax6ΔPE R: 5′-ATAAGCGGC CGCGAATCCTGAGAGTTTGGGTA GTG-3′). The Pax6ΔDP mice were generated by crossing the Pax6ΔPE/+ mice with the EIIa-Cre mice (stock number #003724, Jackson Laboratory, Bar Harbor, ME) to further remove both the DE and EE enhancer elements in the germline, and confirmed by both Southern blots and genotype-PCR (Pax6ΔDP F: AAAGTGGTGGAC AAGATTGC) and (Pax6ΔDP R: 5′-TTAGGGACAGAG CCCTCAGA-3′).
Figure 1. Generation of Pax6ΔPE/ΔPE and Pax6ΔDP/ΔDP enhancer knockouts.

(A) A 450 bp and a 3.1 kb Pax6 genomic sequence were deleted by gene targeting in Pax6ΔPE and Pax6ΔDP, respectively. Frt and LoxP sites are represented by open arrows and solid triangles. PE, the Pax6 promoter-proximal enhancer; EE, the ectodermal enhancer; DE, the promoter-distal enhancer; Sh, Sph I; Sp, Spe I. (B) Southern Blots using the 5′ Sph I or the 3′ Spe I probes confirmed the Pax6Neo allele in the ES cell lysates and the Pax6ΔPE and Pax6ΔDP alleles in mouse tail extracts. (C) Genotyping confirmation of the Pax6ΔPE and Pax6ΔDP mice.
The Pax6Neo, the Pax6ΔPE and the Pax6ΔDP mice were back crossed six generations into C57BL/6 background. The presence of a vaginal plug was considered 0.5 days post coitum, or E0.5. All experimental procedures involving mice were humanely performed in accordance with the Laboratory Animal Research Center at Indiana University (LARC).
Generation of the Prep1flox mice
For the construction of the Prep1flox targeting vector, a 9.2 kb sequence containing the Prep1 genomic region was retrieved from a BAC clone (BACPAC Resources Center at Children’s Hospital Oakland Research Institute, catalogue number RPCI23-2k17) by gap repair into pPL253. This plasmid was further modified with recombineering method to insert two loxP sites and an frt-flanked Neo selection cassette next to the Prep1 exon 8 sequences. Two new restriction sites, EcoR V and Kpn I, were also added to allow the Southern blots identification of correctly targeted ES cell clones, which were used to generate the Prep1Neo mice. The Neo cassette was subsequently removed by mating the Prep1Neo mice with the FLP mice, which left only the two loxP sites flanking the Prep1 exon 9 sequences in the Prep1flox mice. To validate the exon 8 can be cleaved via Cre-mediated recombination, the Prep1flox mice were also crossed with the EIIa-Cre strain to generate the Prep1Δ mice, which were used in Western blot and genotype-PCR confirmation (primers: Prep1f F: 5′-ACAGGAGAAGCAGGCAAAGA -3′ and Prep1f R: 5′-CTGTCCATCACTCCCTGTCC -3′; Prep1Δ F: 5′-AGCTGCTTCAGGGCTGTCT-3′). The Prep1flox mice were back crossed six generations into C57BL/6 background before mated with the Le-Cre (kindly provided by Dr. Ruth Ashery-Padan, Tel Aviv University, Tel Aviv, Israel and Dr. Richard Lang, Children’s Hospital Research Foundation, Cincinnati, OH) and Ap2α-Cre (kindly provided by Dr. Ann Moon, University of Utah, Salt Lake City, UT) to ablate Prep1 in the lens and the pancreas.
Quantitative real time RT-PCR
E15.5 pancreata were quickly collected from litters of Pax6ΔPE/+ x Pax6ΔPE/+ and Pax6ΔDP+ x Pax6ΔDP/+ matings in ice cold DEPC-treated PBS, before placed in 100μl of ice cold RNAlater™ (SIGMA) and briefly chopped into fine pieces while kept on ice. Immediately following dissection of all embryos, pancreas RNA was isolated using the RNeasy® Mini Kit (QIAGEN). Quantitative PCR was carried out in two steps. First, pancreatic cDNA was synthesized using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems), followed by quantitative RT PCR utilizing the TaqMan® Gene Expression assay (Applied Biosystems) in the StepOnePlus™ Real-Time PCR instrument (Applied Biosystems). Primers and probe used for amplification of Pax6 cDNA are: Pax6 F: 5′-CTACCAGCCAATCCCACAGC-3′, Pax6 R: 5′-TTCGGCCCAACATGGAAC-3′ and probe 5′-(6-FAM)CACCACACCTGTCTCCTCCTTCACATCA-3′ (Zhang et al., 2002). The β-actin gene served as the internal control and its primers included: Actb F: 5′-GGC TCCTAGCACCATGAA-3′, Actb R: 5′-ACCGATCCACACAGAGTACT and probe 5′-(6-FAM)TCAAGATCATTGCTCCTCCTGAGCGC. Pax6 gene expression in the Pax6ΔPE/ΔPE and Pax6ΔDP/ΔDP pancreata normalized to the internal control Actb, relative to Pax6 expression in the wild type pancreas was analyzed with StepOne™Software (Applied Biosystems) using the 2−ΔΔCT method. Experiments were repeated three times, each in triplicate.
Immunohistochemistry
Fluorescence immunohistochemical analysis of cryo- and paraffin sections were performed as previously described (Carbe and Zhang, 2011; Qu et al., 2011a). For immunohistochemistry of pancreatic hormones, E15 embryos were paraffin embedded and sectioned transversely to collect all adjacent pancreas sections. Following deparaffination, antigen unmasking and blocking, pancreatic paraffin sections were incubated with both rabbit polyclonal anti-glucagon (1:500) (BioGenex, Sam Ramon, CA) and guinea pig anti-human insulin (1:1000) (Linco, St Charles, MO). Immunofluorescent images of sections were captured and processed with a SPOT RT KE color camera and accompanying SPOT software (Diagnostic Instruments) on a Leica DM500 compound microscope. Embryonic eye cryo-sections underwent antigen retrieval and were processed for fluorescence immunohistochemistry using mouse monoclonal anti-Pax6 (1:10) (the Developmental Studies Hybridoma Bank, Iowa City, Iowa), rabbit polyclonal anti-Pax6 (1:250) and anti-Prox1(1:500), (both from Covance, Berkeley, California); and anti-α, and β crystallins (1:1000), both kindly provided by Sam Zigler (National Eye Institute, Bethesda, MD). Anti-secondary antibodies for all experiments were either Alexa Fluor-488 (1:250) or Alexa Fluor-555 (1:500) conjugated anti-mouse and/or anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA). At least three embryos were tested for each genotype.
Histology
Histology of embryonic eye sections was performed as previously reported (Pan et al., 2010). Briefly, paraffin sections were melted, and deparaffinized in 3 changes of xylene followed by rehydration in absolute ethanol, 95% ethanol and finally 70% ethanol prior to water wash and staining in Harris hematoxylin (Fisher Scientific). Sections were then washed in running tap water followed by counterstaining in Eosin Y (Fisher Scientific), followed by ethanol dehydration and xylene clearing. Following slide mounting in a xylene based medium (Fisher Scientific) and drying, eye and lacrimal gland sections were photographed and processed with a SPOT RT KE color camera, and accompanying SPOT software (Diagnostic Instruments) on a Leica DM500 compound microscope. The lens size was quantified by the ImageJ program (National Institute of Health) and the statistical significance was calculated using Student’s t test.
RNA in situ hybridization
RNA in situ hybridization for whole-mount embryos was performed as previously described (Pan et al., 2006). The Pax6 antisense probe was generated from Pax6 cDNA. Embryos were photographed with a Leica DFC400 camera mounted on a Leica M165FC dissecting microscope. At least three embryos were tested for each genotype.
Western blot
E14 embryos were snap frozen in 1.5 mL test tubes in liquid nitrogen. Ice cold RIPA was then added to the test tube and embryos were quickly chopped with ice cold scissors before homogenized for one minute. Samples were placed on ice for 10 minutes and then centrifuged at 13,200 rpm for 20 minutes at 4°C. Protein concentrations were determined using a BCA Protein Assay Kit (Pierce). Protein bands were separated using an SDS-polyacrylamide gel and transferred to a PVDF membrane (Millipore). After blocking (Licor), the membrane was submerged in the Prep1 antibody (Santa Cruz #6245, 1:200 in 5%BSA) overnight at 4°C and a secondary fluorescent antibody for two hours at room temperature. Protein bands were then scanned using a Licor Odyssey.
Data Analysis
Immunofluorescent hormone positive area was quantified as a proportion of pancreatic epithelial area. In our study every 10th section of E15.5 pancreas was immunostained as described above in Materials and Methods with anti-insulin and anti-glucagon followed by counting the total number of insulin+ and glucagon+ cells per immunostained section. Next the area of each hormone-analyzed pancreatic epithelial section was measured using ImageJ software (National Institutes of Health, Bethesda, Maryland). The proportion of glucagon+ cells and insulin+ cells per epithelial area was expressed as the number of hormone-positive cells / pancreas section area. Differences among groups were determined using the unpaired t-test where P-values less than 0.05 were considered significant. At least three samples were used for each genotype.
RESULTS
The 450 bp proximal enhancer (PE) is required for the Pax6 pancreatic expression
To determine the functional significance of the three known enhancers upstream to the Pax6 P0 promoter, we constructed two strains of knockout mice. In Pax6ΔPE, the 450 bp promoter-proximal enhancer (PE) was deleted from the mouse genome by homologous recombination (Fig. 1A). This is followed by a Cre-mediate recombination to remove a 3.1 kb sequence in Pax6ΔDP mice, which further deleted both the promoter-distal enhancer (DE) and the ectodermal enhancer (EE). By Southern blots, we verified the correct targeting of Pax6 locus in ES cells, and demonstrated the progressive deletion of the three enhancer elements in the Pax6ΔPE and Pax6ΔDP mice (Fig. 1B and C). Monitored by genotyping PCR (Fig. 1D), we backcrossed these mice to the C57BL/6 strain for six generations to ensure a uniform genetic background.
Quantitative real time RT-PCR in the E15.5 Pax6ΔPE/ΔPE pancreas, null for the 450 bp PE element, show a 36.5% decrease in Pax6 expression compared to wild type (P = 0.0016) (Fig. 2A). This is similar to the 40% decrease in Pax6 expression in the E15.5 Pax6ΔDP/ΔDP pancreas, which is null for both the PE and DE elements (P < 0.0001) (Fig. 2A). Thus, deletion of the DE element did not further disrupt the Pax6 pancreatic expression. Since Pax6 is a key regulator of pancreatic islet development, we next examined the number of α and β cells by fluorescent immunohistochemistry. For the glucagon-producing α cells, the Pax6ΔPE/ΔPE mutants showed a 39.4% (P = 0.0009) reduction compared to the wild type, while the Pax6ΔDP/ΔDP mutants showed a 47.8% (P = 0.0022) loss (Fig. 2B and C). Similarly, the insulin-producing β cells were reduced by 44.1% in the Pax6ΔPE/ΔPE mutants and 30.4% in the Pax6ΔDP/ΔDP mutants. The differences of α and β cell numbers between mutants, however, were not significant. Taken together, these results demonstrate that the PE enhancer is necessary for the full Pax6 expression in pancreas. On the other hand, deletion of the putative distal enhancer DE failed to enhance the PE-deletion phenotype, suggesting that the DE enhancer does not play a significant role in regulating the Pax6 pancreatic transcription.
Figure 2. Pax6ΔPE mutation disrupted Pax6 and hormonal expressions in the pancreas.

(A) qPCR analysis showed that Pax6 expression was down regulated to the similar extent in the Pax6ΔPE/ΔPE and Pax6ΔDP/ΔDP pancreata. (B–C) The percentages of insulin and glucagon producing cells were both reduced in the Pax6ΔPE/ΔPE and Pax6ΔDP/ΔDP pancreata. There was no statistical significant difference between Pax6ΔPE/ΔPE and Pax6ΔDP/ΔDP mutants.
Combined deletion of PE, EE and DE down regulated Pax6 expression in the lens and the lacrimal gland
We next examined eye development in the Pax6ΔPE/ΔPE mutants. At E14.5, the Pax6ΔPE/ΔPE embryos appeared grossly normal, without any reduction in both lens and lacrimal gland sizes (Fig. 3A–F). Consistent with this, Pax6 immunostaining was also unchanged in the lens, retina and surface ectoderm (Fig. 3G and H). Prox1 is homeobox transcription factor required for lens fiber elongation and α crystallin is a lens terminal differentiation marker. Similar levels of Prox1 and α crystallin expression were also observed in the Pax6ΔPE/ΔPE and wild type lens (Fig. 3I–L). Finally, the postnatal day 21 (P21) Pax6ΔPE/ΔPE animals have similarly sized eyeballs as the wild type controls and their lenses did not exhibit any cataracts (Fig. 3M–P). Therefore, deletion of the PE element alone did not affect eye development.
Figure 3. Lack of ocular phenotype in Pax6ΔPE/ΔPE mutant.

(A–F) At E14.5, Pax6ΔPE/ΔPE mutant displayed normal eye morphology, lens histology and lacrimal gland budding. le, lens; lg, lacrimal gland. (G–L) Pax6, Prox1 and α crystallin expression was unaffected in the Pax6ΔPE/ΔPE mutant lens. (M–P) The adult Pax6ΔPE/ΔPE mutant at P21 has normal eye size and clear lens.
In contrast to the normal appearance of the Pax6ΔPE/ΔPE mutant eyes, the Pax6ΔDP/ΔDP mutants exhibit significant ocular defects. At E14.5, the Pax6ΔDP/ΔDP mutant eye was already conspicuously smaller than the wild type controls and the retinal pigmented epithelium of the mutant optic cup presented with an optic coloboma (Fig. 4A–B, arrows). Histological analysis of this mutant further revealed a hypoplastic lens, with the posterior lens fiber failing to rise up to the anterior lens epithelium. Importantly, the Pax6ΔDP/ΔDP mutant lens remained attached to the overlying surface ectoderm, which resembled the Peter’s anomaly phenotype in human (Fig. 4C–D, arrows). Pax6 expression in the conjunctival epithelium is also known to be required for normal lacrimal gland budding (Makarenkova et al., 2000). As expected, the Pax6ΔPE/ΔPE lacrimal gland was considerably shorter in length than that of the wild type (Fig. 4E–F, arrows). It has also been previously reported that the deletion of the EE element alone disrupted Pax6 expression in early lens development, but it led to only moderate reduction in lens size at E17.5 (Dimanlig et al., 2001). In contrast, the Pax6ΔDP/ΔDP lens was not only significantly smaller than the wild type at E14.5, but also completely degenerated by P6, leaving behind a disorganized neural retina (Fig. 4G–I). Therefore, the Pax6ΔDP/ΔDP mutant exhibited much more severe ocular defects than the deletion of EE element alone.
Figure 4. Defective lens and lacrimal gland development in Pax6ΔDP/ΔDP mutants.
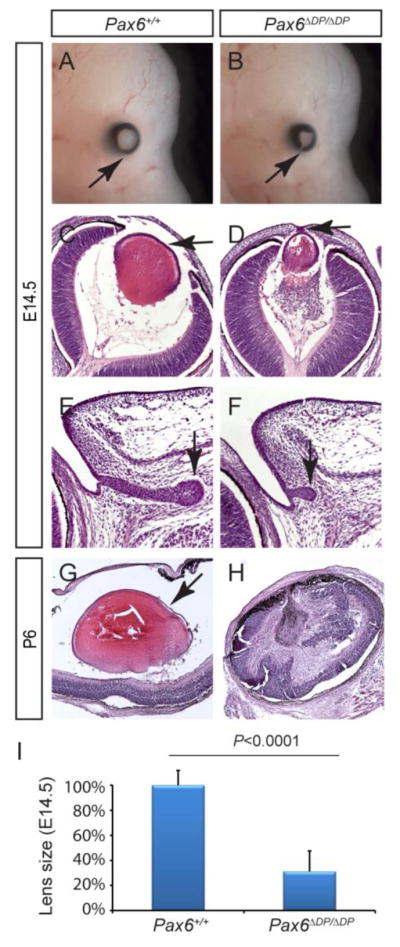
(AB) The E14.5 Pax6ΔDP/ΔDP mutant exhibited coloboma, due to a failure of optic cup closure (arrows). (C–D) Compared to the wild type, the Pax6ΔDP/ΔDP mutant lens was much reduced in size and remained attached to the surface ectoderm (arrows). (E–F) The lacrimal gland budding was stunted in Pax6ΔDP/ΔDP mutant. (G–H) By P6, the lens completely degenerated in Pax6ΔDP/ΔDP mutant. (I) Quantification of lens size at E14.5. [Student’s t test: P<0.0001 for the Pax6ΔDP/ΔDP mutants (n=4) compared to the wild type (n=6).]
Expression analysis further confirmed that Pax6 was indeed down regulated in the Pax6ΔDP/ΔDP mutants. At E10.5 when lens development was initiated, whole mount RNA in situ hybridization with a Pax6 antisense RNA probe revealed a striking reduction in Pax6 expression within the Pax6ΔDP/ΔDP lens vesicle compared to the wild type (Fig. 4A–D, dotted circles). In contrast, optic vesicle Pax6 expression remained unchanged (Fig. 4A–D, arrows). Similarly, Pax6 protein expression as shown by immunostaining was clearly diminished in the Pax6ΔDP/ΔDP mutant lens at E14.5 (Fig. 5E and F, arrows). This was in contrast with the previously reported deletion of the EE element, which affected Pax6 expression only at E9.5 but not at E13.5 and later stages (Dimanlig et al., 2001). Consistent with the persistent corneal-lenticular adhesion, Prox1 was also down regulated in the central lens epithelium (Fig. 5G and H, arrows). The rest of the lens, however, still exhibited normal expression pattern of Prox1, α and β crystallins (Fig. 5G–L). This agrees with a recent report that conditional knockout of Pax6 at this stage does not affect Prox1 and crystallins expressions (Shaham et al., 2009). Taken together, these results suggest that the Pax6ΔDP/ΔDP mutant has deleted additional enhancer(s) that cooperate with the EE element in regulating Pax6 lens expression.
Figure 5. Pax6ΔDP mutation disrupted Pax6 lens expression.

(A–D) Whole mount RNA in situ hybridization showed that Pax6 expression was specifically reduced in the E10.5 Pax6ΔDP/ΔDP lens vesicle (dotted circles). ov, optic vesicle; lv, lens vesicle. (E–F) Pax6 protein expression was also reduced in the E14.5 Pax6ΔDP/ΔDP mutant lens (arrows). (G–L) Prox1 expressed was down regulated in the anterior lens epithelium which was attached to the surface ectoderm (arrows), whereas α and β crystallins were still expressed.
Conditional knockout of Prep1 did not affect Pax6 lens and pancreatic expression
Previous studies have showed that, at least in vitro, the Meis/Prep family homeodomain proteins can bind to all three DE, EE and PE elements (Delporte et al., 2008; Rowan et al., 2010; Zhang et al., 2002; Zhang et al., 2006). This suggests that the cooperative binding of these elements to Meis/Prep proteins may underlie the synergistic interactions among these elements. In agreement with this, the crossing of a Prep1 hypomorphic allele and a null allele was recently shown to disrupt lens induction but not pancreatic development, supporting a dose-dependent role of Prep1 in lens Pax6 regulation (Rowan et al., 2010). To further test this model, we sought to generate a conditional knockout of Prep1 to investigate whether Pax6 is regulated cell autonomously by Prep1 in lens development.
To generate the Prep1flox allele, we employed the standard gene targeting techniques to insert two loxP sites and an frt-flanked Neo selection cassette into the Prep1 locus, which was confirmed by Southern blots (Fig. 6A and B). The Neo gene was subsequently removed via FLP-mediated recombination in germ line, leaving behind only two loxP sites and an frt site next to the exon 8 of the Prep1 gene. The resulting Prep1flox allele was further crossed with a ubiquitous Cre-expressing strain (EIIa-Cre) to generate the Prep1Δ allele, which was confirmed to have lost the Prep1 exon 8 by genotyping PCR (Fig. 6C). Consistent with this, western blots using a Prep1 antibody revealed a 59 kD truncated protein in addition to the 64 kD wild type protein in the Prep1Δ/+ lysates (Fig. 6D). The Prep1 exon 8 encodes the N-terminal portion of the homeodomain, which is critical for Prep1 DNA binding activity. As expected, no homozygous E12.5 Prep1Δ/Δ mutants were ever uncovered in the Prep1Δ/+ heterozygous intercrosses (Fig. 6E), which agrees with the previous report that the Prep1 null embryos died in utero soon after implantation (Fernandez-Diaz et al., 2010).
Figure 6. Generation of Prep1flox allele.

(A) Through homologous recombination, two loxP sites were inserted next to the Prep1 exon 8 to create the Prep1flox allele. This allows subsequent deletion of exon 8 via Cre-mediate recombination to generate the Prep1Δ allele. The shaded boxes in Exon 8–10 code for the Prep1 homoedomain. K, Kpn I; Ev, EcoRV. (B) Confirmation of the gene targeting in ES cells by Southern blots using the 5′ and 3′ probes. (C) Genotyping confirmation of the Prep1flox and Prep1Δ mice. (D) Western blots showed the truncated protein in the Prep1Δ/+ mutant mice. (E) No E12.5 Prep1Δ/ Δ mutants were recovered in heterozygous Prep1Δ/+ mating.
We first ablated Prep1 during early lens development using the Ap2α-Cre driver, which was generated by inserting a Cre cDNA sequence into the endogenous Ap2α locus (Macatee et al., 2003). Previous studies have shown that Ap2α-Cre begins to express Cre recombinase in the presumptive lens ectoderm at E8.0, preceding the onset of Pax6 expression at E8.5 (Grindley et al., 1995; Song et al., 2007). Interestingly, the E10.5 Ap2α-Cre;Prep1flox/flox embryos exhibited normal lens vesicle invagination, and their Pax6 expression was also indistinguishable from that of the wild type (Fig. 7A, B). To further confirm this finding, we next employed a widely used lens and pancreatic Cre driver, Le-Cre transgene, which is controlled by a 6.5 kb Pax6 promoter fragment that contains the DE, EE and PE elements (Ashery-Padan et al., 2000). Active as early as E9.5, this Cre deletor line has been previously used to generate a conditional knockout of Pax6 in lens and pancreas progenitor cells, resulting in a complete abrogation of lens formation and early postnatal lethality due to defective β cell function (Ashery-Padan et al., 2000; Ashery-Padan et al., 2004). Therefore, the Le-Cre transgene is an effective Cre drive to test the maintenance of Pax6 expression, which is required for lens and pancreatic maturation. Le-Cre also contains a bicistronic GFP reporter, which can be used to monitor the enhancer activity of the 6.5 kb Pax6 genomic fragment that includes the DE, EE and PE elements (Pan et al., 2008). At E14.5, identical GFP expression was again observed in the Le-Cre and Le-Cre;Prep1flox/flox cornea, lens and lacrimal gland buds (Fig. 7C and D). Consistent with this, the Le-Cre;Prep1flox/flox did not exhibit any changes in Pax6 and α crystallin immunostaining at E14.5 or histological abnormality at E16.5 (Fig. 7E–J). Furthermore, the adult Le-Cre;Prep1flox/flox mice were also healthy and fertile, without any overt endocrine phenotype. Indeed, quantitative real time RT-PCR confirmed that Pax6 was expressed at the same level in both the wild type and Le-Cre;Prep1flox/flox E15.5 pancreata (Fig. 7K). Therefore, Prep1 ablation failed to disrupt Pax6 regulation in pancreatic and lens development.
Figure 7. Prep1 conditional knockout failed to disrupt lens and pancreas development.

(A–B) Ap2α-Cre mediated Prep1 knockout did not affect lens induction and Pax6 expression. lv, lens vesicle (C–D) Eye and lacrimal gland morphology as represented by the GFP expression from the Le-Cre transgene was normal in the Le-Cre;Prep1flox mutant. co, cornea; le, lens; lg, lacrimal gland. (E–J) The Prep1 conditional knockout did not affect Pax6 and α crystallin expression and lens histology (K) Pax6 expression measured by qPCR was unchanged in the E15.5 Le-Cre;Prep1flox mutant pancreas.
DISCUSSION
To date, the precise location of the Pax6 pancreatic enhancer remains controversial. In their thorough molecular dissection of the regulatory elements within the murine Pax6 locus, Kammandel et al. observed pancreatic expression from transgenic reporter constructs that contained a 5 kb Pax6 genomic fragment, but not a 3 kb sub-fragment (Kammandel et al., 1999). This finding prompted them to attribute the Pax6 pancreatic enhancer to a 124 bp highly conserved sequence (referred to as DE in this study) in the truncated region, located at 3.3 kb upstream of the Pax6 P0 promoter. The in vivo activity of this conserved sequence, however, has never been tested in transgenic analysis by itself. Instead, we have provided evidence that a 450 bp nucleotide conserved sequence (referred to as PE in this study) located approximately 1.9 kb upstream from the Pax6 P0 promoter can independently drive reporter expression in the developing murine pancreas (Zhang et al., 2003; Zhang et al., 2006). More recently, it was observed that the zebrafish pax6b gene, which harbors both the DE and PE elements, is expressed in pancreas (Delporte et al., 2008; Kleinjan et al., 2008). In contrast, the other Pax6 paralog in zebrafish, pax6a, which does not have the DE element, has concomitantly lost its pancreatic expression. This apparent correlation between the zebrafish pax6 DE sequence and pancreatic activity was further supported by promoter exchange experiments, which showed that the PE-containing pax6a promoter can be activated in pancreas when fused with the pax6b DE element (Kleinjan et al., 2008). Nevertheless, this result was recently challenged by further transgenic analyses performed by Delporte et al, who showed that the pax6b PE element, but not DE element, can drive a heterologous promoter in pancreas, although the overall expression level is much lower than that achieved by combining the PE and DE sequences (Delporte et al., 2008). It was therefore proposed that the Pax6 PE element is responsible for the pancreatic specific expression, while the Pax6 DE element is necessary for maintaining a high level of expression.
It is unclear what causes the above discrepancies in locating the Pax6 pancreatic enhancer. While the differences in the species and experimental designs could certainly play a role, it is also clear that these studies were all susceptible to random transgenic insertion events, which was known to negatively affect the consistency of any transient transgenic analysis. In fact, although transgenic analysis is a powerful in vivo tool for expression studies, it nevertheless takes an enhancer out of its larger genomic context. This is especially problematic for investigating the mammalian Pax6 regulation, because additional regulatory elements, such as locus control regions, have been identified hundreds kb away from the Pax6 promoter region (Kleinjan et al., 2001; Lauderdale et al., 2000). Therefore, we have employed the genomic deletion approach in this study to examine the functional significance of the previously reported murine pancreatic enhancer regions. We showed that the removal of the 450 bp PE element alone significantly reduced, but did not abolish, pancreatic Pax6 expression and hormonal production, suggesting that there exists additional pancreatic enhancer(s) in the Pax6 locus. Nevertheless, a larger genomic deletion that encompassed both the DE and PE elements failed to cause additional Pax6 reduction or pancreatic defects. These results underlie the importance of the genomic deletion approach as a functional test of transcriptional regulatory elements, and they support our contention that the 237 bp PE element, but not the upstream DE element, is necessary for the full Pax6 expression in murine pancreatic cells.
Our enhancer-knockout experiments further revealed the complexity of the Pax6 ocular regulation. Whereas targeted elimination of the 450 bp PE element in our Pax6ΔPE/ΔPE mutants appeared to have no deleterious effect on eye development, combined deletion of DE, EE and PE sequences in the Pax6ΔDP/ΔDP embryos resulted in optic colobomas and a hypoplastic lens that failed to separate from the surface ectoderm. Clearly these phenotypes are reminiscent of the Pax6ΔEE/ΔEE mutants (lens hypoplasia and failure of lens detachment), which targeted the EE element, and the Pax6sey (small eye) heterozygous nulls (lens defects and optic colobomas) (Dimanlig et al., 2001; Hill et al., 1991). However, the lens defects in the Pax6ΔDP/ΔDP embryos also appeared to be more severe than that of the Pax6ΔEE/ΔEE mutants. For example, although the Pax6ΔEE/ΔEE mutants exhibited distinct defects in early lens development, its Pax6 expression and lens size became largely normal during late gestation (Dimanlig et al., 2001). In contrast, the Pax6ΔDP mutation led to significant reduction in lens Pax6 expression at E14.5, and complete lens degeneration after birth. Based on distinct lens phenotype in the Pax6ΔEE/ΔEE mutants, Dimanlig et al has proposed there are two phases of Pax6 expressions in lens development, one pre-placodal and one placodal. Here, the severe lens defects in our Pax6ΔDP/ΔDP embryos suggest another post-placodal phase of Pax6 expression, which must be separately regulated from the earlier two phases to support the late-stage lens development. Since Pax6 expression was diminished, but not eliminated, in the Pax6ΔDP/ΔDP lens, enhancers outside the 3.1 kb deletion region are still important for these complex phases of Pax6 lens expression. However, it is tempting to speculate that DE, EE and/or PE elements, the only sequences within the 3.1 kb that are evolutionarily conserved throughout vertebrates, may synergistically act to establish the precise control of the Pax6 late-stage lens expression.
Despite their general lack of sequence similarity, the DE, EE and PE elements are all capable of binding to the Meis/Prep family homeodomain proteins in vitro (Delporte et al., 2008; Rowan et al., 2010; Zhang et al., 2002; Zhang et al., 2006). This striking commonality suggests that Meis/Prep proteins may be conserved upstream regulators of Pax6 in both lens and pancreas development. To test this model, we generated conditional knockouts of Prep1 using the Ap2α-Cre and Le-Cre transgenes, targeting the pre-placodal and placodal phases of lens induction, respectively (Ashery-Padan et al., 2000; Macatee et al., 2003; Song et al., 2007). To our surprise, neither Prep1 conditional mutants displayed any lens or pancreatic defects or Pax6 expression changes. Rowan et al recently showed that a compound Prep1 mutant carrying one hypomorphic allele and one null allele also failed to disrupt pancreatic Pax6 expression, but this mutant nevertheless showed significant lens induction defects (Rowan et al., 2010). One possible explanation for the discrepancy is that Prep1 was systemically disrupted throughout the compound Prep1 mutant, whose lens defects may thus be an indirect consequence of multiple tissue defects. Such a phenomenon is not without precedence for a transcription factor, as the lens specific deletion of Ap2α also failed to recapitulate the lens induction defects observed in the germ-line Ap2α knockout (Pontoriero et al., 2008). Furthermore, there are five members of the Meis/Prep family protein expressed in overlapping patterns in lens and pancreas. Since they recognize the same consensus DNA binding site, it is conceivable that the genetic redundancy within the Meis/Prep family may obscure the function of Prep1 in later lens and pancreas development. Finally, even though Cre-mediated conditional knockouts may exhibit a delay in protein loss after the initial gene deletion (Pan et al., 2010; Qu et al., 2011b), the complete lack of lens defect in our two Prep1 mutants demonstrates that Prep1 alone is at least dispensable for the maintenance of Pax6 expression in lens and pancreatic development.
In summary, we have used gene targeting technology to conclusively demonstrate the importance of three upstream enhancers in Pax6 lens and pancreas regulation. To date, the molecular diagnosis of human aniridia patients have primarily depended on the scrutiny of PAX6 exon mutations. Our study of these Pax6 regulatory sequences may help to guide the search for Pax6 functional mutations into the much wider non-coding regions. We also showed that a conditional knockout of Prep1 failed to disrupt Pax6 expression in lens and pancreatic development, raising the question of Prep1 tissue autonomy in lens induction and/or redundancy in Meis/Prep gene functions. Further studies should focus on the combined deletion of multiple Meis/Prep genes to study their roles in lens and pancreas development.
HIGHLIGHTS.
Genomic deletion analysis identifies an essential pancreatic enhancer for Pax6.
The Pax6 ectodermal enhancer cooperates with its adjacent sequences to regulate Pax6 lens expression.
Prep1 is dispensable for the Pax6 lens and pancreas expression.
Acknowledgments
The authors thank Drs. Ruth Ashery-Padan, Richard Lang, Ann Moon and Samuel Zigler for mice and reagents, and Sheldon Rowan for comments. Christian Carbe was supported by a recipient of DeVault Diabetes Fellowship. This work was supported by grants from the American Diabetes Foundation (1-04-RA-117) and the NIH (EY017061 and EY018868).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen FG, Jensen J, Heller RS, Petersen HV, Larsson LI, Madsen OD, Serup P. Pax6 and Pdx1 form a functional complex on the rat somatostatin gene upstream enhancer. FEBS Lett. 1999;445:315–320. doi: 10.1016/s0014-5793(99)00144-1. [DOI] [PubMed] [Google Scholar]
- Ashery-Padan R, Marquardt T, Zhou X, Gruss P. Pax6 activity in the lens primordium is required for lens formation and for correct placement of a single retina in the eye. Genes Dev. 2000;14:2701–2711. doi: 10.1101/gad.184000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashery-Padan R, Zhou X, Marquardt T, Herrera P, Toube L, Berry A, Gruss P. Conditional inactivation of Pax6 in the pancreas causes early onset of diabetes. Dev Biol. 2004;269:479–488. doi: 10.1016/j.ydbio.2004.01.040. [DOI] [PubMed] [Google Scholar]
- Carbe C, Zhang X. Lens induction requires attenuation of ERK signaling by Nf1. Hum Mol Genet. 2011;20:1315–1323. doi: 10.1093/hmg/ddr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delporte FM, Pasque V, Devos N, Manfroid I, Voz ML, Motte P, Biemar F, Martial JA, Peers B. Expression of zebrafish pax6b in pancreas is regulated by two enhancers containing highly conserved cis-elements bound by PDX1, PBX and PREP factors. BMC Dev Biol. 2008;8:53. doi: 10.1186/1471-213X-8-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimanlig PV, Faber SC, Auerbach W, Makarenkova HP, Lang RA. The upstream ectoderm enhancer in Pax6 has an important role in lens induction. Development. 2001;128:4415–4424. doi: 10.1242/dev.128.22.4415. [DOI] [PubMed] [Google Scholar]
- Fernandez-Diaz LC, Laurent A, Girasoli S, Turco M, Longobardi E, Iotti G, Jenkins NA, Fiorenza MT, Copeland NG, Blasi F. The absence of Prep1 causes p53-dependent apoptosis of mouse pluripotent epiblast cells. Development. 2010;137:3393–3403. doi: 10.1242/dev.050567. [DOI] [PubMed] [Google Scholar]
- Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet. 1994;7:463–471. doi: 10.1038/ng0894-463. [DOI] [PubMed] [Google Scholar]
- Grindley JC, Davidson DR, Hill RE. The Role of Pax-6 in Eye and Nasal Development. Development. 1995;121:1433–1442. doi: 10.1242/dev.121.5.1433. [DOI] [PubMed] [Google Scholar]
- Hill RE, Favor J, Hogan BL, Ton CC, Saunders GF, Hanson IM, Prosser J, Jordan T, Hastie ND, van Heyningen V. Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature. 1991;354:522–525. doi: 10.1038/354522a0. [DOI] [PubMed] [Google Scholar]
- Hogan BL, Horsburgh G, Cohen J, Hetherington CM, Fisher G, Lyon MF. Small eyes (Sey): a homozygous lethal mutation on chromosome 2 which affects the differentiation of both lens and nasal placodes in the mouse. J Embryol Exp Morphol. 1986;97:95–110. [PubMed] [Google Scholar]
- Kammandel B, Chowdhury K, Stoykova A, Aparicio S, Brenner S, Gruss P. Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity. Dev Biol. 1999;205:79–97. doi: 10.1006/dbio.1998.9128. [DOI] [PubMed] [Google Scholar]
- Kim J, Lauderdale JD. Analysis of Pax6 expression using a BAC transgene reveals the presence of a paired-less isoform of Pax6 in the eye and olfactory bulb. Dev Biol. 2006;292:486–505. doi: 10.1016/j.ydbio.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Kleinjan DA, Bancewicz RM, Gautier P, Dahm R, Schonthaler HB, Damante G, Seawright A, Hever AM, Yeyati PL, van Heyningen V, Coutinho P. Subfunctionalization of duplicated zebrafish pax6 genes by cis-regulatory divergence. PLoS Genet. 2008;4:e29. doi: 10.1371/journal.pgen.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan DA, Seawright A, Mella S, Carr CB, Tyas DA, Simpson TI, Mason JO, Price DJ, van Heyningen V. Long-range downstream enhancers are essential for Pax6 expression. Dev Biol. 2006;299:563–581. doi: 10.1016/j.ydbio.2006.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan DA, Seawright A, Schedl A, Quinlan RA, Danes S, van Heyningen V. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum Mol Genet. 2001;10:2049–2059. doi: 10.1093/hmg/10.19.2049. [DOI] [PubMed] [Google Scholar]
- Kokotas H, Petersen MB. Clinical and molecular aspects of aniridia. Clin Genet. 2010;77:409–420. doi: 10.1111/j.1399-0004.2010.01372.x. [DOI] [PubMed] [Google Scholar]
- Lauderdale JD, Wilensky JS, Oliver ER, Walton DS, Glaser T. 3′ deletions cause aniridia by preventing PAX6 gene expression. Proc Natl Acad Sci U S A. 2000;97:13755–13759. doi: 10.1073/pnas.240398797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macatee TL, Hammond BP, Arenkiel BR, Francis L, Frank DU, Moon AM. Ablation of specific expression domains reveals discrete functions of ectoderm- and endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development. 2003;130:6361–6374. doi: 10.1242/dev.00850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarenkova HP, Ito M, Govindarajan V, Faber SC, Sun L, McMahon G, Overbeek PA, Lang RA. FGF10 is an inducer and Pax6 a competence factor for lacrimal gland development. Development. 2000;127:2563–2572. doi: 10.1242/dev.127.12.2563. [DOI] [PubMed] [Google Scholar]
- Pan Y, Carbe C, Powers A, Feng GS, Zhang X. Sprouty2-modulated Kras signaling rescues Shp2 deficiency during lens and lacrimal gland development. Development. 2010;137:1085–1093. doi: 10.1242/dev.042820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Carbe C, Powers A, Zhang EE, Esko JD, Grobe K, Feng GS, Zhang X. Bud specific N-sulfation of heparan sulfate regulates Shp2-dependent FGF signaling during lacrimal gland induction. Development. 2008;135:301–310. doi: 10.1242/dev.014829. [DOI] [PubMed] [Google Scholar]
- Pan Y, Woodbury A, Esko JD, Grobe K, Zhang X. Heparan sulfate biosynthetic gene Ndst1 is required for FGF signaling in early lens development. Development. 2006;133:4933–4944. doi: 10.1242/dev.02679. [DOI] [PubMed] [Google Scholar]
- Pontoriero GF, Deschamps P, Ashery-Padan R, Wong R, Yang Y, Zavadil J, Cvekl A, Sullivan S, Williams T, West-Mays JA. Cell autonomous roles for AP-2alpha in lens vesicle separation and maintenance of the lens epithelial cell phenotype. Developmental dynamics : an official publication of the American Association of Anatomists. 2008;237:602–617. doi: 10.1002/dvdy.21445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Carbe C, Tao C, Powers A, Lawrence R, van Kuppevelt TH, Cardoso WV, Grobe K, Esko JD, Zhang X. Lacrimal Gland Development and Fgf10-Fgfr2b Signaling Are Controlled by 2-O- and 6-O-sulfated Heparan Sulfate. J Biol Chem. 2011a;286:14435–14444. doi: 10.1074/jbc.M111.225003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Hertzler K, Pan Y, Grobe K, Robinson ML, Zhang X. Genetic epistasis between heparan sulfate and FGF-Ras signaling controls lens development. Dev Biol. 2011b;355:12–20. doi: 10.1016/j.ydbio.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz-Laser B, Estreicher A, Klages N, Saule S, Philippe J. Pax-6 and Cdx-2/3 interact to activate glucagon gene expression on the G1 control element. J Biol Chem. 1999;274:4124–4132. doi: 10.1074/jbc.274.7.4124. [DOI] [PubMed] [Google Scholar]
- Rowan S, Siggers T, Lachke SA, Yue Y, Bulyk ML, Maas RL. Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes Dev. 2010;24:980–985. doi: 10.1101/gad.1890410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M, Neubuser A, Kalamaras J, Ee HC, Martin GR, German MS. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev. 1997;11:1662–1673. doi: 10.1101/gad.11.13.1662. [DOI] [PubMed] [Google Scholar]
- Shaham O, Smith AN, Robinson ML, Taketo MM, Lang RA, Ashery-Padan R. Pax6 is essential for lens fiber cell differentiation. Development. 2009;136:2567–2578. doi: 10.1242/dev.032888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song N, Schwab KR, Patterson LT, Yamaguchi T, Lin X, Potter SS, Lang RA. pygopus 2 has a crucial, Wnt pathway-independent function in lens induction. Development. 2007;134:1873–1885. doi: 10.1242/dev.001495. [DOI] [PubMed] [Google Scholar]
- St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, Gruss P. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature. 1997;387:406–409. doi: 10.1038/387406a0. [DOI] [PubMed] [Google Scholar]
- Williams SC, Altmann CR, Chow RL, Hemmati-Brivanlou A, Lang RA. A highly conserved lens transcriptional control element from the Pax-6 gene. Mech Dev. 1998;73:225–229. doi: 10.1016/s0925-4773(98)00057-4. [DOI] [PubMed] [Google Scholar]
- Yasuda T, Kajimoto Y, Fujitani Y, Watada H, Yamamoto S, Watarai T, Umayahara Y, Matsuhisa M, Gorogawa S, Kuwayama Y, Tano Y, Yamasaki Y, Hori M. PAX6 mutation as a genetic factor common to aniridia and glucose intolerance. Diabetes. 2002;51:224–230. doi: 10.2337/diabetes.51.1.224. [DOI] [PubMed] [Google Scholar]
- Zhang X, Friedman A, Heaney S, Purcell P, Maas RL. Meis homeoproteins directly regulate Pax6 during vertebrate lens morphogenesis. Genes Dev. 2002;16:2097–2107. doi: 10.1101/gad.1007602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Heaney S, Maas RL. Cre-loxp fate-mapping of Pax6 enhancer active retinal and pancreatic progenitors. Genesis. 2003;35:22–30. doi: 10.1002/gene.10160. [DOI] [PubMed] [Google Scholar]
- Zhang X, Rowan S, Yue Y, Heaney S, Pan Y, Brendolan A, Selleri L, Maas RL. Pax6 is regulated by Meis and Pbx homeoproteins during pancreatic development. Dev Biol. 2006;300:748–757. doi: 10.1016/j.ydbio.2006.06.030. [DOI] [PubMed] [Google Scholar]