

Abstract

α,β-Unsaturated 2-imidazolyl ketones undergo [2+2] cycloaddition with a variety of Michael acceptors upon irradiation with visible light in the presence of Ru(bpy)32+. Cleavage of the imidazolyl auxiliary from the cycloadducts affords cyclobutane carboxamides, esters, thioesters, and acids that would not be accessible from direct cycloaddition of the corresponding unsaturated carbonyl compounds.
Cyclobutanes are synthetically interesting both because of the diverse structures of cyclobutane-containing natural products1 and because of the utility of strain-releasing ring-fragmentations in the preparation of more complex medium-sized ring systems.2 Conventional photochemical methods3 for the synthesis of cyclobutanes are generally efficient only when cyclic enones are utilized; the triplet excited state of acyclic enones undergoes a rapid, energy-wasting olefin isomerization that outcompetes productive intermolecular cyclizations.4 We recently reported that Ru(bpy)32+ complexes are useful photocatalysts for the [2+2] cycloadditions of aryl enones upon irradiation with visible light.5,6 This method avoids the formation of the problematic triplet excited state of the enone and thus works well with acyclic enones. However, we found the scope of this method to be limited; the involvement of an aryl enone in the reaction was found to be a strict requirement for successful cycloaddition.
We proposed a mechanism for the cycloaddition that rationalizes this constraint (Scheme 1). The key reactive intermediate in this process is an enone radical anion generated by single electron transfer from a photogenerated Ru(bpy)3+ complex7 to a Lewis acid activated enone. The one-electron reduction of aryl enones is significantly more facile than the corresponding reduction of less-conjugated enone substrates. Enoate esters, for example, possess reduction potentials ca. 700 mV more negative than aryl enones,8 which precludes formation of the corresponding enoate radical anions under these photocatalytic conditions.
Scheme 1.

Mechanism of radical anion [2+2] cycloaddition.
We wondered if we might circumvent this limitation in scope by installing a cleavable auxiliary group onto the enone substrate that (1) would facilitate one-electron reduction and subsequent cycloaddition of the enone substrate and (2) could be transformed into a carboxylic acid, ester, amide, or similar carbonyl-containing functional group after the cycloaddition. This cleavable group might be considered a “redox auxiliary”9,10 that temporarily modulates the reduction potential of an otherwise redox-inactive enoate substrate, just as a chiral auxiliary temporarily differentiates the prochiral faces of an otherwise achiral substrate.
Table 1 summarizes our studies to identify a suitable redox auxiliary for the [2+2] cycloaddition. We examined the homodimerization of a number of α,β-unsaturated carbonyl compounds that have been validated as surrogates of carboxylate esters in other synthetic methods. Upon exposure to the conditions we had optimized for intermolecular [2+2] cycloaddition of aryl enones, unsaturated acyl phosphonates11 underwent rapid decomposition (entry 1). N-Acyl pyrroles12 and pyrazoles13 reacted sluggishly and gave unsatisfactory yields of the corresponding dimerized cyclobutanes (entries 2 and 3). On the other hand, α,β-unsaturated 2-acylimidazoles14 reacted smoothly and furnished the desired [2+2] cyclodimer in 82% yield.15 We therefore elected to continue our studies using enones bearing an N-methylimidazol-2-yl auxiliary group.
Table 1.
Dimerizations of candidate enonesa
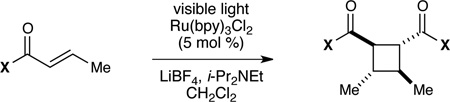 | ||
|---|---|---|
| entry | substrate | yield |
| 1 | 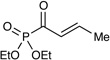 |
0% |
| 2 | 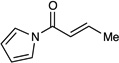 |
16% |
| 3 | 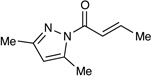 |
<5% |
| 4 | 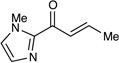 |
82% |
Reactions performed with 5% Ru(bpy)3Cl2, 2.0 equiv LiBF4, 2.0 equiv i-Pr2NEt in 0.1 M MeCN. Molar ratios for intermolecular dimerizations calculated with respect to theoretical yield of product (e.g., 2.5 mol % catalyst with respect to enone).
Next, we studied the crossed intermolecular [2+2] cyclization of acyl imidazole 1 with methyl acrylate (Table 2). The conditions we had previously reported for [2+2] cycloaddition of phenyl enones with methyl acrylate afforded only 43% of the desired crossed cycloadduct in 5:1 dr (entry 1); the undesired homodimerization of 1 was a significant competitive process. Higher concentrations of the Lewis acidic additive (LiBF4) increased the dr without increasing the yield of 3, while lower Lewis acid loadings favored homodimerization (entries 2 and 3). We observed a modest increase in selectivity for the heterodimer when the catalyst loading was lowered to 2.5 mol % (entry 4). The best yield and highest d.r. were obtained when 1 was added slowly via syringe pump to the reaction mixture, which presumably minimizes the homodimerization by minimizing the concentration of 1 with respect to methyl acrylate while keeping the ratio of Lewis acid to substrate high. By using this slow addition protocol, the desired heterodimer 3 could be isolated in 67% yield and with excellent diastereoselectivity (entry 6).
Table 2.
Optimization studies.a
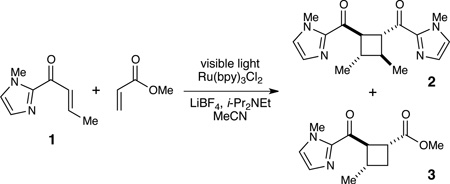 | ||||
|---|---|---|---|---|
| entry | mol % Ru | equiv LiBF4 | % yield 2b | % yield 3 (dr) b |
| 1 | 5.0 | 2 | 24 | 43 (5:1) |
| 2 | 5.0 | 4 | 13 | 42 (10:1) |
| 3c | 5.0 | 0.5 | 50 | 21 (2:1) |
| 4d | 2.5 | 2 | 19 | 51 (5:1) |
| 5e | 2.5 | 2 | <5 | 67 (>10:1) |
Reactions performed with 2.0 equiv i-Pr2NEt in 0.1 M MeCN, and indicated amounts of photocatalyst and LiBF4 with respect to the theoretical yield of product 3 and an irradiation time of 90 min.
Isolated yields with respect to theoretical yield of 2 or 3, respectively.
Irradiated for 120 min.
Irradiated for 150 min.
Aryl enone added dropwise over a 45 min period.
Figure 1 summarizes experiments probing the scope of the crossed intermolecular [2+2] cycloaddition using 2-acyl imidazoles. A variety of Michael acceptors, including α,β-unsaturated esters, thioesters, and ketones, provided good yields and high diastereoselectivites in cycloadditions with 1 (Figure 1, 3–5). As we had observed in our previous studies, high selectivity for the crossed cycloadduct requires the use of a β-unsubstituted Michael acceptor as the reaction partner. However, β-substitution on the acyl imidazole is easily accomodated. Substrates of increased steric demand worked well in this reaction (6–8), and protected heteroatomic functional groups were tolerated under optimized reaction conditions (9–11).
Figure 1.

Scope of the intermolecular coupling reaction.a,b
a Unless otherwise noted, reactions performed with 5.0 equiv Michael acceptor with respect to 1.0 equiv of aryl enone, 2.5 mol % Ru(bpy)3Cl2, 2.0 equiv LiBF4, 2.0 equiv i-Pr2NEt in 0.1 M MeCN; aryl enone added dropwise over a 45 min period. b Isolated yields and diastereomer ratios are the averaged results of two reproducible experiments. c 0.5 equiv LiBF4. d 4.0 equiv LiBF4; aryl enone was added in one portion.
We also explored intramolecular [2+2] cycloadditions of 2-acylimidazoles. In these experiments, we observed somewhat higher yields when the loading of LiBF4 was reduced to 0.5 equiv. These conditions enabled intramolecular cycloadditions with a variety of acceptor moieties, including esters, ketones, and amides (Figure 2, 12–15). The use of an α-substituted Michael acceptor required prolonged reaction times, but the expected cycloadduct bearing a quaternary stereocenter (16) was produced with excellent diastereoselectivity.
Figure 2.

Scope of intramolecular [2+2] reaction.a,b
a Unless otherwise noted, reactions performed using 2.5 mol % Ru(bpy)3Cl2, 0.5 equiv LiBF4, 2.0 equiv i-Pr2NEt in 0.1 M MeCN. b Isolated yields and diastereomer ratios are the averaged results of two reproducible experiments. c Reaction conducted using 0.5 equiv i-Pr2NEt.
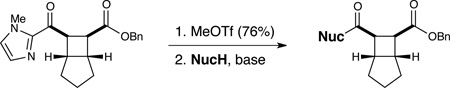
Finally, we investigated conditions for transformation of the 2-acylimidazole moiety into carboxylic acid derivatives14 (Table 3). The auxiliary group of cycloadduct 12 can easily be N-alkylated with MeOTf to afford the corresponding imidazolium salt. Upon recrystallization, this white crystalline material is stable to prolonged storage on the bench for at least six months.16 Displacement of the imidazolyl group proceeds smoothly with a variety of oxygen nucleophiles without loss of stereochemical integrity (entries 1–3). While bulky tertiary alcohols did not react with the imidazolium salt (entry 4), the more nucleophilic t-butyl thiol produced the corresponding thioester in quantitative yield (entry 5). Finally, the 2-acylimidazolium moiety could be transformed into an amide functional group upon treatment with either primary or secondary amines (entries 6 and 7). Thus, the use of this redox auxiliary strategy enables the synthesis of a variety of cyclobutane carboxylic acid derivatives that would not otherwise be accessible using our previously reported photocatalytic [2+2] cycloaddition methodology.
Table 3.
Cleavage of the redox auxiliary.a
 | |||
|---|---|---|---|
| entry | NucH | yieldb | dr |
| 1c | H2O | 52%c | >10:1 |
| 2c | MeOH | 86% c | >10:1 |
| 3 | i-PrOH | 88% | >10:1 |
| 4 | t-BuOH | 0% | n.d. |
| 5 | t-BuSH | 99% | >10:1 |
| 6d | BnNH2 | 98% | >10:1 |
| 7d | pyrrolidine | 75% | >10:1 |
Unless otherwise noted, cleavage of the imidazolium group was conducted using an excess of the nucleophile and 3.5 equiv of DBU in CH2Cl2.
Isolated yields.
Cleavage conducted in Et2O.
No DBU added.
In conclusion, we have circumvented a limitation in the scope of the photocatalytic [2+2] cycloaddition developed in our laboratory by using unsaturated 2-acylimidazole groups as redox auxiliaries. These heteroaryl groups facilitate the reduction of the enone substrate to the key radical anion intermediate required for cycloaddition and are then susceptible to cleavage with a variety of nucleophiles under mild conditions. This redox auxiliary approach could be applied to other reactions that involve the reduction of carbonyl compounds to the corresponding radical anions. Continued studies in our laboratory will apply these concepts to other reactions of photogenerated radical ions.
Supplementary Material
Acknowledgment
Financial support from the NIH (GM095666) and Sloan Foundation is gratefully acknowledged. The NMR facilities at UW-Madison are funded by the NSF (CHE-9208463, CHE-9629688) and NIH (RR08389-01).
Footnotes
Supporting Information Available Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Hansen TV, Stenstrøm Y. Naturally Occurring Cyclobutanes. In: Hudlicky T, editor. Organic Synthesis: Theory and Applications. Vol. 5. Elsevier; Oxford, U.K.: 2001. pp. 1–38. [Google Scholar]; (b) Dembitsky VM. J. Nat. Med. 2008;62:1–33. doi: 10.1007/s11418-007-0166-3. [DOI] [PubMed] [Google Scholar]
- 2.(a) Oppolzer W. Acc. Chem. Res. 1982;15:135–141. [Google Scholar]; (b) Winkler JD, Bowen CM, Liotta F. Chem. Rev. 1995;95:2003–2020. [Google Scholar]; (c) Lee-Ruff E, Mladenova G. Chem. Rev. 2003;103:1449–1483. doi: 10.1021/cr010013a. [DOI] [PubMed] [Google Scholar]; (d) Namyslo JC, Kaufmann DE. Chem. Rev. 2003;103:1485–1537. doi: 10.1021/cr010010y. [DOI] [PubMed] [Google Scholar]
- 3.For reviews of [2+2] enone photocycloadditions, see: de Mayo P. Acc. Chem. Res. 1971;4:41–47. Baldwin SW. Org. Photochem. 1981;5:123–225. Crimmins MT. Chem. Rev. 1988;88:1453–1473. Demuth M, Mikhail G. Synthesis. 1989:145–162. Schuster DI, Lem G, Kaprinidis NA. Chem. Rev. 1993;93:3–22. Iriondo-Alberdi J, Greaney MF. Eur. J. Org. Chem. 2007:4801–4815. Bach T, Hehn JP. Angew. Chem. Int. Ed. 2011;50:1000–1045. doi: 10.1002/anie.201002845.
- 4.Morrison H, Rodriguez O. J. Photochem. 1974;3:471–474. [Google Scholar]
- 5.(a) Ischay MA, Anzovino ME, Du J, Yoon TP. J. Am. Chem. Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]; (b) Du J, Yoon TP. J. Am. Chem. Soc. 2009;131:14604–14605. doi: 10.1021/ja903732v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For reviews on recent developments in transition metal photoredox catalysis in organic synthesis, see: Zeitler K. Angew. Chem., Int. Ed. 2009;48:9785–9789. doi: 10.1002/anie.200904056. Yoon TP, Ischay MA, Du J. Nat. Chem. 2010;2:527–532. doi: 10.1038/nchem.687. Narayanam JMR, Stephenson CRJ. Chem. Soc. Rev. 2011;40:102–113. doi: 10.1039/b913880n. Teply F. Collect. Czech. Chem. Commun. 2011;76:859–917.
- 7.The reduction potential of Ru(bpy)3+, which we presume to be the catalytically relevant photoreductant in this process, is −1.2 V vs SCE. For a review of the photoelectrochemistry of Ru(bpy)32+ see: Kalyanasundaram K. Coord. Chem. Rev. 1982;46:159–244.
- 8.House HO, Huber LE, Umen MJ. J. Am. Chem. Soc. 1972;94:8471–8475. [Google Scholar]
- 9.Facilitation of electrochemical reactions using a non-cleavable redox-active group has been termed a "redox tag" strategy by Chiba. See: Okada Y, Akaba R, Chiba K. Org. Lett. 2009;11:1033–1035. doi: 10.1021/ol802984n. Okada Y, Nishimoto A, Akaba R, Chiba K. J. Org. Chem. 2011;76:3470–3476. doi: 10.1021/jo200490q.
- 10.Similarly, facilitation of electrochemical reactions using a silyl or stannyl electrofugal group has been termed an "electroauxiliary" approach by Yoshida. See: Yoshida J, Takada K, Ishichi Y, Isoe S. J. Chem. Soc. Chem. Commun. 1994:2361–2362. Yoshida J, Nishiwaki K. J. Chem. Soc. Dalton Trans. 1998:2589–2596.
- 11.(a) Evans DA, Johnson JS. J. Am. Chem. Soc. 1998;120:4895–4896. [Google Scholar]; (b) Evans DA, Scheidt KA, Fandrick KR, Lam HW, Wu J. J. Am. Chem. Soc. 2003;125:10780–10781. doi: 10.1021/ja036985c. [DOI] [PubMed] [Google Scholar]; (c) Takenaka N, Abell JP, Yamamoto H. J. Am. Chem. Soc. 2007;129:742–743. doi: 10.1021/ja0668320. [DOI] [PubMed] [Google Scholar]; (d) Samanta S, Zhao C-G. J. Am. Chem. Soc. 2006;128:7442–7443. doi: 10.1021/ja062091r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Jiang H, Paixão MW, Monge D, Jørgensen KA. J. Am. Chem. Soc. 2010;132:2775–2783. doi: 10.1021/ja9097803. [DOI] [PubMed] [Google Scholar]
- 12.(a) Lee SD, Brook MA, Chan TH. Tetrahedron Lett. 1983;24:1569–1572. [Google Scholar]; (b) Kinoshita T, Okada S, Park SR, Matsunaga S, Shibasaki M. Angew. Chem. Int. Ed. 2003;42:4680–4684. doi: 10.1002/anie.200352509. [DOI] [PubMed] [Google Scholar]; (c) Shaghafi MB, Kohn BL, Jarvo ER. Org. Lett. 2008;10:4743–4746. doi: 10.1021/ol801830h. [DOI] [PubMed] [Google Scholar]
- 13.(a) Sibi MP, Shay JJ, Liu M, Jasperse CP. J. Am. Chem. Soc. 1998;120:6615–6616. [Google Scholar]; (b) Itoh K, Kanemasa S. J. Am. Chem. Soc. 2002;124:13394–13395. doi: 10.1021/ja027313+. [DOI] [PubMed] [Google Scholar]; (c) Ishihara K, Fushimi M. Org. Lett. 2006;8:1921–1924. doi: 10.1021/ol060651l. [DOI] [PubMed] [Google Scholar]; (d) Sibi MP, Itoh K. J. Am. Chem. Soc. 2007;129:8064–8065. doi: 10.1021/ja071739c. [DOI] [PubMed] [Google Scholar]
- 14.(a) Davies DH, Haire NA, Hall J, Smith EH. Tetrahedron. 1992;48:7839–7856. [Google Scholar]; (b) Evans DA, Song H-J, Fandrick KR. Org. Lett. 2006;8:3351–3354. doi: 10.1021/ol061223i. [DOI] [PubMed] [Google Scholar]; (c) Andrus MB, Christiansen MA, Hicken EJ, Gainer MJ, Bedke DK, Harper SR, Dodson DS, Harris DT. Org. Lett. 2007;9:4865–4868. doi: 10.1021/ol702197r. [DOI] [PubMed] [Google Scholar]; (d) Evans DA, Fandrick KR, Song HJ, Scheidt KA, Xu R. J. Am. Chem. Soc. 2007;129:10029–10041. doi: 10.1021/ja072976i. [DOI] [PubMed] [Google Scholar]; (e) Trost BM, Lehr K, Michaelis DJ, Xu J, Buckl AK. J. Am. Chem. Soc. 2010;132:8915–8917. doi: 10.1021/ja103771w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Consistent with this observation, cyclic voltammetry revealed that the α,β-unsaturated 2-acylimidazole reduces at a significantly less negative peak potential than the other test substrates depicted in Table 1. See the Supporting Information for details of these electrochemical measurements.
- 16.The subsequent cleavage of the imidazolyl group could also be achieved without isolation of the acylimidazolium salt; however, we found that the yields of the cleavage products were somewhat lower when this one-pot protocol was utilized.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.