

Abstract
Structure-based studies led to the identification of a constrained derivative of S-trityl-L-cysteine (STLC) scaffold as a candidate inhibitor of hepatitis C virus (HCV) NS5B polymerase. A panel of STLC derivatives were synthesized and investigated for their activity against HCV NS5B. Three STLC derivatives, 9, F-3070, and F-3065, were identified as modest HCV NS5B inhibitors with IC50 values between 22.3 to 39.7 μM. F-3070 and F-3065 displayed potent inhibition of intracellular NS5B activity in the BHK-NS5B-FRLuc reporter and also inhibited HCV RNA replication in the Huh7/Rep-Feo1b reporter system. Binding mode investigations suggested that the STLC scaffold can be used to develop new NS5B inhibitors by further chemical modification at one of the trityl phenyl group.
Keywords: antiviral agents, hepatitis C, HCV NS5B polymerase, inhibitors, STLC derivatives
1. Introduction
Hepatitis C virus (HCV) infection represents a major public-health concern. It is estimated that over 200 million people, ~3% of the world population, are chronically infected with the virus [1–3]. HCV has an array of immune evasion strategies and can persist in the host for years. Individuals with chronic HCV infection are at increased risk of developing cirrhosis and hepatocellular carcinoma [3–7]. Currently, HCV infections are treated by a combination of pegylated-interferon, the nucleoside analog ribavirin, and one of two recently approved HCV protease inhibitors, Boceprevir or Telaprevir [8–13]. However, this therapy is limited in efficacy against the various HCV genotypes Furthermore, in addition to its high cost, the current treatment is associated with severe side effects and a complicated dosing regimen that may limit patient compliance [11, 12]. Also the possibility of selecting drug resistant HCV variants remains [12, 13]. Therefore, the development of additional, efficacious and more cost effective HCV antiviral therapies that target viral proteins and have limited effects on host biological processes is a priority.
HCV is a member of the Flaviviridae family. The positive sense, 9.6 kb RNA genome is translated into a single 3000 amino acid polyprotein via an IRES sequence located within the 5' non-translated region (NTR) of the viral genome [14, 15]. The viral polyprotein is processed by both host and viral proteases into individual viral proteins consisting of four structural (core, E1, E2, and p7) and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) [16]. HCV replicates exclusively in the cytoplasm of host cells. Replication of the viral RNA genome is mediated by the RNA-dependent RNA polymerase (RdRp) activity of the HCV nonstructural protein NS5B [17–19]. Because of the absolute requirement of NS5B to synthesize nascent HCV RNA, NS5B represents an attractive target for the development of anti-HCV inhibitors [20, 21]. Furthermore, host cells lack RdRp. Therefore, an inhibitor that blocks RdRp activity should, in theory, have minimal or no effect on host biological processes. Though, a number of NIs and NNIs with potent in vitro anti-NS5B activity have been identified in recent years, they have presented challenges of toxicity and selection of resistant viruses, thus necessitating identification of better NS5B inhibitor scaffolds.
The structure of NS5B has been extensively characterized. The 66 kDa viral polymerase resembles a “right hand” with the active site contained in the palm domain and the RNA interacting region in the finger and thumb domains [22–25]. Current NS5B inhibitors can be divided into two classes, nucleoside inhibitors (NI) and non-nucleoside inhibitors (NNI). Once converted by host proteins into nucleotides, NIs cause RNA-chain termination upon incorporation by NS5B into the nascent RNA chains. NNIs bind to one of the five allosteric sites on NS5B and inhibit the initiation step of RNA synthesis.
Recently, we reported on the utility of three-dimensional quantitative structure-activity relationship (3D-QSAR) in combination with ligand-based and structure-based alignment procedures for in silico screening of new HCV NS5B polymerase inhibitors [26]. This investigation identified four new NS5B inhibitors from forty candidates examined from the NCI diversity set [26]. The most interesting hit, NSC123526 (Fig. 1), has been reported to be active against other viruses [27] and can be simply viewed as a constrained derivative of the S-trityl-L-cysteine (STLC) scaffold. STLC derivatives are versatile compounds endowed with antileukemic activity [28] and are also reported to inhibit the human mitotic kinesin Eg5 (HMKEg) by a non-competitive mechanism [29].
Fig. 1.
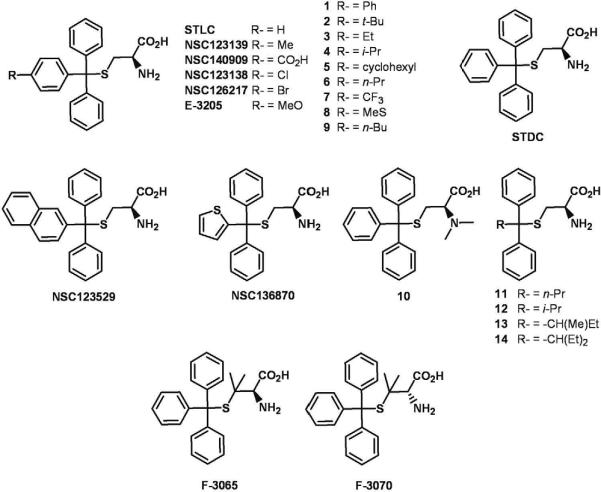
Structures of NSC123526, STLC, STDC and STLC derivatives.
Herein, we describe molecular modeling studies that led us to explore the potential of STLC and its derivatives to inhibit HCV NS5B RdRp activity in vitro. Further, we examined the effect of STLC derivatives on intracellular HCV NS5B RdRp activity and on HCV RNA replication. Among the tested STLC derivatives, we identified three compounds as novel HCV NS5B inhibitor leads. These compounds merit further optimization through classical medicinal chemistry and virtual screening.
2. Results and Discussion
2.1. Molecular Modeling
Recently, we utilized structure-based 3-D QSAR modeling to identify NS5B thumb-binding inhibitors and reported on the identification of NSC123526 as a modest HCV NS5B inhibitor [26]. NSC123526 can be considered as a constrained STLC derivative (Fig. 1). Since STLC derivative NSC123139 (Fig. 1) was found to be most potent in inhibiting HMKEg, we performed cross-docking experiments to investigate whether it could also bind the HCV-NS5B thumb domain [26, 29]. Figure 2 depicts the docked conformation of NSC123139 in HCV-NS5B and HMKEg.
Fig. 2.

Molecular docking of STLC derivatives in NS5B. Panel A: Docked conformations of NSC123139 (red-colored carbon atoms) and NSC123526 (green-colored carbon atoms) within NS5B thumb domain. Panel B: HMKEg (PDB entry code 2fme) with docked conformation of NSC123139 (red-colored carbon atoms) and the experimental bound NSC123526 (green-colored carbon atoms).
The activity of the docked NSC123139 (Predicted pIC50=5.64) was predicted by our 3-D QSAR model in the same range of NSC123526 (Experimental pIC50=4.33, predicted pIC50=5.4) [26]. However, NSC123139 exhibited a much weaker inhibition of NS5B RdRp activity in vitro (Table 1), compared to NSC123526, as previously reported [26].
Table 1.
Anti-HCV NS5B RdRp activity of STLC derivatives.
| Compound | % Inhibitiona | IC50 (μM)b |
|---|---|---|
| STLC | 12.6±2.3 | n.d. |
| STDC | 17.0±0.6 | n.d. |
| NSC123139 | 23.1±1.6 | n.d. |
| NSC136870 | 23.4±2.9 | n.d. |
| NSC140909 | 30.9±3.7 | n.d. |
| NSC123529 | 14.7±1.5 | n.d. |
| NSC123138 | 28.3±5.9 | n.d. |
| NSC126217 | 20.2±2.5 | n.d. |
| 1 | 22.4±5.4 | n.d. |
| 2 | 22.6±2.0 | n.d. |
| 3 | 36.8±2.4 | n.d. |
| 4 | 31.2±1.5 | n.d. |
| 5 | 20.5±1.1 | n.d. |
| 6 | 36.9±2.5 | n.d. |
| 7 | 43.5±0.8 | n.d. |
| 8 | 44.3±3.0 | n.d. |
| 9 | 60.0±3.4 | 39.7±0.9 |
| 10 | 17.2±2.9 | n.d. |
| 11 | 19.1±1.7 | n.d. |
| 12 | 22.5±2.2 | n.d. |
| 13 | 34.0±1.1 | n.d. |
| 14 | 33.1±0.7 | n.d. |
| 17a | 31.7±1.8 | n.d. |
| 17b | 28.7±2.1 | n.d. |
| 17c | 27.4±4.2 | n.d. |
| 17d | 24.0±4.5 | n.d. |
| 17e | 36.7±2.1 | n.d. |
| 17f | 36.0±1.0 | n.d. |
| 17g | 33.3±2.3 | n.d. |
| 17h | 28.3±5.9 | n.d. |
| 17i | 16.1±3.0 | n.d. |
| 17j | 14.0±3.3 | n.d. |
| 17k | 22.0±1.4 | n.d. |
| 17l | n.i. | n.d. |
| F-3070 | 82.8±1.3 | 22.3±5.9 |
| E-3205 | 40.2±0.7 | n.d. |
| F-3065 | 76.7±2.4 | 24.6±6.0 |
Percent inhibition was determined at 100 μM concentration of the indicated compound and represents an average of at least two independent measurements in duplicate.
The IC50 values of the compounds were determined from dose-response curves employing 8–12 concentrations of each compound in duplicate in two independent experiments. Curves were fitted to data points using nonlinear regression analysis and IC50 values were interpolated from the resulting curves using GraphPad Prism 3.03 software.
n.d., not determined.
n.i., no inhibition.
Based on the above partial results, we tested a series of STLC derivatives for their ability to inhibit NS5B, with the objective of identifying new lead scaffolds. While our investigations with additional STLC derivatives was still ongoing, the co-crystal structures of HMKEg with NSC123139 (pdb entry code 2wog and 2xae) and other STLCs (2xr2 and 3ken) were released [30]. Nevertheless, docking calculations performed through Autodock Vina, were in good agreement with the experimental results (rmsd= 0.44) and with similar docking calculations previously reported [31], thus supporting our protocol.
The above docking protocol was also applied to the other STLCs. In addition, we analyzed the Autodock Vina proposed binding mode of the two most active compounds, F-3065 and F-3070, cross-docked into the 15 NS5B-NNI co-crystal structures as previously described by us [26]. As expected, docked conformations of F-3065 and F-3070 ((R) and (S) enantiomers of the same compound, respectively) exhibited the lowest binding energy in the PDB entry 2d3u. Further, the bound conformations of F-3065 and F-3070, in agreement with the biological data, revealed that the cysteine stereocenter does not affect the overall binding mode, wherein the terminal amino acid group is involved in a hydrogen bonding network, as shown in the ligplot diagrams in Figure 3. In particular the α-amino acid portion of F-3065 makes two hydrogen bonds, one between its amino group and the carbonyl group of Trp528 (N---O distance = 3.01 Å) and the other between a carboxy oxygen and the ε-amino group of Lys533 (O---N distance = 2.98 Å) (Fig. 3A).The ligplot diagram of the (S) enantiomer F-3070, that forms two hydrogen bonds with its two carboxy oxygens, one with the guanidinic nitrogen of Arg501 (O---N distance = 3.21 Å) and the other with ε-amino group of Lys533 (O---N distance 3.05–3.22 Å), is shown in Figure 3B. This type of hydrogen bonding network was observed in all other STLC derivatives (Fig. 5) suggesting that hydrogen bonds are the leading interactions.
Fig. 3.
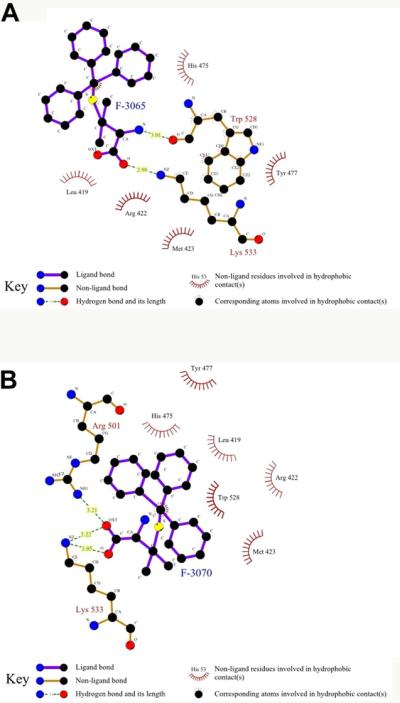
Panel A: Ligplot diagram for F-3065 docked in NS5B (PDB ID 2d3u). Panel B: Ligplot diagram for F-3070 docked in NS5B (PDB ID 2d3u).
Fig. 5.

STLC analogues docked within the HCV NS5B (in pink ribbon) thumb allosteric surface. The compounds overlap in this pocket.
Other notable interactions are hydrophobic in nature, and the trityl moieties are buried in the thumb allosteric binding side (Fig. 4). For both F-3065 and F-3070, one phenyl is placed in a pocket formed by Leu419, Arg422, Met423 and Trp528, while the other two benzenes fill-up two depressions on the enzyme surface. By comparing the binding mode of the most active STLCs with that of the experimental co-crystallized compound found in 2d3u and considering the conserved binding modes shown in Figure 5, we believe the STLC can be used as a starting scaffold, whose activity could be improved by inserting a side chain in one of the two surface bound benzene rings to better fill the binding cleft formed by Leu419, Met423, Ile482, Val485, Ala486, Leu489 and Leu497 (Fig. 6) and occupied by a 2-(4-cyanophenyl)thiophene group in the original complex (PDB ID 2d3u). As expected and within the limit of any predictive model, the application of our 3-D QSAR to all the new STLCs, predicted these compounds to have activities between the 10–100 μM range (data not shown).
Fig. 4.
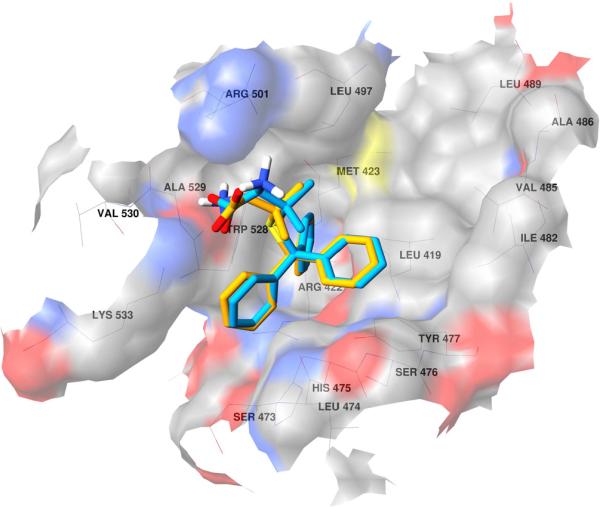
Docked conformation of F-3065 (orange) and F-3070 (cyan) in NS5B (PDB ID 2d3u). The enzyme surface is shown in atom type color.
Fig. 6.

F-3065 (orange) and F-3070 (cyan) overlapped on the 2d3u co-crystallized ligand (green). HCV NS5B (PDB ID 2d3u, in pink ribbon) and the thumb allosteric surface (in atom type color) are also shown.
2.2. Chemistry
A total of 35 STLC derivatives were utilized in this study (Fig. 1 and Scheme 1). STLC and 14 derivatives (1–14) have been reported previously [29, 31]. Compounds F-3070 and F-3065 were purchased from Bachem, while STDC (NSC123676), NSC123139, NSC136870, NSC140909, NSC123529, NSC123138, and NSC126217 were procured from NCI/NIH. In accordance with published literature, 12 STLC derivatives were newly synthesized for this investigation (Scheme 1). Starting aryldiphenylmethanol compounds 15 were prepared in good yields from appropriate esters (ArCO2Me) and phenylmagnesium chloride (data not shown) [32]. Condensation of cysteamine.HCl (16) with Ar(Ph)2COH (Ar = 4-Me-Ph, 4-Et-Ph, 4-n-Pr-Ph, 4-MeS-Ph, 4-I-Ph, 4-(Ph)-Ph and 2-naphthyl) 15a–g in TFA gave final compounds 17a–g in 29–47% yield (Scheme 1). Treatment of L-cysteine (18) or L-penicillamine (19) with Ar(Ph)2COH (Ar = 4-(n-C5H11)-Ph, 4-(n-C6H13)-Ph, 4-PrO-Ph and 4-n-Bu-Ph) 15h–k in the presence of BF3.Et2O afforded target compounds 17h–l in 30–55% yield (Scheme 1).
Scheme 1.

Synthesis of STLC derivatives 17a-l. Reagents and conditions: (a) TFA, rt, 3 h; (b) BF3.Et2O, AcOH, rt, 2 h
2.3. Biological Studies
With the objective of identifying novel HCV NS5B inhibitors, we investigated STLC and its derivatives employing the in vitro NS5B RdRp inhibition assay as described previously [33–35]. Recombinant HCV NS5B (genotype 1b) carrying an N-terminal His-tag and C-terminal 21-amino acid truncation (NS5BCΔ21) was purified to homogeneity by Ni-NTA chromatography and used as a source of enzyme [33–35]. Wedelolactone, a documented NS5B inhibitor, was employed as an internal reference standard, and yielded an IC50 value of 36.0 μM (data not shown), consistent with our previously reported value [34]. In order to identify a wider range of NS5B inhibitor candidates, preliminary screening of STLC and its derivatives was conducted at 100 μM compound concentration. While the parent STLC molecule yielded only ~12% inhibition of NS5B RdRp activity during preliminary screening, its thirty-five derivatives, with the exception of 17l, exhibited a much higher inhibition ranging from 14 to 83% (Table 1). Of these, three compounds 9, F-3070, and F-3065 having ≥60% anti-NS5B activity at 100 μM were further pursued for their IC50 value determination. This analysis resulted in the identification of F-3070 and F-3065 with near similar IC50 values, as the two most potent of the 36 STLC derivatives examined in this investigation, while 9 exhibited ~1.6–1.8-fold higher IC50 value compared to the two afore-mentioned compounds. Together, these data suggest that STLC scaffold may offer further scope for improvement of its anti-NS5B activity.
To evaluate the anti-HCV activity of STLC compounds in a more biologically relevant setting, we employed the BHK-NS5B-FRLuc reporter and the Huh7/Rep-Feo1b reporter systems [36, 37]. The former reporter system carries stably transfected NS5B and a bicistronic reporter gene, (+)FLuc-(−)UTR-RLuc for cell based investigations of HCV NS5B RdRp inhibitors [36]. The advantage of this system is that it can simultaneously measure intracellular HCV NS5B RdRp activity as reflected by the ratio of Renilla to firefly luciferase luminescence and cellular viability which is reflected by the firefly luciferase luminescence, thus enabling the identification of potent non-toxic inhibitors. The Huh7/Rep-Feo1b reporter system, on the other hand, autonomously replicates the subgenomic HCV genotype 1b replicon RNA carrying the firefly luciferase reporter as an indicator of HCV RNA replication, and has been widely employed to identify inhibitors of HCV RNA replication [37].
Only three STLC derivatives F-3070, F-3065, and E-3205 inhibited intracellular NS5B RdRp activity in the BHK-NS5B-FRLuc reporter at 100 μM concentration (Table 2). The two more potent of these, F-3070 and F-3065 exhibited ≥84% inhibition while E-3205 displayed only ~44% inhibition of NS5B RdRp activity, consistent with the in vitro data. In terms of their cytotoxicity parameters, F-3070 and F-3065 did not affect cell viability at 100 μM, as was evident from equivalent levels of firefly luciferase luminescence in compound treated cells versus DMSO controls. Treatment with E-3205 however, decreased cell viability by ~70% at 100 μM concentration. The remaining thirty-three STLC derivatives as well as the parent molecule, exhibited ≥50% reduction in cell viability at 100 μM, with only a marginal 15–30% decrease in intracellular NS5B activity (data not shown), consistent with the in vitro RdRp data.
Table 2.
Anti-HCV effects of STLC derivatives in cell based reporter assay.
| Compound | BHK-NS5B-FRLuca |
Huh7/Rep-Feolbb |
||
|---|---|---|---|---|
| Viability (%) | Inhibition (%) | Viability (%) | Inhibition (%) | |
| F-3070 | 100.0 | 85.4 | 72.6 | 89.5 |
| E-3205 | 30.2 | 44.4 | 44.2 | 88.6 |
| F-3065 | 100.0 | 84.3 | 74.1 | 91.2 |
BHK-NS5B-FRLuc and
Huh7/Rep-Feolb reporter cells were treated with the indicated compounds at 100 μM concentration for 42 h.
Cell viability in the BHK-NS5B-FRLuc reportera was estimated as the relative levels of Firefly luciferase in compound treated cells versus DMSO controls, while mat in the Huh7/Rep-Feo lb cellsb was evaluated by the MTS assay. The inhibitory effect of the compounds on NS5B RdRp activitya and HCV RNA replicationb is presented as percent of DMSO treated controls. Data represents an average of three independent experiments in duplicate.
In the Huh7/Rep-Feo1b reporter system, compounds F-3070 and F-3065 exhibited an overall similar pattern of cell viability and HCV RNA replication inhibition, corresponding to ~73–74% and ~89–91%, respectively at 100 μM concentration (Table 2). E-3205, however, exhibited decreased cell viability (44%) compared to the other two compounds, though its inhibition of HCV RNA replication (~89%) was similar. It is worth noting here that the inhibition observed in this system may be partly attributed to the cellular toxicity effects of these compounds.
The results in this present study suggest that STLC derivatives inhibit HCV RNA replication by targeting the NS5B polymerase. It is possible that other host factors such as HMKEg are also targeted by STLCs in the HCV replicase complex and needs to be elucidated. These studies provide a platform to optimize the STLC scaffold as a potent anti-NS5B inhibitor. An extensive focused virtual screening approach is ongoing on a database constituted of more than 500K trityl cysteine analogues to optimize the newly reported lead compounds.
3. Conclusion
In summary, STLC derivatives were identified as novel inhibitors of HCV NS5B polymerase activity in vitro and in cell-based assays. This study validates structure-based molecular modeling coupled with 3-D QSAR prediction, as a viable strategy for identification of new structural scaffolds targeting NS5B. STLC binding mode analysis revealed a common way by which STLCs bind to the HCV NS5B thumb allosteric site and further suggested that improved STLC derivatives may be achieved by chemical modification at one of the trityl phenyl ring.
4. Experimental section
4.1. Molecular Modeling
All molecules were generated by means of molecular mechanics of Chemaxon Marvin software (http://www.chemaxon.com/). Molecular graphics images were produced using UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco on a 3 GHz AMD CPU equipped IBM-compatible workstation with the Debian 5.0 version of the Linux operating system. Different from the previous protocol, the faster Autodock Vina [38] docking program was used in place of Autodock for all docking studies. Docking assessment was conducted via re-docking, re-docking modeled, cross-docking and cross-docking modeled as previously reported [26]. Autodock Vina proved to be as good as Autodock (data not shown), but much faster in calculations. The compounds were then submitted for structure-based molecular alignment through cross-docking protocols as previously reported [26]. For activity predictions, the previously developed SB 3-D QSAR model was applied without any modification [26]. The program ligplot v. 4.0 was used [39] to generate the ligand/NS5B interaction maps.
4.2. Chemistry
General methods Melting points were determined using a Büchi capillary instrument and are uncorrected. Optical rotations were measured at the sodium D line (589 nm) at 25 °C with a Perkin-Elmer 241 polarimeter using a 1 dm path length cell. 1H and 13C NMR spectra were recorded on a Bruker 300, 400 or 500 MHz spectrometers. Chemical shifts (δ) are in parts per million. The following abbreviations were used to designate the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Mass spectra were recorded with a Perkin-Elmer SCIEX API spectrometer. Elemental analyses were performed on a Thermoquest Flash 1112 series EA analyzer. Elemental analyses were found to be within ± 0.4 of the theoritical values. Purity of tested compounds was >95%. All commercially available reagents and solvents were used without further purification. STLC and derivatives 1–14 have been previously described [29]. E-3205, F-3070 and F-3065 were purchased from Bachem. STDC (NSC124676), NSC123139, NSC136870, NSC140909, NSC123529, NSC123138, and NSC126217 were procured from NCI/NIH.
4.2.1. General procedure for preparation of compounds 17a–g
At 0°C and under argon atmosphere, a solution of cysteamine.HCl (16) (1.33 mmol) was added dropwise to a solution of appropriate alcohol 15 (1.33 mmol) in TFA (5 mL). The reaction mixture was stirred at room temperature for 1 h, evaporated and extracted with a saturated solution of NaHCO3(aq) and EtOAc. The organic phase was dried over MgSO4, filtered, and evaporated under vacuum. The oil was crystallized from Et2O or Et2O/pentane 1:1. The desired compounds 17a–g were obtained by filtration in the range of 29 to 47% yield.
4.2.1.1. 2-[1-(4-Ethylphenyl)-1,1-Diphenylmethylthio]ethanamine (17a)
Starting alcohol = 1-(4-ethylphenyl)-1,1-diphenylmethanol (15a). Yield: 31%; mp 138–140 °C; 1H NMR (300 MHz, CD3OD + D2O): δ 1.23 (t, 3H, J = 7.5 Hz, CH3), 2.45–2.59 (m, 4H, 2 CH2), 2.58 (q, 2H, J = 7.5 Hz, CH2), 7.17 (d, 2H, J = 8.5 Hz, HAr), 7.22–7.35 (m, 8H, HAr), 7.43–7.46 (m, 4H, HAr); 13C NMR (100 MHz, DMSO-d6): δ 15.3 (CH3), 27.6 (CH2), 28.6 (CH2), 37.7 (CH2), 66.2 (Cq), 126.9 (2 × CH), 127.5 (2 × CH), 128.2 (4 × CH), 129.0 (6 × CH), 141.3 (Cq), 142.3 (Cq), 144.2 (2 × Cq); MS (ESI): m/z 370 [M + Na]+; Anal. Calcd for C23H25NS: C 79.49, H 7.25, N 4.03, found: C, 79.47, H 7.20, N 3.97.
4.2.1.2. 2-[1,1-Diphenyl-4-(phenyl)phenylmethylthio]ethanamine (17b)
Starting alcohol = 1-(4-phenylphenyl)-1,1-diphenylmethanol (15b). Yield: 30%; mp 160–162 °C; 1H NMR (300 MHz, CD3OD + D2O): δ 2.50–2.62 (s, 4H, 2 CH2), 7.27–7.63 (m, 19H, HAr); 13C NMR (125 MHz, DMSO-d6): δ 28.7 (CH2), 37.8 (CH2), 66.2 (Cq), 126.4 (2 × CH), 126.6 (2 × CH), 127.0 (2 × CH), 127.6 (CH), 128.3 (4 × CH), 128.9 (2 × CH), 129.0 (4 × CH), 129.6 (2 × CH), 138.6 (Cq), 139.2 (Cq), 143.2 (Cq), 143.9 (2 × Cq); MS (ESI): m/z 418 [M + Na]+; Anal. Calcd for C27H25NS: C 81.98, H 6.37; N 3.54, found: C 82.26, H 6.44, N 3.73.
4.2.1.3. 2-[1,1-Diphenyl-4-(propyl)phenylmethylthio]ethanamine (17c)
Starting alcohol = 1,1-diphenyl-1-(4-propylphenyl)methanol (15c). Yield: 45%; mp 133–135 °C; 1H NMR (300 MHz, CD3OD + D2O): δ 0.94 (t, 3H, J = 7.3 Hz, CH3), 1.58–1.70 (m, 2H, CH2), 2.45–2.49 (m, 2H, CH2), 2.50–2.60 (m, 4H, CH2), 7.14 (d, 2H, J = 8.3 Hz, HAr), 7.22–7.34 (m, 8H, HAr), 7.42–7.45 (m, 4H, HAr); 13C NMR (100 MHz, DMSO-d6): δ 13.8 (CH3), 23.9 (CH2), 28.6 (CH2), 36.7 (CH2), 37.7 (CH2), 66.2 (Cq), 126.9 (2 × CH), 128.1 (2 × CH), 128.2 (4 × CH), 128.9 (2 × CH), 129.0 (4 × CH), 140.8 (Cq), 141.3 (Cq), 144.2 (2 × Cq); MS (ESI): m/z 384 [M + Na]+; Anal. Calcd for C24H27NS: C, 79.73, H 7.53, N 3.87, found: C 79.55, H 7.40, N 3.82.
4.2.1.4. 2-[1,1-Diphenyl-4-(methylthio)phenylmethylthio]ethanamine (17a)
Starting alcohol = 1,1-diphenyl-1-(4-methylthiophenyl)methanol (15d). Yield: 40%; mp 142–144 °C; 1H NMR (300 MHz, CD3 + D2O): δ 2.47 (s, 3H, CH3), 2.50–2.59 (m, 4H, CH2), 7.20–7.37 (m, 10H, HAr), 7.43–7.46 (m, 4H, HAr); 13C NMR (125 MHz, DMSO-d6): δ 14.3 (CH3), 28.6 (CH2), 37.7 (CH2), 66.0 (Cq), 125.3 (2 × CH), 127.0 (2 × CH), 128.2 (4 × CH), 128.9 (4 × CH), 129.6 (2 × CH), 137.0 (Cq), 140.3 (Cq), 143.9 (2 × Cq); MS (ESI): m/z 388 [M + Na]+; Anal. Calcd for C22H23NS2: C 72.28, H 6.34, N 3.83, found: C 72.00, H 6.35, N 3.77.
4.2.1.5. 2-[1-(4-Iodophenyl)-1,1-Diphenylmethylthio]ethanamine (7e)
Starting alcohol = 1-(4-iodophenyl)-1,1-diphenylmethanol (15e). Yield: 47%; mp 150–152 °C; 1H NMR (300 MHz, CD3OD + D2O): δ 2.54 (s, 4H, 2 CH2), 7.21–7.36 (m, 8H, HAr), 7.41–7.44 (m, 4H, HAr), 7.68 (d, 2H, J = 10.8 Hz, HAr); 13C NMR (125 MHz, DMSO-d6): δ 28.8 (CH2), 37.7 (CH2), 66.0 (Cq), 93.4 (Cq), 127.1 (2 × CH), 128.3 (4 × CH), 128.9 (4 × CH), 131.4 (2 × CH), 137.0 (2 × CH), 143.5 (2 × Cq), 143.8 (Cq); MS (ESI): m/z 468 [M + Na]+; Anal. Calcd for C21H20INS: C 56.63, H 4.53, N 3.15, found: C 56.60, H 4.61, N 3.22.
4.2.1.6. 2-[1-(4-Methylphenyl)-1,1-Diphenylmethylthio]ethanamine (17f)
Starting alcohol = 1-(4-methylphenyl)-1,1-diphenylmethanol (15f). Yield: 29%; mp 138–140 °C; 1H NMR (300 MHz, CD3OD + D2O): δ 2.32 (s, 3H, CH3), 2.48–2.55 (m, 4H, 2 CH2), 7.13 (d, 2H, J = 8.1 Hz, HAr), 7.24–7.34 (m, 8H, HAr), 7.41–7.45 (m, 4H, HAr); 13C NMR (100 MHz, DMSO-d6): δ 20.5 (CH3), 29.0 (CH2), 37.9 (CH2), 66.2 (Cq), 126.9 (2 × CH), 128.2 (4 × CH), 128.7 (2 × CH), 129.0 (6 × CH), 136.1 (Cq), 141.1 (Cq), 144.2 (2 × Cq); MS (ESI): m/z 356 [M + Na]+; Anal. Calcd for C22H23NS: C 79.23, H 6.95, N 4.20, found: C 78.88, H 7.03, N 4.19.
4.2.1.7. 2-[1-(2-Naphthyl)-1,1-(diphenyl)methylthio]ethanamine (17g)
Starting alcohol = 1-(2-naphthyl)-1,1-diphenylmethanol (15g) [32, 40]. Yield: 32%; mp 126–128 °C; 1H NMR (300 MHz, CD3OD + D2O): δ 2.34–2.47 (s, 4H, 2 CH2), 7.21–7.34 (m, 6H, HAr), 7.43–7.49 (m, 6H, HAr), 7.55 (dd, 1H, J = 1.9, 8.9 Hz, HAr), 7.70–7.83 (m, 4H, HAr); 13C NMR (100 MHz, DMSO-d6): δ 35.6 (CH2), 40.8 (CH2), 66.0 (Cq), 126.4 (CH), 126.5 (CH), 126.8 (2 × CH), 127.2 (CH), 127.3 (CH), 127.5 (CH), 128.1 (5 × CH), 128.2 (CH), 129.1 (4 × CH), 131.6 (Cq), 132.3 (Cq), 141.9 (Cq), 144.5 (2 × Cq); MS (ESI): m/z 392 [M + Na]+; Anal. Calcd for C25H23NS: C 81.26, H 6.27, N 3.79, found: C 81.38, H 6.31, N 3.89.
4.2.2. General procedure for preparation of compounds 17h–17l
At 0 °C and under argon atmosphere, a solution of BF3.Et2O (1.33 mmol) was added dropwise to a solution of appropriate alcohol 15 (0.86 mmol), L-cysteine (18) or L-penicillamine (19) (0.77 mmol) in AcOH (1 mL). The reaction mixture was stirred at room temperature for 3 h. Addition of 10% solution of NaOAc (2 mL), then H2O (2 mL) led to the formation of a gum. After elimination of the supernatant, the final compound was precipitated by addition of pentane or Et2O. The desired compounds 17h–17l were obtained by filtration in the range of 30 to 55% yield.
4.2.2.1. S-[1-(4-Pentylphenyl)-1,1-diphenylmethyl]-L-cysteine (17h)
Starting alcohol = 1-(4-pentylphenyl)-1,1-diphenylmethanol (15h). Yield: 55%; mp 127–129 °C; = +61 (c = 0.52 in MeOH); 1H NMR (300 MHz, CD3OD + D2O): δ 0.91 (t, 3H, J = 6.7 Hz, CH3), 1.32–1.39 (m, 4H, 2 CH2), 1.56–1.66 (m, 2H, CH2), 2.59 (broad t, 2H, J = 7.9 Hz, CH2), 2.70 (dd, 1H, J = 9.2, 13.5 Hz, CH2), 2.82 (dd, 1H, J = 4.2, 13.5 Hz, CH2), 3.04 (dd, 1H, J = 4.2, 9.2 Hz, CH), 7.13 (d, 2H, J = 8.5 Hz, HAr), 7.20–7.35 (m, 8H, HAr), 7.43–7.46 (m, 4H, HAr); 13C NMR (100 MHz, CD3OD): δ 14.4 (CH3), 23.6 (CH2), 32.3 (CH2), 32.7 (CH2), 34.0 (CH2), 36.4 (CH2), 54.6 (CH), 68.0 (Cq), 128.0 (2 × CH), 129.1 (6 × CH), 130.6 (2 × CH), 130.7 (4 × CH), 142.8 (Cq), 143.0 (Cq), 145.8 (2 × Cq), 172.0 (CO); MS (ESI): m/z 456 [M + Na]+; Anal. Calcd for C27HH31NO2S: C 74.79, H 7.21, N 3.23, found: C 74.79, H 7.17, N 3.25.
4.2.2.2. S-[1-(4-Hexylphenyl)-1,1-diphenylmethyl]-L-cysteine (17i)
Starting alcohol = 1-(4-hexylphenyl)-1,1-diphenylmethanol (15i). Yield: 40%; mp 129–131 °C; = +53 (c = 0.54 in MeOH). 1H NMR (300 MHz, CD3OD + D2O): δ 0.90 (t, 3H, J = 6.7 Hz, CH3), 1.30–1.42 (m, 6H, 3 CH2), 1.54–1.66 (m, 2H, CH2), 2.59 (broad t, 2H, J = 7.9 Hz, CH2), 2.70 (dd, 1H, J = 9.2, 13.5 Hz, CH2), 2.82 (dd, 1H, J = 4.2, 13.5 Hz, CH2), 3.03 (dd, 1H, J = 4.2, 9.2 Hz, CH), 7.12 (d, 2H, J = 8.3 Hz, HAr), 7.21–7.35 (m, 8H, HAr), 7.43–7.46 (m, 4H, HAr); 13C NMR (100 MHz, CD3OD): δ 14.4 (CH3), 23.7 (CH2), 30.1 (CH2), 32.5 (CH2), 32.8 (CH2), 34.0 (CH2), 36.4 (CH2), 54.7 (CH), 68.0 (Cq), 128.0 (2 × CH), 129.1 (6 × CH), 130.6 (2 × CH), 130.7 (4 × CH), 142.8 (Cq), 143.0 (Cq), 145.8 (2 × Cq), 172.0 (CO); MS (ESI): m/z 470 [M + Na]+; Anal. Calcd for C28H33NO2S: C 75.13, H 7.43, N 3.13, found: C 74.87, H 7.30, N 3.02.
4.2.2.3. S-[1,1-Diphenyl-1-(4-propoxyphenyl)methyl]-L-cysteine (17j)
Starting alcohol = 1,1-diphenyl-1-(4-propoxyphenyl)methanol (15j) [41]. Yield: 43%; mp 144 146 °C; = +60 (c = 0.51 in MeOH); 1H NMR (300 MHz, CD3OD + D2O): δ 1.03 (t, 3H, J = 7.3 Hz, CH3), 1.72–1.84 (m, 2H, CH2), 2.68 (dd, 1H, J = 9.2, 13.4 Hz, CH2), 2.83 (dd, 1H, J = 4.0, 13.4 Hz, CH2), 3.04 (dd, 1H, J = 4.0, 9.2 Hz, CH), 3.92 (t, 2H, J = 6.4 Hz, CH2), 6.92 (d, 2H, J = 8.8 Hz, HAr), 7.22–7.33 (m, 8H, HAr), 7.43–7.45 (m, 4H, HAr); 13C NMR (100 MHz, CD3OD): δ 10.8 (CH3), 23.6 (CH2), 34.2 (CH2), 55.0 (CH), 67.7 (Cq), 70.5 (CH2), 114.9 (2 × CH), 127.9 (2 × CH) 129.1 (4 × CH), 130.5 (4 × CH), 132.0 (2 × CH), 137.2 (Cq), 146.0 (Cq), 146.1 (Cq), 159.3 (Cq), 172.3 (CO); MS (ESI): m/z 444 [M + Na]+; Anal. Calcd for C25H27NO3S: C 71.23, H 6.46, N 3.32, found: C 71.44, H 6.54, N 3.30.
4.2.2.4. S-[1-(4-Butylphenyl)-1,1-diphenylmethyl]-L-penicillamine (17k)
Starting alcohol = 1-(4-butylphenyl)-1,1-diphenylmethanol (15k). Yield: 30%; mp 123–125 °C; = +171 (c = 0.54 MeOH); 1H NMR (300 MHz, CD3OD + D2O): δ 0.93 (t, 3H, J = 7.3 Hz, CH3), 1.30 (s, 3H, CH3), 1.32–1.40 (m, 2H, CH2), 1.42 (s, 3H, CH3), 1.54–1.64 (m, 2H, CH2), 1.85 (s, 1H, CH), 2.59 (t, 2H, J = 7.5 Hz, CH2), 7.14 (d, 2H, J = 8.3 Hz, HAr), 7.19–7.34 (m, 6H, HAr), 7.56 (d, 2H, J = 8.5 Hz, HAr), 7.67–7.70 (m, 4H, HAr); 13C NMR (100 MHz, CD3OD): δ 14.3 (CH3), 23.4 (CH2), 25.9 (CH3), 27.9 (CH3), 34.7 (CH2), 36.1 (CH2), 53.5 (Cq), 61.9 (CH), 69.2 (Cq), 127.9 (2 × CH), 129.0 (6 × CH) 130.7 (2 × CH), 130.8 (2 × CH), 130.9 (2 × CH), 142.9 (Cq), 143.2 (Cq), 146.0 (Cq), 146.1 (Cq), 170.6 (CO); MS (ESI): m/z 470 [M + Na]+; Anal. Calcd for C28H33NO2S: C 75.13, H 7.43, N 3.13, found: C 75.45, H, 7.53, N 3.32.
4.2.2.5. S-[1,1-Diphenyl-1-(4-propoxyphenyl)methyl]-L-penicillamine (17l)
Starting alcohol = 1,1-diphenyl-1-(4-propoxyphenyl)methanol (15j). Yield: 34%; mp 133–135 °C; = +69 (c = 0.15 in MeOH); 1H NMR (300 MHz, CD3OD + D2O): δ 1.03 (t, 3H, J = 7.1 Hz, CH3), 1.31 (s, 3H, CH3), 1.43 (s, 3H, CH3), 1.74–1.81 (m, 2H, CH2), 1.92 (s, 1H, CH), 3.93 (t, 2H, J = 6.0 Hz, CH2), 6.87 (d, 2H, J = 8.4 Hz, HAr), 7.20–7.30 (m, 6H, HAr), 7.56 (d, 2H, J = 8.4 Hz, HAr), 7.64–7.72 (m, 4H, HAr); 13C NMR (100 MHz, CD3OD): δ 10.8 (CH3), 23.7 (CH2), 25.9 (CH3), 28.4 (CH3), 53.2 (Cq), 62.0 (CH), 69.2 (Cq), 70.5 (CH2), 114.6 (2 × CH), 127.8 (2 × CH) 128.7 (4 × CH), 129.5 (4 × CH), 131.6 (2 × CH), 137.3 (Cq), 146.0 (2 × Cq), 159.6 (Cq), 172.6 (CO); MS (ESI): m/z 472 [M + Na]+; Anal. Calcd for C27H31NO3S: C 72.13, H 6.95, N 3.12, found: C 71.99, H 7.00, N 3.11.
4.3 Biological Studies
4.3.1 NS5B inhibition assay
Recombinant NS5B carrying the N-terminal histidine-tag was purified from the plasmid pThNS5BCΔ21 expressed in Escherichia coli DH5α by Ni-NTA chromatography [33, 34]. The compounds were dissolved in dimethylsulfoxide (DMSO) as a 10 mM stock solution and stored at −20 °C. Serial dilutions were made in DMSO immediately prior to the assay. The activity of the compounds against HCV NS5B was evaluated by the standard primer dependent elongation assay as previously described [33, 34]. Briefly, preliminary screening was performed in the presence or absence of 100 μM STLC or the indicated derivative in a reaction buffer containing 20 mM Tris-HCl (pH 7.0), 100 mM NaCl, 100 mM Na-glutamate, 0.1 mM DTT, 0.01% BSA, 0.01% Tween-20, 5% glycerol, 20 U/mL of RNasin, 20 μM UTP, 2 μCi [α-32P]UTP, 0.25 μM polyrA/U12, 100 ng NS5BCΔ21 and 1 mM MnCl2. Following 60 minutes incubation at 30 °C, reactions were terminated by the addition of chilled 5% trichloroacetic acid (TCA) containing 0.5 mM sodium pyrophosphate. Reaction products were precipitated on GF-B filters and quantified on a liquid scintillation counter. NS5B activity in the presence of DMSO control was set at 100% and that in the presence of the STLC derivatives was determined relative to this control. Compounds exhibiting greater than 50% inhibition at 100 μM were evaluated for their IC50 values dose-response from curves employing 8–12 concentrations of the compounds in duplicate in two independent experiments. Curves were fitted to data points using nonlinear regression analysis and IC50 values were interpolated from the dose response curves using GraphPad Prism 3.03 software.
4.3.2 Cell culture
BHK-NS5B-FRLuc reporter cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% heat-inactivated fetal bovine serum, 5% antibiotic-antimycotic, 5% nonessential amino acid, 1 mg/mL G418 and 10 μg/mL blasticidin. Huh7/Rep-Feo1b replicon reporter cells were cultivated in DMEM containing 10% fetal calf serum, 5% antibiotic and 0.5 mg/mL G418. All cell lines were incubated at 37 °C in the presence of 5% CO2 supplement.
4.3.3. BHK-NS5B-FRLuc reporter assay
The effect of the compounds on intracellular NS5B RdRp activity was screened employing the BHK-NS5B-FRLuc reporter system as previously described [36]. Briefly, BHK-NS5B-FRLuc reporter cells were plated at a confluence of 1 × 104 cells/well in 96 well plates and incubated with DMSO (1%) or the indicated compound (100 μM) for 42 h. Reporter gene expression was measured with a Dual-Glo Luciferase Assay Kit (Promega, USA) in accordance with the manufacturer's instructions. Effect of the compounds on cell viability was estimated as the relative levels of firefly luciferase in compound treated cells versus DMSO controls. The inhibitory effect of the compounds on the intracellular NS5B RdRp activity was evaluated from the percent reduction in RLuc to FLuc luminescence signal in compound treated cells versus DMSO controls.
4.3.4. Huh7/Rep-Feo1b reporter system
The effect of the compounds on HCV RNA replication was screened employing the Huh7/Rep-Feo1b replicon reporter cells as previously described [42]. Briefly, 1 × 104 Huh7/Rep-Feo1b cells were plated in 96 well plates and treated with 100 μM concentration of the indicated compound or DMSO for 42 h. The concentration of DMSO in cell culture was kept constant at 1.0%. Cell viability was measured by the colorimetric MTS assay employing the CellTiter 96AQueous One Solution assay reagent (Promega, USA). Inhibitory effect of the compounds on HCV RNA replication was measured as the relative levels of firefly luciferase signals in compound treated cells versus DMSO controls.
Research Highlights
HCV NS5B inhibitors were designed employing structure-based protocol.
Some of the designed compounds were obtained via a facile synthetic approach.
Enzyme and cell based assays proved STLC as leads against HCV NS5B.
Acknowledgements
This work was supported by the National Institute of Health Research Grants DK066837 and CA153147 to N.K.-B. We would like to thank Prof. Garland R. Marshall (Washington University of St. Louis - MO) for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Wasley A, Alter MJ. Epidemiology of hepatitis C: geographic differences and temporal trends. Semin Liver Dis. 2000;20:1–16. doi: 10.1055/s-2000-9506. [DOI] [PubMed] [Google Scholar]
- [2].Grakoui A, Hanson HL, Rice CM. Bad time for Bonzo? Experimental models of hepatitis C virus infection, replication, and pathogenesis. Hepatology. 2001;33:489–495. doi: 10.1053/jhep.2001.23041. [DOI] [PubMed] [Google Scholar]
- [3].Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med. 2001;345:41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- [4].Saito I, Miyamura T, Ohbayashi A, Harada H, Katayama T, Kikuchi S, Watanabe Y, Koi S, Onji M, Ohta Y, et al. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc Natl Acad Sci U S A. 1990;87:6547–6549. doi: 10.1073/pnas.87.17.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ruiz J, Sangro B, Cuende JI, Beloqui O, Riezu-Boj JI, Herrero JI, Prieto J. Hepatitis B and C viral infections in patients with hepatocellular carcinoma. Hepatology. 1992;16:637–641. doi: 10.1002/hep.1840160305. [DOI] [PubMed] [Google Scholar]
- [6].Hoofnagle JH, di Bisceglie AM. The treatment of chronic viral hepatitis. N Engl J Med. 1997;336:347–356. doi: 10.1056/NEJM199701303360507. [DOI] [PubMed] [Google Scholar]
- [7].Pawlotsky JM. Pathophysiology of hepatitis C virus infection and related liver disease. Trends Microbiol. 2004;12:96–102. doi: 10.1016/j.tim.2003.12.005. [DOI] [PubMed] [Google Scholar]
- [8].Feld JJ, Liang TJ. HCV persistence: cure is still a four letter word. Hepatology. 2005;41:23–25. doi: 10.1002/hep.20561. [DOI] [PubMed] [Google Scholar]
- [9].Ferenci P. Pegylated interferon plus ribavirin for chronic hepatitis C: the role of combination therapy today, tomorrow and in the future. Minerva Gastroenterol Dietol. 2006;52:157–174. [PubMed] [Google Scholar]
- [10].Pawlotsky JM. Virology of hepatitis B and C viruses and antiviral targets. J Hepatol. 2006;44:S10–13. doi: 10.1016/j.jhep.2005.11.005. [DOI] [PubMed] [Google Scholar]
- [11].Sheridan C. New Merck and Vertex drugs raise standard of care in hepatitis C. Nat Biotechnol. 2011;29:553–554. doi: 10.1038/nbt0711-553. [DOI] [PubMed] [Google Scholar]
- [12].Rice C. Perspective: miles to go before we sleep. Nature. 2011;474:S8. doi: 10.1038/474S8a. [DOI] [PubMed] [Google Scholar]
- [13].Garber K. Hepatitis C: move over interferon. Nat Biotechnol. 2011;29:963–966. doi: 10.1038/nbt.2031. [DOI] [PubMed] [Google Scholar]
- [14].Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- [15].Tsukiyama-Kohara K, Iizuka N, Kohara M, Nomoto A. Internal ribosome entry site within hepatitis C virus RNA. J Virol. 1992;66:1476–1483. doi: 10.1128/jvi.66.3.1476-1483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brass V, Moradpour D, Blum HE. Molecular virology of hepatitis C virus (HCV): 2006 update. Int J Med Sci. 2006;3:29–34. doi: 10.7150/ijms.3.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Behrens SE, Tomei L, De Francesco R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. Embo J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- [18].Hagedorn CH, van Beers EH, De Staercke C. Hepatitis C virus RNA-dependent RNA polymerase (NS5B polymerase) Curr Top Microbiol Immunol. 2000;242:225–260. doi: 10.1007/978-3-642-59605-6_11. [DOI] [PubMed] [Google Scholar]
- [19].Moradpour D, Brass V, Bieck E, Friebe P, Gosert R, Blum HE, Bartenschlager R, Penin F, Lohmann V. Membrane association of the RNA-dependent RNA polymerase is essential for hepatitis C virus RNA replication. J. Virol. 2004;78:13278–13284. doi: 10.1128/JVI.78.23.13278-13284.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].De Francesco R, Rice CM. New therapies on the horizon for hepatitis C: are we close? Clin Liver Dis. 2003;7:211–242. xi. doi: 10.1016/s1089-3261(02)00069-7. [DOI] [PubMed] [Google Scholar]
- [21].Kwong AD, McNair L, Jacobson I, George S. Recent progress in the development of selected hepatitis C virus NS3.4A protease and NS5B polymerase inhibitors. Curr Opin Pharmacol. 2008;8:522–531. doi: 10.1016/j.coph.2008.09.007. [DOI] [PubMed] [Google Scholar]
- [22].Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey FA. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci U S A. 1999;96:13034–13039. doi: 10.1073/pnas.96.23.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol. 1999;6:937–943. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- [24].Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, Miyano M. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure. 1999;7:1417–1426. doi: 10.1016/s0969-2126(00)80031-3. [DOI] [PubMed] [Google Scholar]
- [25].Bressanelli S, Tomei L, Rey FA, De Francesco R. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J. Virol. 2002;76:3482–3492. doi: 10.1128/JVI.76.7.3482-3492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Musmuca I, Caroli A, Mai A, Kaushik-Basu N, Arora P, Ragno R. Combining 3-D quantitative structure-activity relationship with ligand based and structure based alignment procedures for in silico screening of new hepatitis C virus NS5B polymerase inhibitors. J Chem Inf Model. 2010;50:662–676. doi: 10.1021/ci9004749. [DOI] [PubMed] [Google Scholar]
- [27].Silverman JE, Ciustea M, Shudofsky AM, Bender F, Shoemaker RH, Ricciardi RP. Identification of polymerase and processivity inhibitors of vaccinia DNA synthesis using a stepwise screening approach. Antiviral Res. 2008;80:114–123. doi: 10.1016/j.antiviral.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zee-Cheng KY, Cheng CC. Experimental antileukemic agents. Preparation and structure-activity study of S-tritylcysteine and related compounds. J Med Chem. 1970;13:414–418. doi: 10.1021/jm00297a019. [DOI] [PubMed] [Google Scholar]
- [29].Debonis S, Skoufias DA, Indorato RL, Liger F, Marquet B, Laggner C, Joseph B, Kozielski F. Structure-activity relationship of S-trityl-L-cysteine analogues as inhibitors of the human mitotic kinesin Eg5. J Med Chem. 2008;51:1115–1125. doi: 10.1021/jm070606z. [DOI] [PubMed] [Google Scholar]
- [30].Kaan HY, Hackney DD, Kozielski F. The structure of the kinesin-1 motor-tail complex reveals the mechanism of autoinhibition. Science (New York, N.Y. 333:883–885. doi: 10.1126/science.1204824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kaan HY, Weiss J, Menger D, Ulaganathan V, Tkocz K, Laggner C, Popowycz F, Joseph B, Kozielski F. Structure-activity relationship and multidrug resistance study of new S-trityl-L-cysteine derivatives as inhibitors of Eg5. Journal of medicinal chemistry. 54:1576–1586. doi: 10.1021/jm100991m. [DOI] [PubMed] [Google Scholar]
- [32].Weber ES, Wierig K, A., Goldberg I. Triarylmethanol host compounds. Synthesis, crystalline complex formation and X-ray crystal structures of three inclusions species. J. Inclus. Phenom. Mol. 1997;28:163–179. [Google Scholar]
- [33].Kaushik-Basu N, Bopda-Waffo A, Talele TT, Basu A, Chen Y, Kucukguzel SG. 4-Thiazolidinones: a novel class of hepatitis C virus NS5B polymerase inhibitors. Front Biosci. 2008;13:3857–3868. doi: 10.2741/2974. [DOI] [PubMed] [Google Scholar]
- [34].Kaushik-Basu N, Bopda-Waffo A, Talele TT, Basu A, Costa PR, da Silva AJ, Sarafianos SG, Noel F. Identification and characterization of coumestans as novel HCV NS5B polymerase inhibitors. Nucleic Acids Res. 2008;36:1482–1496. doi: 10.1093/nar/gkm1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen Y, Bopda-Waffo A, Basu A, Krishnan R, Silberstein E, Taylor DR, Talele TT, Arora P, Kaushik-Basu N. Characterization of aurintricarboxylic acid as a potent hepatitis C virus replicase inhibitor. Antivir Chem Chemother. 2009;20:19–36. doi: 10.3851/IMP1286. [DOI] [PubMed] [Google Scholar]
- [36].Lee JC, Tseng CK, Chen KJ, Huang KJ, Lin CK, Lin YT. A cell-based reporter assay for inhibitor screening of hepatitis C virus RNA-dependent RNA polymerase. Anal Biochem. 403:52–62. doi: 10.1016/j.ab.2010.04.004. [DOI] [PubMed] [Google Scholar]
- [37].Itsui Y, Sakamoto N, Kakinuma S, Nakagawa M, Sekine-Osajima Y, Tasaka-Fujita M, Nishimura-Sakurai Y, Suda G, Karakama Y, Mishima K, Yamamoto M, Watanabe T, Ueyama M, Funaoka Y, Azuma S, Watanabe M. Antiviral effects of the interferon-induced protein guanylate binding protein 1 and its interaction with the hepatitis C virus NS5B protein. Hepatology (Baltimore, Md. 2009;50:1727–1737. doi: 10.1002/hep.23195. [DOI] [PubMed] [Google Scholar]
- [38].Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- [40].Barluenga J, Fananas FJ, Sanz R, Marcos C, Trabada M. On the reactivity of o-lithioaryl ethers: tandem anion translocation and Wittig rearrangement. Org Lett. 2002;4:1587–1590. doi: 10.1021/ol0258029. [DOI] [PubMed] [Google Scholar]
- [41].Kawakami YS, Y., Okada A. Synthesis and polymerization of mono-p-alkoxy-substituted triphenylmethyl acrylate. Polymer J. 1990;22:705–718. [Google Scholar]
- [42].Kim K, Kim KH, Kim HY, Cho HK, Sakamoto N, Cheong J. Curcumin inhibits hepatitis C virus replication via suppressing the Akt-SREBP-1 pathway. FEBS Lett. 584:707–712. doi: 10.1016/j.febslet.2009.12.019. [DOI] [PubMed] [Google Scholar]