

Abstract
Multiple sclerosis (MS) is an immune-mediated demyelinating disease of the central nervous system (CNS). Here we document for the first time that the cytokine IL-33 is upregulated in both the periphery and the CNS of MS patients. Plasma IL-33 was elevated in MS patients compared to normal subjects and a three-month treatment of MS patients with interferon β-1a resulted in significant decrease of IL-33 levels. Similarly, stimulated cultured lymphocytes and macrophages from MS patients had elevated IL-33 levels compared to normal subjects. In parallel, the transcription factor NF-κB that mediates IL-33 transcription was also elevated in leukocytes of MS patients. IL-33 was elevated in normal-appearing white matter and plaque areas from MS brains and astrocytes were identified as an important source of IL-33 expression in the CNS. In summary, IL-33 levels are elevated in the periphery and CNS of MS patients, implicating IL-33 in the pathogenesis of MS.
Keywords: multiple sclerosis, autoimmunity, demyelination, cytokines, IL-1, IL-33, st2, interferon therapy, inflammation, NF-kappa B, macrophages, T-cells, macrophages, mast cells, plasma, central nervous system, astrocytes, microglia, EAE, TMEV
1. INTRODUCTION
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system (CNS) characterized by multifocal white matter lesions and progressive neurologic disability (1). Several studies have demonstrated that MS lesions contain multiple leukocyte cell types including lymphocytes, macrophages, and dendritic cells, all of which are believed to contribute to lesion formation by various distinct and interacting mechanisms (2, 3). Interestingly, both leukocytes and CNS lesions of MS patients have elevated levels of cytokines and contain activated transcription factors including NF-κB (4, 5), STAT1 (5–7) and STAT6 (8–10), which can lead to enhanced expression of inflammatory genes and toxic molecules known to promote demyelination (11–17).
Interleukin-33 (IL-33) is a novel cytokine of the IL-1 cytokine family that has been recently implicated in several inflammatory and autoimmune diseases like Lupus Erythematosus, rheumatoid arthritis, ulcerative colitis, psoriasis, and cardiovascular disease (18–20). IL-33 transcription is mediated by NF-κB activation secondary to Toll-like receptor (TLR) signaling or stimulation with TNF-α or IL-1β. (21–24). The receptor for IL-33, ST2L is differentially expressed on multiple leukocyte subsets and signals to activate both MAPK and NF-κB pathways through primarily TNF receptor-associated Factor 6 (TRAF6) (21, 25). ST2L is expressed on endothelial cells, mast cells, basophils, macrophages, and dendritic cells in vivo and are thus believed to be main targets for IL-33 inflammatory activity, including upregulation of the cytokines IL-6, IL-8, and IL-13 (18, 19, 23, 26–29).
Administration of exogenous IL-33 in vivo leads to pronounced splenomegaly and eosinophilia, increased mucous production, and hypertrophy of the digestive and respiratory tracts. These pathological changes occur in conjunction with increases in Th2 molecules such as IL-4, IL-5, and IL-13 and IgE (23). Other studies emphasize the importance of IL-33 in amplifying innate immunity and possibly serving as an alarmin to activate the immune system following cell necrosis or apoptosis (30, 31)
IL-33 has been shown to have important biologic functions in several immune mediated diseases including asthma, dermatitis, rheumatoid arthritis, inflammatory bowel disease, autoimmune hepatitis, and cardiovascular disease among others (18, 19, 31). IL-33 can act to modulate several pathways including Th2 skewing and mast cell, eosinophil, and macrophage activation (32). Regarding CNS disease, our group was the first to show that IL-33 is expressed in mouse CNS astrocytes and that CNS-derived IL-33 is functionally active (33). Recently, another study showed that IL-33 is induced in the CNS in the experimental allergic encephalomyelitis EAE mouse model of MS (34). Specifically, mouse IL-33 is predominantly expressed in the CNS by endothelial cells and astrocytes where it affects astrocyte activation and ultimately results in microglia proliferation and secretion of cytokines and chemokines (34). Alternatively, IL-33 can activate CNS mast cells through degranulation and secretion of other pro-inflammatory mediators including IL-1, IL-6, and MCP-1 (33, 35–37).
This is the first study that explores the expression of the cytokine IL-33 in MS. Here we demonstrate that IL-33 is elevated in the plasma of patients with active relapsing remitting (RR) MS. Interestingly, in vivo therapeutic treatment with interferon β-1a suppresses IL-33 levels in MS patients, suggesting a possible mode of interferon action. Furthermore, stimulation of IL-2 expanded PBMCs (peripheral blood mononuclear cells) and macrophages from MS patients resulted in significantly increased levels of IL-33 compared to normal controls. Furthermore, we demonstrate that IL-33 elevation correlates with increased activation of the transcription factor NF-κB, which has been previously shown to induce IL-33 expression (21–23). Lastly, we show that the CNS – both in normal appearing white matter (NAWM) and plaque tissue – of MS patients, expresses higher levels of IL-33 compared to normal subjects. In summary, these findings suggest that IL-33 may play a potential role in the pathogenesis of MS both in the periphery and within the CNS.
2. MATERIALS AND METHODS
2.1. Patient selection for blood
All patients selected were clinically diagnosed as having definite relapsing-remitting (RR) MS (38), and none received any disease modifying treatment including IFN-β, glatiramir acetate, steroids, or other immunosuppressive agents for at least two months prior to donating blood. RR MS group donated blood before and after a three-month treatment with recombinant interferon β-1a (39). Table I provides additional information on the patients and normal subjects used in this study. The Institutional Review Board of SUNY Upstate University approved all studies, and both patients and normal controls granted informed consent before providing blood.
Table I. Biometric data of MS patients and normal subjects who donated blood for the study.
Il-33 protein and mRNA was quantified in the plasma, and freshly isolated PBMCs, cultured IL-2 expanded PBMCs, and cultured macrophages of those subjects. RR MS are untreated Relapsing Remitting multiple sclerosis patients. The data are shown in mean value ± SD. For the gender, F stands for females and M stands for males. The age at onset of clinically diagnosed MS and disease duration are shown in years. The clinical symptoms were measured on Kurtzuke’s Expanded Disability Status Scale (EDSS) at the time the initial blood was drawn. Patients with RR MS donated blood before and after a three-month treatment with recombinant interferon β-1a and EDSS scores shown here were calculated just before treatment with interferon β-1a.
| Patient Category | Number of Subjects | Age (Years) | Gender | Age at Onset | Disease Duration | EDSS Score |
|---|---|---|---|---|---|---|
| Normal Subjects | 32 | 41±12 | 22F, 10M | --- | --- | --- |
| RR MS | 32 | 41±9 | 22F, 10M | 36±9 | 5.9± 5 | 2.5±1.4 |
| IFN β-1a Tx | 20 | 40±8 | 14F, 6M | 34±9 | 5.4±4 | 2.4±1.2 |
2.2. PBMC isolation and expansion
Patients and normal subjects donated 60 ml of blood which was collected in heparinized tubes. Blood was diluted 1:1 with HBSS and overlaid onto lymphocyte separation medium (Cellgro, Herndon, VA). After centrifugation, the plasma was collected and used to quantify cytokine levels, while the 10 ml of the interface containing the PBMCs was collected and washed twice with HBSS. For the in vivo studies several samples of the freshly isolated cells were either resuspended in STAT- 60 (Tel-Test, Friendswood, TX) for RNA analysis, RIPA buffer for protein analysis (40), or fixed for intracellular flow cytometry analysis. For the in vitro studies, the remaining PBMCs were cultured for one week in RPMI media with 20 units/ml IL-2 (R & D Systems), and 10% fetal bovine serum. After one week, cells were treated with cytokines and analyzed as outlined in the text. Cultured PBMCs were stained with antibodies against CD3, CD19, and CD14 were used to for differentiating T-cells, B-cells, and monocytes respectively. The proportion of cell types making up the PBMCs of MS patients and controls were the same (80% T-cells, 5% B-cells, 3% monocytes, and 12% of negative cells). A majority of negative cells probably represent natural killer (NK) cells based on their scatter properties (41).
2.3. Human peripheral blood monocyte-derived macrophages
Patients and normal subjects donated 50 ml of blood collected in heparinized tubes. Blood was diluted 1:1 with Hank’s balanced salt solution (HBSS) and overlaid onto lymphocyte separation medium (Cellgro, Herndon, VA). After centrifugation, the 10 ml of the interface containing the PBMCs was collected and washed twice with HBSS. Adherent monocytes were cultured for one week in RPMI media, 15% fetal bovine serum, and 50ng/mL GM-CSF as previously described (42, 43). Medium was replenished every 3 days and non-adherent cells were removed at the second feeding (at 6 days). For experiments, cells were cultured for an additional day in the absence of GM-CSF, washed twice with PBS, and adherent cells were lysed in STAT- 60 (Tel-Test, Friendswood, TX) for RNA analysis or in RIPA buffer (44) for protein analysis. Flow cytometry cells were detached by incubation in 5.0mM EDTA for 10 minutes at 37 degrees C. The method consistently yielded more than 95% pure macrophages assessed by both morphological criteria as described (45) and expression of the myeloid lineage marker CD14 as determined by flow cytometric analysis.
2.4. Brain samples
Fresh frozen tissue specimens were obtained from the Human Brain and Spinal Fluid Resource Center, Veteran’s Administration, West Los Angeles Healthcare Center, Los Angeles, CA 90073, which is sponsored by NINDS/NIMH, the National Multiple Sclerosis Society, and the Department of Veterans Affairs. Frozen tissue samples from normal controls and MS patients were screened by a neuropathologist to confirm the clinical diagnosis of MS and exclude confounding pathologies. None of the subjects had ischemic brain injury. Tissue from MS patients consisted of either normal appearing white matter (NAWM) that had no apparent histopathologic changes, or plaque that contained white matter tissue with areas of focal demyelination and infiltration of immune cells. The tissue quality was determined with tissue pH with a cutoff pH of 6.7 (Table 2). The quantity of the purified RNA was assessed using the RNA Pico Lab Chip Kit with the Agilent Technologies Bioanalyzer and agarose gel electrophoresis. In addition, RNA was quantified spectrophotometrically and only samples with sufficient quantity (500ng total yield) and appropriate optical density (OD 260/280 ratio = 1.7–2.1) were used for subsequent analysis (46).
Table II. Biometric data of MS patients and normal subjects that were used to examine Il-33 expression in the CNS.
Brain tissue was used to quantify IL-33 mRNA and protein and brain IL-33 expression and localization was determined by immunohistochemisty and immunofluorescence. Fresh frozen brain sections were received from the Brain Bank. Brain white matter was used from normal subjects. The brains of MS patients had two district separate blocks from normal appearing white matter (NAWM) and plaque areas. The data are shown as the mean value ± SD. For the gender, F stands for females and M stands for males.
| Patient Category | Number of Subjects | Age (Years) | Gender | Lysis Time | Tissue pH |
|---|---|---|---|---|---|
| Normal Subjects | 20 | 63±13 | 11F, 9M | 15±4 | 7.1±0.2 |
| MS | 9 | 60±12 | 6F, 3M | 19±7 | 7.0±0.3 |
2.5. Real-Time RT-PCR
Total RNA was isolated using STAT-60 RNA isolation kit (Tel-test). RNA was quantified spectrophotometrically and 0.5 μg of total RNA was converted into cDNA. Briefly, to generate cDNA 0.5μg total RNA and random primers (Invitrogen, Carlsbad, CA) were incubated at 72 degrees for 10 minutes. Reverse transcription was performed using the Superscript II RT enzyme (Invitrogen, Carlsbad, CA) and following the manufacturer’s specifications. cDNA was diluted to 200 μL with water and 4μL was used for quantitative real time PCR using a SYBR Green kit (Abgene, Epson, UK). The following forward and reverse primers were used at 10 nM: IL-33 AAGGCAAAGCACTCCACAGT and GAAAGAAGTTTGCCCCATGT, MCP-1 GCTCATAGCAGCCACCTTC and GCTTCTTTGGGACACTTGC, IP-10 TTCAAGGAGTACCTCTCTCTAG and CTGGATTCAGACATCTCTTCTC, TARC CGAGGGACCAATGTGGGC and GGGTGAGGAGGCTTCAAGACC, GAPDH ACCACCATGGAGAAGGC and GGCATGGACTGTGGTCATGA, β-Actin AGGCACCAGGGCGTGAT and GCCCACATAGGAATCCTTCTGAC. The PCR parameters were 15 minutes for 95 degrees, 35 cycles at 95 degrees for 15 seconds, and 60 degrees for 1 minute in an ABI Prism 700 thermocycler (Applied Biosystems, Foster City, CA). Serial dilutions of cDNA containing a known copy number of each gene were used in each quantitative PCR run in order to generate a standard curve relating copy number to threshold amplification cycle (47). Gene expression levels were calculated during the logarithmic amplification phase by determining the initial mRNA copy number using the standard curve. Amplification of each gene specific fragment was confirmed both by examination of melting peaks and by agarose gel electrophoresis.
2.6. Cytokine ELISA
The levels of the cytokine IL-33, IL-6, IFN-γ, and IL-13 were measured using R&D Systems DuoSet ELISA kits (R&D Systems) following the manufacturer’s protocol.
2.7 NF-κB DNA-binding activity assay
NF-κB DNA-binding activity was analyzed using the TransAMNF-κB p65 transcription factor assay kit (Active Motif, Carlsbad, CA) following the manufacturer’s instructions and as previously described (48–50). Briefly, nuclear extracts were prepared (51) from macrophages of normal subjects and MS patients that were treated with media alone or 5μg/mL of LPS for 1 hour. Alternatively, nuclear extracts were prepared from freshly isolated PBMCs that were isolated from normal subjects and MS patients before and after a three-month treatment with IFN-β1a. Protein levels of the nuclear extracts were quantified with the Bradford assay (Pierce Chemicals, Rockford, IL) and 10 μg were incubated in a 96-well plate coated with oligonucleotide containing the NF-κB consensus-binding sequence 5′-GGGACTTTCC-3′. Bound NF-κB was then detected by a p65-specific primary antibody. An HRP-conjugated secondary antibody was then applied to detect the bound primary antibody and provided the basis for colorimetric quantification. The enzymatic product was measured at 450 nm with a reference wavelength of 650 nm by a microplate reader. To quantify the amount of NF-κB, serial dilutions of purified p65 recombinant protein (20ng - 0.16ng) were measured to provide a calibration curve between p65 binding and absorbance. The specificity of the assay was further tested by the addition of wild type or mutated NF-κB consensus oligonucleotide in the competitive or mutated competitive control wells before the addition of nuclear extracts. The addition of the wild-type NF-κB consensus oligonucleotide completely abolished NF-κB binding.
2.8 Immunohistochemistry
Fresh frozen tissue was sectioned at 7 micrometers, placed onto glass slides and fixed in −20 °C acetone for 15 minutes. Endogenous peroxidase was inactivated using 0.5% hydrogen peroxide in PBS for 10 minutes. Endogenous biotin was quenched using the Dako Biotin blocking system (Dako, Carpinteria, CA). Tissue was blocked for 2 hours in PBS containing 10% horse serum at room temperature (RT). After blocking, tissue sections were incubated with primary mouse monoclonal antibodies to IL-33 (Nessy-1, Axxora, San Diego, USA) or control mouse IgG antibodies (Dako, Carpinteria, CA) in PBS containing 10% horse serum overnight at 4°C. Subsequently, tissue sections were incubated with 5ug/mL of the biotinylated anti-mouse IgG antibody followed by streptavadin-HRP (Dako, Carpinteria, CA). Red color precipitate was produced using AEC according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). Sections were coverslipped using aqueous mounting medium (Electron Microscopy Sciences, Hatfield, PA, USA). Images were taken at 200X magnification on a Nikon Eclipse E800 microscope equipped with a spot camera. Bar in the figure represents 40 micrometers.
2.9 Double Immunofluorescence of IL-33 and GFAP
Fresh frozen tissue was sectioned at 7 micrometers, placed onto glass slides and fixed in -20 °C acetone for 15 minutes. Sections were incubated with the primary antibodies, IL-33 (Axxora, San Diego, USA), at 4°C overnight followed by GFAP (Serotec, Oxford, England) for 1 h room temperature. Secondary antibodies (Jackson Immunoreasearch, West Grove, PA, USA) were incubated for 2 hr at room temperature. Slides were mounted with gelvatol and kept at 4° C in the dark. Images were taken on a SPOT RT Slider CCD camera using a Nikon Eclipse TE 2000-U microscope.
2.10 Statistical Analysis
Histograms contain statistical means with standard error values. The number of MS patients and normal subjects used in each experiment are shown in Table I & II. The p-values were generated using the unpaired Student’s t-test and a p-value of less than 0.05 was chosen to indicate statistical significance between two sample means.
3. RESULTS
3.1. Plasma IL-33 is elevated MS patients
The levels of IL-33 protein were measured in plasma of RRMS patients and normal subjects (Figure 1). Plasma was isolated from blood of 32 normal subjects and 32 untreated RRMS patients, and the levels of IL-33 protein were quantified with ELISA (Table I). The average level of IL-33 in normal subjects was 60 pg/mL. In contrast, active RRMS patients had significantly higher levels of IL-33 with an average concentration of 280 pg/mL (Figure 1A). Interestingly, in vivo treatment with recombinant IFN-β 1a for three-months significantly suppressed the levels of plasma IL-33 compared to untreated RRMS patients. In order to corroborate the ELISA findings and examine whether IL-33 was transcriptionally regulated, we also quantified IL-33 mRNA in freshly isolated PBMCs by real-time RT-PCR (Figure 1B). Freshly isolated PBMCs demonstrated a significant 15-fold higher level of IL-33 compared to normal subject PBMCs. Importantly, as with protein levels in the blood, IFN-β-1a significantly suppressed IL-33 RNA levels in PBMC. To control for mRNA loading we examined the expression of the housekeeping genes GAPDH and β-actin by real time PCR in the samples and demonstrated similar expression levels (data not shown).
Figure 1.

IL-33 protein in plasma and mRNA expression in freshly drawn PBMCs and plasma levels of IL-6, IFN-γ, and IL-13. Cytokines levels were quantified in plasma PBMCs of normal subjects (C), untreated RR MS patients (MS), and in vivo in RR MS patients following three month treatment with interferon β-1a (MS-β). A. IL-33 was quantified by ELISA in plasma. B. Il-33 mRNA levels were quantified in freshly isolated PBMCs. The absolute mRNA transcript copy numbers per 10ng of total RNA were quantified using real time RT-PCR and were normalized to the levels of normal subjects that are shown as 1. C – E: The levels of IL-6, IFN-γ, and IL-13 were quantified in the plasma of the same samples of normal subjects and MS patient before and after three month treatment with interferon β-1a.
Additionally we quantified the expression of the plasma cytokines IL-6, IFN-γ, and IL-13 in the same samples of normal subjects, and RRMS before and following in vivo treatment with IFN-β-1a (Figure 1C–E). IL-6, IFN-γ, and IL-13 were elevated in untreated RRMS patients compared to normal subjects (52–54). IFN-β 1a treatment significantly reduced the levels of the NF-κB-responsive cytokine but not the levels of IFN-γ. Importantly, IL-13 that has been shown to be induced following IL-33 stimulation was also reduced in plasma of RRMS following in vivo IFN-β 1a treatment (23, 29).
3.2. IL-33 expression in cultured lymphocytes and macrophages of MS patients
Next we examined the expression of IL-33 in cultured PBMCs from untreated RRMS patients and normal control subjects. First PBMCs were cultured in the presence of IL-2 for one week to expand the lymphocyte population (Figure 2A). The proportion of cell types making up the PBMCs of MS patients and controls were 80% T cells, 5% B cells, and 3% monocytes as previously documented (10). We further stimulated cultured PBMCs with phorbol myristate acetate (PMA) and ionomycin for 24 hours. Following stimulation, the levels of IL-33 measured by ELISA were increased, and importantly PBMCs from untreated RRMS patients secreted significantly higher levels compared to normal subjects (Figure 2A).
Figure 2.
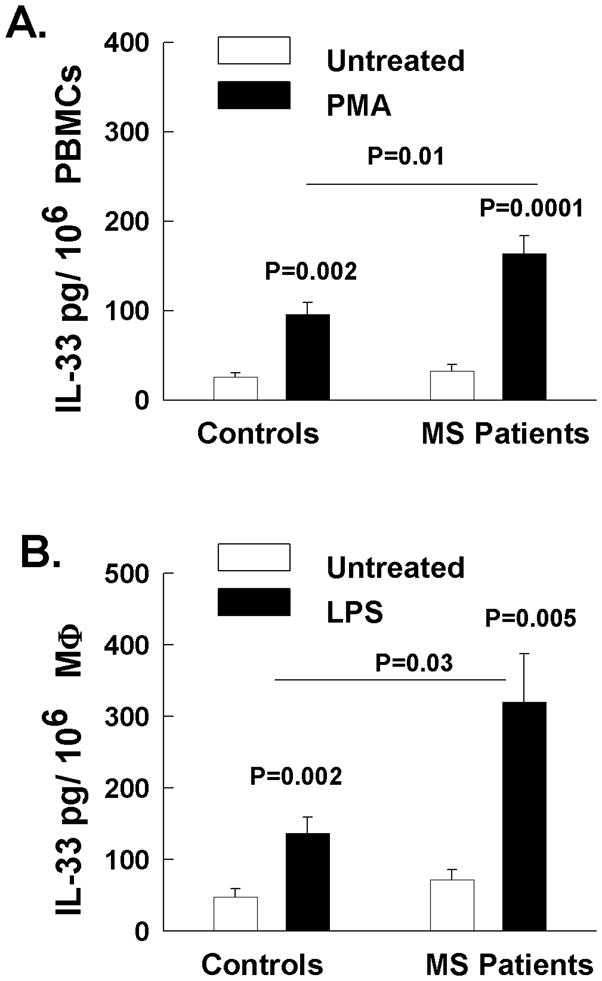
IL-33 was quantified by ELISA in the supernatants of cultured PBMCs or macrophages of normal subjects and MS patients. A. PBMCs were cultured in the presence of IL-2 for one week and then stimulated with Phorbol myristate acetate and and ionomycin (PMA) for 24 hours or media alone. Then IL-33 was quantified in the supernatants B. Adherent monocytes were expanded in the presences of GM-CSF for one week. Macrophages were then stimulated with LPS for 24 hours and secreted IL-33 protein levels were determined with ELISA.
Additionally, we examined the expression of IL-33 in macrophages of normal subjects and RRMS patients (Figure 2B). Blood monocytes were expanded in the presence of GM-CSF for one week resulting in more than 95% adherent macrophages determined by morphology and CD14 staining (55). Macrophages were then stimulated with LPS for 24 hours and secreted IL-33 protein levels were determined with ELISA (Figure 2B) as previously shown (56). Importantly, the stimulated macrophages from MS patients secreted significantly higher levels of IL-33 compared to macrophages from normal subjects.
3.3. NF-κB expression activation correlates with IL-33 levels
To examine possible mechanisms of elevated IL-33 in leukocytes of MS patients, we quantified the activation of the transcription factor NF-κB, which has been shown previously to mediate IL-33 induction (21–24). Nuclear extracts from freshly isolated PBMCs or cultured macrophages were prepared and allowed to bind an NF-κB consensus oligonucleotide sequence. Bound NF-κB was then detected by a p65 (RelA)-specific antibody and quantified based on a calibration using purified p65 recombinant protein. NF-κB DNA binding activity was significantly increased in PBMCs of MS patients compared to normal subjects and IFN-β treatment resulted in a significant decrease in NF-κB activation (Figure 3A) (4, 5, 54, 55). Furthermore, LPS treatment significantly induced NF-κB activation and was significantly more elevated in macrophages from MS patients compared to normal subjects (Figure 3B). Thus, NF-κB activation correlated with IL-33 expression levels suggesting a possible transcriptional mechanism by which IL-33 is elevated in MS.
Figure 3.

NF-κB activation in macrophages of MS patients and normal subjects. Nuclear extracts were isolated from freshly isolated PBMCs or cultured macrophages and allowed to bind an NF-κB consensus-binding sequence. Bound NF-κB was then detected by a p65 specific antibody and quantified based on a calibration curve generated by using a purified p65 recombinant protein. The NF-κB DNA binding activity is reported as ng of bound p65 protein per 10μg of total protein in nuclear extracts. A. NF-κB activation in freshly isolated PBMCs from normal subjects (C), untreated MS patients (MS), and MS patients following three month in vivo IFN-β treatment (MS-β). B. NF-κB activation in cultured macrophages from normal subjects and MS patients following treatment with media alone or LPS for 1 hour.
3.4. IL-33 is elevated in the CNS of MS patients
In mice, in addition to immune cells, CNS resident cells, and especially astrocytes express high levels of IL-33 (19, 33). Also, IL-33 is induced in the CNS of EAE and TMEV mouse models of MS. Therefore, it was important to examine the expression of IL-33 in the CNS of MS patients (Figure 4). We used fresh frozen sectioned brains in which both the diagnosis and differentiation of normal appearing white matter (NAWM) or plaque was confirmed by a neuropathologist (Table II). The tissue quality was determined by tissue pH with a cutoff of 6.7 and appropriate RNA quality was determined as previously described (46). First, we examined the tissue expression of IL-33 protein by ELISA and mRNA by RT-PCR in MS NAWM that contained CNS resident cells but no apparent infiltrating immune cells. IL-33 protein was significantly elevated in the NAWM of MS patients compared to normal subject white matter (Figure 4A). IL-33 mRNA was correspondingly increased in the NAWM from MS patients compared to white matter of normal subjects (Figure 4B). Additionally, both the IL-33 mRNA and protein were elevated in white matter containing areas of focal demyelination and immune cell infiltration (plaque) of MS patients compared to white matter from normal subjects.
Figure 4.

IL-33 expression along with cytokine and chemokine expression in white matter from brains of normal subjects (C), normal appearing white matter of MS patients (NAWM), and white matter plaque areas of MS patients (Plaque). A. IL-33 protein expression by ELISA quantified as pg Il-33/100μg of total protein in brain lysate. B. IL-33 mRNA expression determined by real time RT-PCR and results were normalized to the levels of normal subjects that are shown as 1. C – E: The levels of IL-6, IFN-γ, and IL-13 were quantified in the brain of the same samples by ELISA and displayed as pg cytokine/100μg of brain protein. F – H: The levels of the chemokines MCP-1, IP-10, and TARC were also quantified by real time RT-PCR and results were normalized to the levels of normal subjects that are shown as 1
Furthermore, we quantified the expression of the cytokines IL-6, IFN-γ, and IL-13 by ELISA (Figure 5C–E) and the mRNA expression of the chemokines MCP-1, IP-10, and TARC by RT-PCR (Figure 5F–H) in the same samples to examine how expression correlates with IL-33 and evaluate the degree of inflammation in MS brain NAWM or plaque areas. In NAWM there was significant elevated levels of IL-6, IL-13, MCP-1, IP-10, and TARC suggesting that moderate inflammation was present and was originated from CNS resident cells. IFN-γ was not elevated in NAWM suggesting that the primary source was infiltrating immune cells. In the plaque areas involving active areas of demyelination, dense leukocyte infiltrate, and peri-plaque white matter, showed increase levels of IL-6, IFN-γ, IL-13 and MCP-1, IP-10, and TARC compared to normal subjects.
Figure 5.

Imunohistochemical staining of IL-33 the CNS of MS patients. A–C: Hematoxylin and eosin stain of A. white matter from normal subjects, B. normal appearing white matter from MS patients, and C. plaque region with focal demyelination and immune cell infiltration from MS patient. D–F: Negative control IgG immunohistochemical staining with red precipitate of D. white matter from normal subjects, E. normal appearing white matter from MS subject, and F. plaque region with focal demyelination and immune cell infiltration from MS patient. G–I: Representative immunohistochemical staining of IL-33 with red precipitate of G. white matter from normal subjects, H. normal appearing white matter from MS patients, and I. plaque region with focal demyelination and immune cell infiltration from MS patient. Images were taken at 200x magnification and the bar indicates a length of 40uM.
3.5 CNS IL-33 immunohistochemistry and localization in astrocytes
Furthermore, to determine the distribution and cellular localization of IL-33, brain tissue was immunohistochemically stained with IL-33 antibody (Figure 5). For negative control we used a control mouse IgG that did not reveal any staining pattern. IL-33 was present in nuclei of cells both in plaques, periplaque regions, and in normal appearing white matter cells of MS brains. In distinct contrast, IL-33 staining of cells in normal subject white matter showed only trace amounts of IL-33 as well as light staining around blood vessels (Figure 5). Double immunofluorescence staining showed a high incidence of IL-33 within GFAP+ astrocytes of MS patients confirming previous findings in mouse astrocytes both in vivo and in vitro (19, 33). Taken together, IL-33 expression in CNS glia of MS subjects is much higher than in normal subject brain and may therefore play an important role in the pathophysiology of MS (Figure 6).
Figure 6. Double immunofluorescence of IL-33 and GFAP in MS brain.

Representative images from fresh-frozen normal appearing white matter of MS patients. Tissue was stained with A. glial fibrillary acidic protein (GFAP) a specific marker for astrocytes and appears red in the image, B: IL-33 cytokine staining that appears green. C. The images for GFAP and IL-33 were merged. D. DAPI staining showing cell neuclei Arrow indicates the identical cell through the images. Bar indicates 20 μM.
4. DISCUSSION
This is the first study to describe the expression of the cytokine IL-33 in MS. We have shown that the levels of IL-33 are elevated in the plasma and in freshly isolated PBMCs of RRMS patients. Furthermore, therapeutic treatment with IFN-β-1a significantly suppressed IL-33 expression in the blood of MS patients. In vitro, activation of IL-2-expanded PBMCs and cultured macrophages led to a significantly higher secretion of IL-33 in MS patients compared to normal subjects. Importantly, we have shown that both NAWM and plaques of MS patients have significantly higher levels of IL-33 compared to white matter in brains of normal subjects. These data suggest a possible role for IL-33 in the immune response in the periphery and within the CNS in MS subjects that may be relevant to the demyelinating process.
Several cytokines are elevated in MS, which can lead to elevated transcription factors including NF-κB in blood leukocytes (54). Importantly, NF-κB signaling plays a central role in MS pathogenesis in modulating neural cell survival and by inducing numerous inflammatory genes including cytokines, proteases, and reactive oxygen species (57, 58) in CNS resident cells and infiltrating immune cells. We have previously shown that a deficiency in the protein tyrosine phosphatase SHP-1, a master negative regulator of inflammatory signaling, is at least partly responsible for enhanced NF-κB activation in PBMCs, macrophages, and CNS resident cells of MS patients (54, 55). Importantly, IL-33 transcription is mediated by NF-κB activation (21–24) and here we demonstrate that NF-κB activation correlates with IL-33 expression suggesting one of the possible mechanisms of the transcriptional upregulation of IL-33 in leukocytes of MS subjects.
An important consideration arising from these studies is the source and functional role for IL-33 in the pathogenesis of MS. The present studies indicate two potential sites from which IL-33 may be derived. The first is CNS tissue in which we have previously demonstrated nuclear IL-33 is induced and released by astrocytes treated with various TLR ligands (59). As with other IL-1-family cytokines, astrocytes required both a priming step with TLR ligands and subsequent triggering step such as treatment with ATP (60) for maximal release of IL-33 (59). IL-33 release occurs by an incompletely defined mechanism in which these cytokines are liberated from the nucleus and/or cytosol to the extracellular environment through the plasma membrane. In the case of IL-1β and IL-18, proteolytic processing of the pro-forms in the cytoplasm occurs via caspase-1 activity within assembled inflammasomes (61). However, in the case of IL-33, release does not require proteolytic processing by inflammasomes (62). Rather, nuclear IL-33 release from cells occurs following necrosis in which the plasma membrane becomes extensively damaged (63). Thus, increased IL-33 activity in tissues follows a pattern of induction and release that combines mechanisms seen with IL-1 family cytokines (ie. priming) and that of alarmins such as HMGB1 which require cell necrosis for release (62). Alarmins such as IL-33 signal tissue damage to innate effectors that promote further inflammation for elimination of necrotic tissue.
In addition to having an important role as a pro-inflammatory NF-κB-inducing cytokine, IL-33 is extensively involved in promoting Th2 and allergic immune responses and mediating activation of mast cells and eosinophils (33, 64–66). There is a documented elevation in both Th1 and Th2 cytokines in MS and IL-33 secretion in the periphery and from activated astrocytes in the CNS of MS patients which might be partially responsible for promoting the Th2 response (52, 54, 55, 67). IL-33 has also been shown to activate mast cells, which is an important inflammatory cell in MS (36, 68–70). Therefore, the elevated IL-33 expression of IL-33 in MS might play a critical role in mast cell activation that can mediate secretion of neurotoxic molecules, enhance blood-brain barrier entry, and modulate activation of CNS and immune cells. Importantly, and additional function of IL-33 is to facilitate trafficking of immune cells into the CNS by specifically attracting Th2 cells and increasing adhesion to fibronectin of human mast cells (71, 72).
Macrophages play an important role in MS through direct phagocytosis of myelin and through secretion of inflammatory molecules that cause oligodendrocyte and neuronal death (55, 73, 74). IL-33 has been shown to act on monocytes, dendritic cells, and macrophages to induce the expression of cytokines, chemokines, and other proinflammatory genes (56, 75, 76). Therefore, elevated IL-33 expression levels documented here are expected to contribute to inflammatory demyelination through macrophage and dentritic cell activation. More work is needed to examine if macrophages from MS subjects have altered sensitivity to IL-33 stimulation.
We demonstrated that in vivo three-month IFN-β treatment significantly decreases IL-33 expression in plasma and fresh PBMCs of MS patients and correlated with significant reduction in NF-κB activation and IL-6 and IL-13 expression. We have previously shown that IFN-β treatment both in vivo and in vitro induces the expression of the protein tyrosine phosphatase SHP-1 that can negatively regulate NF-κB activation and NF-κB responsive gene expression (51, 54, 55). Additionally, we have previously shown that anti-SHP-1 siRNA prevented IFN-β mediated NF-κB suppression. Based on our findings, it is possible that IFN- β suppresses IL-33 expression by reducing NF-κB activation, but further experiments are required to establish the exact mechanism.
The CNS has been found to have extremely high expression of IL-33 at the mRNA level and indeed the CNS has the highest expression of IL-33 of all organs in the body (23). Here we demonstrate that IL-33 protein is significantly elevated in brain white matter areas of active demyelination containing dense leukocyte infiltrate along with high levels of inflammatory infiltrate. Importantly, IL-33 is elevated both at the protein and mRNA level in normal appearing white matter of MS patients compared to normal subjects, suggesting that CNS resident cells are an important source of IL-33 in the CNS. Additional inflammatory mediators like IL-6, IL-13, MCP-1, IP-10, and TARC are also elevated in NAWM of MS patients further suggesting a significant inflammatory reaction in CNS resident cells despite absence of infiltrating immune cells (8, 9).
We have previously described the expression of biologically active IL-33 protein by CNS cells, particularly mouse astrocytes (33). Astrocytes are nonhematopoietic epithelial-like cells of the CNS and are known to express both subunits of the IL-33R, ST2L and IL-1R accessory protein (IL-1Rap) (23, 77). Additionally, studies in mice have shown that IL-33 is expressed by both astrocytes and endothelial cells in vivo, which may act on microglia expressing ST2L (34). Within the CNS, microglia are an important target of IL-33, which increases microglia proliferation, secretion of inflammatory cytokines and chemokines, and phagocytosis (34). Importantly, activated microglia are important mediators of demyelination in MS (78). These data point to an possible inflammatory role of IL-33 in the CNS that can contribute to immune cell activation and augment signaling pathways mediating oligodendrocyte and neuronal injury.
Although speculative, a primary role for IL-33 in MS pathogenesis can be easily envisioned based on our present observations and known requirements for IL-33 release from cells. For instance, intracellular IL-33 may be both increased and subsequently released from astrocytes and macrophages by viruses infections (59, 61, 79). This is an attractive pathway as viruses have long been implicated in the pathogenesis of MS and many excellent animal models exist (80). Thus, virus infection provides both priming via viral PAMPs (eg. dsRNA) and release via virus-induced necrosis (81–84). Cell necrosis further results in extracellular increases in danger-associated molecular patterns (DAMPs) (eg. ATP) in which extensive plasma membrane leakage (85, 86) and IL-33 release to the extracellular space is amplified (87, 88).
The targets for liberated IL-33 in MS is another key unanswered question but various sites can be considered that are consistent with MS lesion development. For instance, activation of mast cells by released IL-33 (89, 90) which have been implicated to play a key role in MS pathogenesis(91, 92), secrete large quantities of TNF-α and IL-6 (69). Moreover, IL-33 may be particularly proinflammatory in the CNS as both microglia and astrocytes express IL-33 receptors and respond by proliferating and producing TNF-α and IL-1β (34). These archetypical proinflammatiory cytokines further promote inflammatory mediators and recruitment of lymphocyte and macrophage effectors to the CNS instigating intense inflammatory responses in the white matter (93). Finally, MS lesion development may relate to enhanced systemic IL-33 expression in plasma and peripheral blood leukocytes described in the present studies. As in the CNS, microbial infections may be inducers of IL-33 production and release by peripheral myeloid and lymphoid cells in which IL-33 expression has been described (79, 94). While the exact role of IL-33 in MS remains to be determined, our present data allow consideration of potential pathways in which IL-33 may participate in MS that will be better understood once complex aspects of IL-33 expression, feedback mechanisms (95), and IL-33 responsiveness of immune cell subsets (18) are completely known.
Highlights.
The first study documenting IL-33 expression in multiple sclerosis
IL-33 was elevated in plasma and activated leukocytes of MS patients
Three-month treatment with interferon β-1a suppressed plasma IL-33 levels
IL-33 was elevated in brains of MS patients and astrocytes highly expressed IL-33
IL-33 is elevated in the periphery and CNS, implicating IL-33 in MS pathogenesis
Acknowledgments
We would like to thank Dr. Isobel Scarisbrick at the Mayo Clinic for providing the cDNA plamids used in performing real time RT-PCR. We thank Carol Ozark for collection of normal subject blood samples and Christophoros P. Christophi for helping with RNA isolation. Also we acknowledge the help of Cornelia Mihai and Luis J. Mejico in helping with patient recruitment. We appreciate the help of Richard L. Davis with providing his neuropathology expertise of MS brain. This work was supported in part by research grants from EMD Serono, Inc. and Pfizer, Inc. to Dr. Burk Jubelt, the National Multiple Sclerosis Society (RG2569C5) to Dr. Paul T. Massa, and NIH grant (NS041593) to Dr. Paul T. Massa.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938–52. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 2.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47(6):707–17. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 3.Kornek B, Lassmann H. Neuropathology of multiple sclerosis-new concepts. Brain Res Bull. 2003;61(3):321–6. doi: 10.1016/s0361-9230(03)00095-9. [DOI] [PubMed] [Google Scholar]
- 4.Eggert M, Goertsches R, Seeck U, Dilk S, Neeck G, Zettl UK. Changes in the activation level of NF-kappa B in lymphocytes of MS patients during glucocorticoid pulse therapy. J Neurol Sci. 2008;264(1–2):145–50. doi: 10.1016/j.jns.2007.08.026. [DOI] [PubMed] [Google Scholar]
- 5.Gobin SJ, Montagne L, Van Zutphen M, Van Der Valk P, Van Den Elsen PJ, De Groot CJ. Upregulation of transcription factors controlling MHC expression in multiple sclerosis lesions. Glia. 2001;36(1):68–77. doi: 10.1002/glia.1096. [DOI] [PubMed] [Google Scholar]
- 6.Frisullo G, Angelucci F, Caggiula M, Nociti V, Iorio R, Patanella AK, et al. pSTAT1, pSTAT3, and T-bet expression in peripheral blood mononuclear cells from relapsing-remitting multiple sclerosis patients correlates with disease activity. J Neurosci Res. 2006;84(5):1027–36. doi: 10.1002/jnr.20995. [DOI] [PubMed] [Google Scholar]
- 7.Feng X, Petraglia AL, Chen M, Byskosh PV, Boos MD, Reder AT. Low expression of interferon-stimulated genes in active multiple sclerosis is linked to subnormal phosphorylation of STAT1. J Neuroimmunol. 2002;129(1–2):205–15. doi: 10.1016/s0165-5728(02)00182-0. [DOI] [PubMed] [Google Scholar]
- 8.Zeis T, Graumann U, Reynolds R, Schaeren-Wiemers N. Normal-appearing white matter in multiple sclerosis is in a subtle balance between inflammation and neuroprotection. Brain. 2008;131(Pt 1):288–303. doi: 10.1093/brain/awm291. [DOI] [PubMed] [Google Scholar]
- 9.Cannella B, Raine CS. Multiple sclerosis: cytokine receptors on oligodendrocytes predict innate regulation. Ann Neurol. 2004;55(1):46–57. doi: 10.1002/ana.10764. [DOI] [PubMed] [Google Scholar]
- 10.Christophi GP, Hudson CA, Gruber RC, Christophi CP, Mihai C, Mejico LJ, et al. SHP-1 deficiency and increased inflammatory gene expression in PBMCs of multiple sclerosis patients. Lab Invest. 2008;88(3):243–55. doi: 10.1038/labinvest.3700720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dogan RN, Karpus WJ. Chemokines and chemokine receptors in autoimmune encephalomyelitis as a model for central nervous system inflammatory disease regulation. Front Biosci. 2004;9:1500–5. doi: 10.2741/1284. [DOI] [PubMed] [Google Scholar]
- 12.Ubogu EE, Cossoy MB, Ransohoff RM. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol Sci. 2006;27(1):48–55. doi: 10.1016/j.tips.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Hendriks JJ, Teunissen CE, de Vries HE, Dijkstra CD. Macrophages and neurodegeneration. Brain Res Brain Res Rev. 2005;48(2):185–95. doi: 10.1016/j.brainresrev.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez M. Effectors of demyelination and remyelination in the CNS: implications for multiple sclerosis. Brain Pathol. 2007;17(2):219–29. doi: 10.1111/j.1750-3639.2007.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scarisbrick IA, Linbo R, Vandell AG, Keegan M, Blaber SI, Blaber M, et al. Kallikreins are associated with secondary progressive multiple sclerosis and promote neurodegeneration. Biol Chem. 2008;389(6):739–45. doi: 10.1515/BC.2008.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scarisbrick IA, Blaber SI, Lucchinetti CF, Genain CP, Blaber M, Rodriguez M. Activity of a newly identified serine protease in CNS demyelination. Brain. 2002;125(Pt 6):1283–96. doi: 10.1093/brain/awf142. [DOI] [PubMed] [Google Scholar]
- 17.Karpus WJ, Kennedy KJ, Fife BT, Bennett JL, Dal Canto MC, Kunkel SL, et al. Anti-CCL2 treatment inhibits Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J Neurovirol. 2006;12(4):251–61. doi: 10.1080/13550280600873819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. 2010;10(2):103–10. doi: 10.1038/nri2692. [DOI] [PubMed] [Google Scholar]
- 19.Kurowska-Stolarska M, Hueber A, Stolarski B, McInnes IB. Interleukin-33: a novel mediator with a role in distinct disease pathologies. J Intern Med. 2011;269(1):29–35. doi: 10.1111/j.1365-2796.2010.02316.x. [DOI] [PubMed] [Google Scholar]
- 20.Theoharides TC, Zhang B, Kempuraj D, Tagen M, Vasiadi M, Angelidou A, et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc Natl Acad Sci U S A. 2010;107(9):4448–53. doi: 10.1073/pnas.1000803107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Funakoshi-Tago M, Tago K, Hayakawa M, Tominaga S, Ohshio T, Sonoda Y, et al. TRAF6 is a critical signal transducer in IL-33 signaling pathway. Cell Signal. 2008;20(9):1679–86. doi: 10.1016/j.cellsig.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Lu R, Zhao G, Pflugfelder SC, Li DQ. TLR-mediated induction of pro-allergic cytokine IL-33 in ocular mucosal epithelium. Int J Biochem Cell Biol. 2011;43(9):1383–91. doi: 10.1016/j.biocel.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23(5):479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 24.Nile CJ, Barksby E, Jitprasertwong P, Preshaw PM, Taylor JJ. Expression and regulation of interleukin-33 in human monocytes. Immunology. 2010;130(2):172–80. doi: 10.1111/j.1365-2567.2009.03221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Funakoshi-Tago M, Tago K, Sato Y, Tominaga S, Kasahara T. JAK2 is an important signal transducer in IL-33-induced NF-kappaB activation. Cell Signal. 2011;23(2):363–70. doi: 10.1016/j.cellsig.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Espinassous Q, Garcia-de-Paco E, Garcia-Verdugo I, Synguelakis M, von Aulock S, Sallenave JM, et al. IL-33 enhances lipopolysaccharide-induced inflammatory cytokine production from mouse macrophages by regulating lipopolysaccharide receptor complex. J Immunol. 2009;183(2):1446–55. doi: 10.4049/jimmunol.0803067. [DOI] [PubMed] [Google Scholar]
- 27.Turnquist HR, Thomson AW. IL-33 broadens its repertoire to affect DC. Eur J Immunol. 2009;39(12):3292–5. doi: 10.1002/eji.200940026. [DOI] [PubMed] [Google Scholar]
- 28.Aoki S, Hayakawa M, Ozaki H, Takezako N, Obata H, Ibaraki N, et al. ST2 gene expression is proliferation-dependent and its ligand, IL-33, induces inflammatory reaction in endothelial cells. Mol Cell Biochem. 2010;335(1–2):75–81. doi: 10.1007/s11010-009-0244-9. [DOI] [PubMed] [Google Scholar]
- 29.Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U S A. 2009;106(32):13463–8. doi: 10.1073/pnas.0906988106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A. 2010;107(43):18581–6. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volarevic V, Mitrovic M, Milovanovic M, Zelen I, Nikolic I, Mitrovic S, et al. Protective Role of IL-33/ST2 Axis in Con A-Induced Hepatitis. J Hepatol. 2011 doi: 10.1016/j.jhep.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 32.Smith DE. The biological paths of IL-1 family members IL-18 and IL-33. J Leukoc Biol. 2010;89(3):383–92. doi: 10.1189/jlb.0810470. [DOI] [PubMed] [Google Scholar]
- 33.Hudson CA, Christophi GP, Gruber RC, Wilmore JR, Lawrence DA, Massa PT. Induction of IL-33 expression and activity in central nervous system glia. J Leukoc Biol. 2008;84(3):631–43. doi: 10.1189/jlb.1207830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yasuoka S, Kawanokuchi J, Parajuli B, Jin S, Doi Y, Noda M, et al. Production and functions of IL-33 in the central nervous system. Brain Res. 2011;1385:8–17. doi: 10.1016/j.brainres.2011.02.045. [DOI] [PubMed] [Google Scholar]
- 35.Chakraborty S, Kaushik DK, Gupta M, Basu A. Inflammasome signaling at the heart of central nervous system pathology. J Neurosci Res. 2010;88(8):1615–31. doi: 10.1002/jnr.22343. [DOI] [PubMed] [Google Scholar]
- 36.Theoharides TC, Alysandratos KD, Angelidou A, Delivanis DA, Sismanopoulos N, Zhang B, et al. Mast cells and inflammation. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbadis.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Theoharides TC, Kempuraj D, Kourelis T, Manola A. Human mast cells stimulate activated T cells: implications for multiple sclerosis. Ann N Y Acad Sci. 2008;1144:74–82. doi: 10.1196/annals.1418.029. [DOI] [PubMed] [Google Scholar]
- 38.McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50(1):121–7. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- 39.Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet. 1998;352(9139):1498–504. [PubMed] [Google Scholar]
- 40.Massa PT, Wu H. Interferon regulatory factor element and interferon regulatory factor 1 in the induction of major histocompatibility complex class I genes in neural cells. J Interferon Cytokine Res. 1995;15(9):799–810. doi: 10.1089/jir.1995.15.799. [DOI] [PubMed] [Google Scholar]
- 41.Chang L, Gusewitch GA, Chritton DB, Folz JC, Lebeck LK, Nehlsen-Cannarella SL. Rapid flow cytometric assay for the assessment of natural killer cell activity. J Immunol Methods. 1993;166(1):45–54. doi: 10.1016/0022-1759(93)90327-4. [DOI] [PubMed] [Google Scholar]
- 42.De Nichilo MO, Burns GF. Granulocyte-macrophage and macrophage colony-stimulating factors differentially regulate alpha v integrin expression on cultured human macrophages. Proc Natl Acad Sci U S A. 1993;90(6):2517–21. doi: 10.1073/pnas.90.6.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blystone SD, Graham IL, Lindberg FP, Brown EJ. Integrin alpha v beta 3 differentially regulates adhesive and phagocytic functions of the fibronectin receptor alpha 5 beta 1. J Cell Biol. 1994;127(4):1129–37. doi: 10.1083/jcb.127.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christophi GP, Hudson CA, Gruber R, Christophi CP, Massa PT. Promoter-specific induction of the phosphatase SHP-1 by viral infection and cytokines in CNS glia. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krissansen GW, Elliott MJ, Lucas CM, Stomski FC, Berndt MC, Cheresh DA, et al. Identification of a novel integrin beta subunit expressed on cultured monocytes (macrophages). Evidence that one alpha subunit can associate with multiple beta subunits. J Biol Chem. 1990;265(2):823–30. [PubMed] [Google Scholar]
- 46.Atz M, Walsh D, Cartagena P, Li J, Evans S, Choudary P, et al. Methodological considerations for gene expression profiling of human brain. J Neurosci Methods. 2007;163(2):295–309. doi: 10.1016/j.jneumeth.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christophi GP, Isackson PJ, Blaber S, Blaber M, Rodriguez M, Scarisbrick IA. Distinct promoters regulate tissue-specific and differential expression of kallikrein 6 in CNS demyelinating disease. J Neurochem. 2004;91(6):1439–49. doi: 10.1111/j.1471-4159.2004.02826.x. [DOI] [PubMed] [Google Scholar]
- 48.Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, et al. Development of a sensitive multi-well colorimetric assay for active NFkappaB. Nucleic Acids Res. 2001;29(4):E21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu L, Zou W, Yu C, Lin J, He X. Human recombinant PLD2 can repress p65 activity of guinea pigs of chronic asthma in vivo. Cell Mol Immunol. 2006;3(4):307–10. [PubMed] [Google Scholar]
- 50.Sun J, Ramnath RD, Tamizhselvi R, Bhatia M. Neurokinin A engages neurokinin-1 receptor to induce NF-kappaB-dependent gene expression in murine macrophages: implications of ERK1/2 and PI 3-kinase/Akt pathways. Am J Physiol Cell Physiol. 2008;295(3):C679–91. doi: 10.1152/ajpcell.00042.2008. [DOI] [PubMed] [Google Scholar]
- 51.Massa PT, Wu C. Increased inducible activation of NF-kappaB and responsive genes in astrocytes deficient in the protein tyrosine phosphatase SHP-1. J Interferon Cytokine Res. 1998;18(7):499–507. doi: 10.1089/jir.1998.18.499. [DOI] [PubMed] [Google Scholar]
- 52.Link H. The cytokine storm in multiple sclerosis. Mult Scler. 1998;4(1):12–5. doi: 10.1177/135245859800400104. [DOI] [PubMed] [Google Scholar]
- 53.Hohnoki K, Inoue A, Koh CS. Elevated serum levels of IFN-gamma, IL-4 and TNF-alpha/unelevated serum levels of IL-10 in patients with demyelinating diseases during the acute stage. J Neuroimmunol. 1998;87(1–2):27–32. doi: 10.1016/s0165-5728(98)00053-8. [DOI] [PubMed] [Google Scholar]
- 54.Christophi G, Panos M, Hudson C, Mihai C, Jubelt B, Massa P. Interferon-b Treatment in Multiple Sclerosis Attenuates Inflammatory Gene Expression Through Inducible Activity of the Phosphatase SHP-1. Clinical Immunology. 2009;133(1):27–44. doi: 10.1016/j.clim.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Christophi GP, Hudson CA, Panos M, Gruber RC, Massa PT. Modulation of macrophage infiltration and inflammatory activity by the phosphatase SHP-1 in virus-induced demyelinating disease. J Virol. 2009;83(2):522–39. doi: 10.1128/JVI.01210-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohno T, Oboki K, Morita H, Kajiwara N, Arae K, Tanaka S, et al. Paracrine IL-33 Stimulation Enhances Lipopolysaccharide-Mediated Macrophage Activation. PLoS One. 2011;6(4):e18404. doi: 10.1371/journal.pone.0018404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Massa PT, Aleyasin H, Park DS, Mao X, Barger SW. NFkappaB in neurons? The uncertainty principle in neurobiology. J Neurochem. 2006;97(3):607–18. doi: 10.1111/j.1471-4159.2006.03810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yan J, Greer JM. NF-kappa B, a potential therapeutic target for the treatment of multiple sclerosis. CNS Neurol Disord Drug Targets. 2008;7(6):536–57. doi: 10.2174/187152708787122941. [DOI] [PubMed] [Google Scholar]
- 59.Hudson CA, Christophi GP, Gruber RC, Wilmore JR, Lawrence DA, Massa PT. Induction of IL-33 expression and activity in central nervous system glia. J Leukoc Biol. 2008;84(3):631–643. doi: 10.1189/jlb.1207830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243(1):136–51. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 61.Lamkanfi M, Dixit VM. Modulation of inflammasome pathways by bacterial and viral pathogens. J Immunol. 2011;187(2):597–602. doi: 10.4049/jimmunol.1100229. [DOI] [PubMed] [Google Scholar]
- 62.Lamkanfi M, Dixit VM. IL-33 raises alarm. Immunity. 2009;31(1):5–7. doi: 10.1016/j.immuni.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 63.Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31(1):84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 64.Oboki K, Nakae S, Matsumoto K, Saito H. IL-33 and Airway Inflammation. Allergy Asthma Immunol Res. 2011;3(2):81–8. doi: 10.4168/aair.2011.3.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol. 2010;185(6):3472–80. doi: 10.4049/jimmunol.1000730. [DOI] [PubMed] [Google Scholar]
- 66.Louten J, Rankin AL, Li Y, Murphy EE, Beaumont M, Moon C, et al. Endogenous IL-33 enhances Th2 cytokine production and T-cell responses during allergic airway inflammation. Int Immunol. 2011;23(5):307–15. doi: 10.1093/intimm/dxr006. [DOI] [PubMed] [Google Scholar]
- 67.Hermans G, Stinissen P, Hauben L, Van den Berg-Loonen E, Raus J, Zhang J. Cytokine profile of myelin basic protein-reactive T cells in multiple sclerosis and healthy individuals. Ann Neurol. 1997;42(1):18–27. doi: 10.1002/ana.410420106. [DOI] [PubMed] [Google Scholar]
- 68.Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med. 2000;191(5):813–22. doi: 10.1084/jem.191.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu D, Jiang HR, Kewin P, Li Y, Mu R, Fraser AR, et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci U S A. 2008;105(31):10913–8. doi: 10.1073/pnas.0801898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sayed BA, Walker ME, Brown MA. Cutting edge: mast cells regulate disease severity in a relapsing-remitting model of multiple sclerosis. J Immunol. 2011;186(6):3294–8. doi: 10.4049/jimmunol.1003574. [DOI] [PubMed] [Google Scholar]
- 71.Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY. IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol. 2007;37(10):2779–86. doi: 10.1002/eji.200737547. [DOI] [PubMed] [Google Scholar]
- 72.Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, et al. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab Invest. 2007;87(10):971–8. doi: 10.1038/labinvest.3700663. [DOI] [PubMed] [Google Scholar]
- 73.Breij EC, Brink BP, Veerhuis R, van den Berg C, Vloet R, Yan R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol. 2008;63(1):16–25. doi: 10.1002/ana.21311. [DOI] [PubMed] [Google Scholar]
- 74.Christophi GP, Panos M, Hudson CA, Christophi RL, Gruber RC, Mersich AT, et al. Macrophages of multiple sclerosis patients display deficient SHP-1 expression and enhanced inflammatory phenotype. Lab Invest. 2009;89(7):742–59. doi: 10.1038/labinvest.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Joshi AD, Oak SR, Hartigan AJ, Finn WG, Kunkel SL, Duffy KE, et al. Interleukin-33 contributes to both M1 and M2 chemokine marker expression in human macrophages. BMC Immunol. 2010;11:52. doi: 10.1186/1471-2172-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183(10):6469–77. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 77.Andre R, Lerouet D, Kimber I, Pinteaux E, Rothwell NJ. Regulation of expression of the novel IL-1 receptor family members in the mouse brain. J Neurochem. 2005;95(2):324–30. doi: 10.1111/j.1471-4159.2005.03364.x. [DOI] [PubMed] [Google Scholar]
- 78.Jack C, Ruffini F, Bar-Or A, Antel JP. Microglia and multiple sclerosis. J Neurosci Res. 2005;81(3):363–73. doi: 10.1002/jnr.20482. [DOI] [PubMed] [Google Scholar]
- 79.Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(7):631–8. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lipton HL, Liang Z, Hertzler S, Son KN. A specific viral cause of multiple sclerosis: one virus, one disease. Ann Neurol. 2007;61(6):514–23. doi: 10.1002/ana.21116. [DOI] [PubMed] [Google Scholar]
- 81.Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11(1):63–9. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 82.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11(5):395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pang IK, Iwasaki A. Inflammasomes as mediators of immunity against influenza virus. Trends Immunol. 2011;32(1):34–41. doi: 10.1016/j.it.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rajan JV, Rodriguez D, Miao EA, Aderem A. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J Virol. 2011;85(9):4167–72. doi: 10.1128/JVI.01687-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Delarasse C, Gonnord P, Galante M, Auger R, Daniel H, Motta I, et al. Neural progenitor cell death is induced by extracellular ATP via ligation of P2X7 receptor. J Neurochem. 2009;109(3):846–57. doi: 10.1111/j.1471-4159.2009.06008.x. [DOI] [PubMed] [Google Scholar]
- 86.Domercq M, Perez-Samartin A, Aparicio D, Alberdi E, Pampliega O, Matute C. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia. 2010;58(6):730–40. doi: 10.1002/glia.20958. [DOI] [PubMed] [Google Scholar]
- 87.Marina-Garcia N, Franchi L, Kim YG, Miller D, McDonald C, Boons GJ, et al. Pannexin-1-Mediated Intracellular Delivery of Muramyl Dipeptide Induces Caspase-1 Activation via Cryopyrin/NLRP3 Independently of Nod2. J Immunol. 2008;180(6):4050–7. doi: 10.4049/jimmunol.180.6.4050. [DOI] [PubMed] [Google Scholar]
- 88.Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007;26(4):433–43. doi: 10.1016/j.immuni.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 89.Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. 2007;179(4):2051–4. doi: 10.4049/jimmunol.179.4.2051. [DOI] [PubMed] [Google Scholar]
- 90.Enoksson M, Lyberg K, Moller-Westerberg C, Fallon PG, Nilsson G, Lunderius-Andersson C. Mast cells as sensors of cell injury through IL-33 recognition. J Immunol. 2011;186(4):2523–8. doi: 10.4049/jimmunol.1003383. [DOI] [PubMed] [Google Scholar]
- 91.Gregory GD, Robbie-Ryan M, Secor VH, Sabatino JJ, Jr, Brown MA. Mast cells are required for optimal autoreactive T cell responses in a murine model of multiple sclerosis. Eur J Immunol. 2005;35(12):3478–86. doi: 10.1002/eji.200535271. [DOI] [PubMed] [Google Scholar]
- 92.Theoharides TC, Kempuraj D, Iliopoulou BP. Mast cells, T cells, and inhibition by luteolin: implications for the pathogenesis and treatment of multiple sclerosis. Adv Exp Med Biol. 2007;601:423–30. doi: 10.1007/978-0-387-72005-0_45. [DOI] [PubMed] [Google Scholar]
- 93.Cannella B, Raine CS. The adhesion molecule and cytokine profile of multiple sclerosis lesions [see comments] Annals of Neurology. 1995;37:424–435. doi: 10.1002/ana.410370404. [DOI] [PubMed] [Google Scholar]
- 94.Nile CJ, Barksby E, Jitprasertwong P, Preshaw PM, Taylor JJ. Expression and regulation of interleukin-33 in human monocytes. Immunology. 2010 doi: 10.1111/j.1365-2567.2009.03221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, et al. The dual function cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-kappaB-stimulated gene transcription. J Immunol. 2011;187(4):1609–16. doi: 10.4049/jimmunol.1003080. [DOI] [PubMed] [Google Scholar]